取代的嘌呤核苷酸
文献发布时间:2023-06-19 09:30:39
本申请是申请日为2016年3月7日、申请号为201680021594.1、名称为“用于HCV 治疗的β-D-2′-脱氧-2′α-氟-2′-β-C-取代的-2-改性的-N6-取代的嘌呤核苷酸”的专利申请的分案申请。
优先权
本申请要求2015年3月6日提交的U.S.S.N.62/129,319、2015年11月11日提交的U.S.S.N.62/253,958和2016年1月8日提交的U.S.S.N 62/276,597的优先权,将其中每篇的全部内容并入本文。
发明领域
本发明涉及核苷酸化合物和组合物,以及其用于治疗丙型肝炎病毒(“HCV”)的用途。
发明背景
丙型肝炎(HCV)是一种RNA单链病毒,并且是丙型肝炎病毒属的成员。据估计,75%的所有肝病病例是由HCV引起的。HCV感染可导致肝硬化和肝癌,如果保持发展,可导致需要肝移植的肝衰竭。全世界约1.7-2亿人受到感染,估计美国有3-4百万感染者。
RNA聚合酶是靶向RNA单链病毒的一种关键组分。HCV非结构蛋白NS5B RNA 依赖性RNA聚合酶是一种负责引发和催化病毒RNA合成的关键酶。因此,HCV NS5B 是一个目前抗-HCV药剂的药物研究和开发的有吸引力的靶标。存在两个主要亚类的NS5B 抑制剂:核苷类似物,其被合成代谢为其活性三磷酸盐(其充当聚合酶的可替代底物);和非核苷抑制剂(NNIs),其结合所述蛋白的变构区域。核苷或核苷酸抑制剂模拟天然聚合酶底物,并充当链终止剂。它们抑制RNA转录的启动和初生DNA链的延长。
除了靶向RNA聚合酶之外,在组合治疗中也可靶向其它RNA病毒蛋白质。例如,治疗方法的另一个靶标HCV蛋白是NS3/4A(一种丝氨酸蛋白酶)和NS5A(一种非结构蛋白,其是HCV复制酶的一种必需组分,并且对于细胞途径发挥一系列作用)。.
在2013年12月,批准了第一个核苷NS5B聚合酶抑制剂索非布韦 (sofosbuvir)(
2’-脱氧-2’-α-氟-β-C-甲基尿嘧啶核苷-5’-三磷酸酯。
2014年,美国FDA批准了
2014年,美国FDA还批准了AbbVie’s VIEKIRA Pak
2015年7月,美国FDA批准了Technivie
2015年10月,美国FDA警告HCV治疗Viekira Pak和Technivie可导致严重的肝损伤,主要在患有潜在晚期肝病的患者中,并且要求向说明书中加入关于安全性的附加信息。.
其它目前批准用于HCV的治疗剂包括干扰素α-2b或PEG基化的干扰素α-2b
另一种NS5B聚合酶抑制剂目前正在研发中。默克公司正在开发尿苷核苷酸前药MK-3682(之前的Idenix IDX21437)。该药物目前处于II期组合试验中。
描述了用于治疗黄病毒科(包括HCV)的核苷聚合酶抑制剂的美国专利和WO申请包括如下公司提交的那些:Idenix Pharmaceuticals(6,812,219;6,914,054;7,105,493; 7,138,376;7,148,206;7,157,441;7,163,929;7,169,766;7,192,936;7,365,057;7,384,924; 7,456,155;7,547,704;7,582,618;7,608,597;7,608,600;7,625,875;7,635,689;7,662,798; 7,824,851;7,902,202;7,932,240;7,951,789;8,193,372;8,299,038;8,343,937;8,362,068; 8,507,460;8,637,475;8,674,085;8,680,071;8,691,788,8,742,101,8,951,985;9,109,001;9,243,025;US2016/0002281;US2013/0064794;WO/2015/095305;WO/2015/081133; WO/2015/061683;WO/2013/177219;WO/2013/039920;WO/2014/137930;WO/2014/052638; WO/2012/154321);默克公司(6,777,395;7,105,499;7,125,855;7,202,224;7,323,449; 7,339,054;7,534,767;7,632,821;7,879,815;8,071,568;8,148,349;8,470,834;8,481,712; 8,541,434;8,697,694;8,715,638,9,061,041;9,156,872和WO/2013/009737);埃默里大学(Emory University)(6,348,587;6,911,424;7,307,065;7,495,006;7,662,938;7,772,208; 8,114,994;8,168,583;8,609,627;US 2014/0212382;和WO2014/1244430);吉利德科学/ 药物公司(Gilead Sciences/Pharmasset Inc).(7,842,672;7,973,013;8,008,264;8,012,941; 8,012,942;8,318,682;8,324,179;8,415,308;8,455,451;8,563,530;8,841,275;8,853,171; 8,871,785;8,877,733;8,889,159;8,906,880;8,912,321;8,957,045;8,957,046;9,045,520; 9,085,573;9,090,642;和9,139,604)和(6,908,924;6,949,522;7,094,770;7,211,570;7,429,572; 7,601,820;7,638,502;7,718,790;7,772,208;RE42,015;7,919,247;7,964,580;8,093,380; 8,114,997;8,173,621;8,334,270;8,415,322;8,481,713;8,492,539;8,551,973;8,580,765; 8,618,076;8,629,263;8,633,309;8,642,756;8,716,262;8,716,263;8,735,345;8,735,372; 8,735,569;8,759,510和8,765,710);罗氏公司(Hoffman La La-Roche)(6,660,721),罗氏(Roche)(6,784,166;7,608,599,7,608,601和8,071,567);阿里奥斯生物制药公司(AliosBioPharma Inc)(8,895,723;8,877,731;8,871,737,8,846,896,8,772,474;8,980,865;9,012,427; US 2015/0105341;US 2015/0011497;US 2010/0249068;US2012/0070411;WO2015/054465; WO 2014/209979;WO 2014/100505;WO 2014/100498;WO 2013/142159;WO2013/142157; WO 2013/096680;WO 2013/088155;WO 2010/108135),EnantaPharmaceuticals(US 8,575,119;8,846,638;9,085,599;WO 2013/044030;WO 2012/125900),Biota(7,268,119; 7,285,658;7,713,941;8,119,607;8,415,309;8,501,699和8,802,840),生物