一种多取代异吲哚啉化合物及其制备方法
文献发布时间:2023-06-19 09:49:27
技术领域
本发明属于化学合成技术领域,具体涉及一种多取代吲哚啉化合物及其制备方法。
背景技术
异吲哚啉类化合物是一类十分重要的含氮生物碱,广泛存在于自然界中。研究表明很多异吲哚类化合物显示了广泛的生理和药理活性,如抗肿瘤、消炎等。此外异吲哚类化合物在药物方面也有十分重要的应用,如抗焦虑药物,(R)-JM1232,治疗风湿性关节炎、骨关节炎的药物indoprofen(吲哚布芬),治疗慢性瘙痒的药物serlopitant(司洛匹坦),治疗焦虑症和恐慌症的药物Pagoclone,天然产物Pastlachloride A则显示了较好的抗真菌活性,Taliscanine可用来治疗帕金森综合症等。异吲哚啉类化合物在色素、颜料、光电材料等方面也具有十分重要的应用。
目前,合成简单异吲哚类化合物的方法有很多,但通常需要使用昂贵的过渡金属催化剂如铑催化剂、钌催化剂、金催化剂等,并且合成方法通常需要多个反应步骤。而对于多环结构的异吲哚啉化合物尤其是大环多取代官能团化的异吲哚啉化合物的合成则未见报道。
发明内容
本发明的目的在于解决现有技术中在结构复杂的多取代异吲哚啉化合物方面的不足,提供一种多取代异吲哚啉化合物及其合成方法。本发明采用“一锅法”进行,不仅原料简单、所需的催化剂也比较便宜,而且操作简便、高效,产率较高且反应底物范围广。
技术方案
一种多取代异吲哚啉化合物,其结构式如式(I)所示:
其中,R为取代基团,选自甲基、苯基、卤素取代苯基、硝基取代苯基、烷基取代苯基或甲氧基取代苯基中的任意一种;X为杂原子,选自N、O、S中的至少一种;n为1至12之间的整数。
上述多取代异吲哚啉化合物的制备方法:将N-磺酰基取代的1,6-双炔、β-溴代环烯醛以及钯催化剂、铜催化剂在无水无氧条件下加入到有机溶剂中,以二异丙胺为碱,在65-75℃下搅拌反应,反应结束后,将得到的反应液分离纯化,得到多取代异吲哚啉化合物。
进一步,所述N-磺酰基取代的1,6-双炔的结构式如式(II)所示:
式(II)中R同式(I)中R对应一致。
进一步,所述β-溴代环烯醛的结构式如式(III)所示:
式(III)中X和n同式(I)中X和n对应一致。
进一步,所述钯催化剂选自醋酸钯[Pd(OAc)
进一步,所述的铜催化剂选自碘化亚铜、溴化亚铜、氯化亚铜或三(三苯基膦)溴化亚铜中的任意一种,更优选为三(三苯基膦)溴化亚铜。
进一步,所述有机溶剂为乙腈、二氯乙烷、甲苯、四氢呋喃或乙酸乙酯中的一种,更优选为乙酸乙酯,产率最高。
进一步,所述N-磺酰基取代的1,6-双炔、β-溴代环烯醛、钯催化剂、铜催化剂以及二异丙胺的摩尔比为100:240:1:4:500,该配比下,产率最高。
进一步,所述分离纯化方法为:将反应液冷却至室温后,用饱和氯化铵溶液淬灭,然后乙酸乙酯萃取两次,取有机相用无水硫酸镁干燥,过滤后浓缩,将粗产物通过硅胶柱层析,并以体积比为(2~15):100的乙酸乙酯:石油醚为洗脱剂进行分离纯化。
本发明的反应式如下:
反应机理如下:
首先N-磺酰基取代的1,6-双炔与β-溴代环烯醛在钯和铜催化剂催化作用下经过Sonogashira偶联,随后[4+2]环化得到环状联烯中间体,该中间体随后经过醛基迁移得到多取代异吲哚啉化合物。
本发明的有益效果:本发明以N-磺酰基取代的1,6-双炔和β-溴代环烯醛为原料,合成了一系列结构新颖的多取代异吲哚啉化合物,不仅原料简单、所需的催化剂也比较便宜,而且操作简便、高效,产率较高且反应底物范围广。相对于普通的异吲哚啉化合物,本发明制备的多取代的异吲哚啉化合物具有多个环及醛官能团,包括常规方法所不易制备的大环稠合异吲哚啉化合物,其结构更加复杂多样。本发明的多取代异吲哚啉化合物通过改性可作为二元醇应用于聚醚、聚酯、聚氨酯等高分子材料领域,此外由于具有大的共轭体系,在有机光电材料领域也具有潜在的应用价值。
附图说明
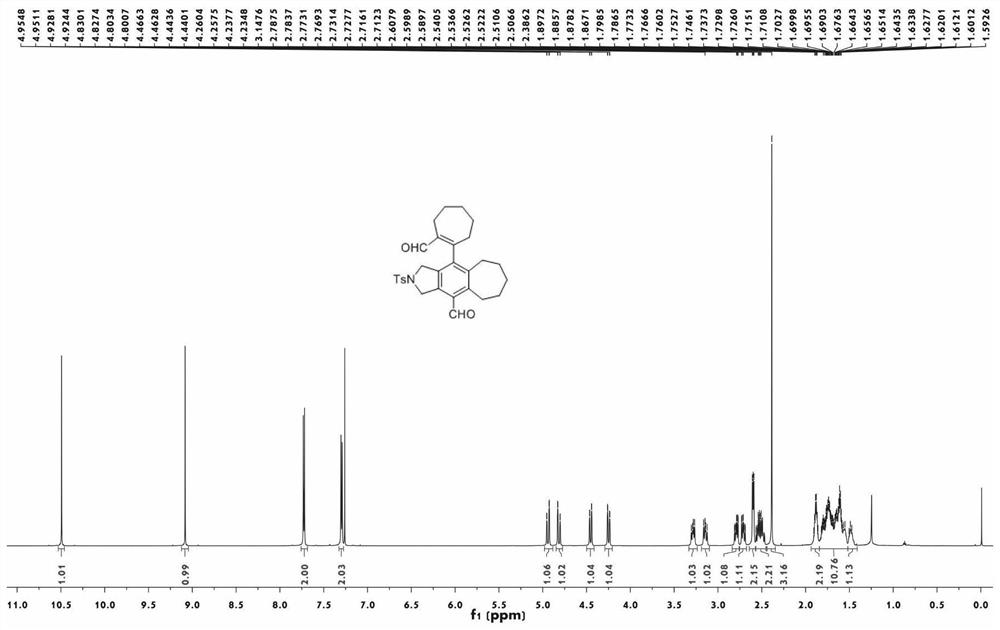
图1为本发明实施例1制备的化合物1aa的核磁共振氢谱图;
图2为本发明实施例1制备的化合物1aa的核磁共振碳谱图;
图3为本发明实施例1制备的化合物1aa的单晶衍射图。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
实施例1
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环庚烯醛3a(0.72mmol),在70℃搅拌反应12小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1aa(115mg,产率78%)。
实施例1制备的化合物1aa的核磁共振氢谱图如图1所示;实施例1制备的化合物1aa的核磁共振碳谱图如图2所示;实施例1制备的化合物1aa的单晶衍射图如图3所示。
实施例2
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对硝基苯磺酰基取代的1,6-双炔2b(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环庚烯醛3a(0.72mmol),在70℃搅拌反应15小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1ba(111mg,产率71%)。
实施例3
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环戊烯醛3b(0.72mmol),在70℃搅拌反应12小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1ab(106mg,产率81%)。
实施例4
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环己环烯醛3c(0.72mmol),在70℃搅拌反应15小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1ac(106mg,产率76%)。
实施例5
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环辛烯醛3d(0.72mmol),在70℃搅拌反应20小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1ad(97mg,产率62%)。
实施例6
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环十二烯醛3e(0.72mmol),在70℃搅拌反应24小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=8:100),即得目标产物1ae(104mg,产率55%)。
实施例7
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代环十五烯醛3f(0.72mmol),在70℃搅拌反应24小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=9:100),即得目标产物1af(112mg,产率52%)。
实施例8
一种多取代异吲哚啉化合物的制备方法:取25mL圆底烧瓶,氮气氛围下,往6mL乙酸乙酯中依次加入N-对甲基苯磺酰基取代的1,6-双炔2a(0.3mmol)、四(三苯基膦)钯(0.003mmol)、三(三苯基膦)溴化亚铜(0.012mmol),随后加入二异丙胺(1.5mmol),β-溴代六元环烯醛3g(0.72mmol),在70℃搅拌反应24小时,待反应结束,冷却至室温,饱和氯化铵溶液淬灭,乙酸乙酯萃取两次,收集有机相,无水硫酸镁干燥,过滤后浓缩,通过硅胶柱层析分离(洗脱剂为乙酸乙酯:石油醚=15:100),即得目标产物1ag(69mg,产率49%)。
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。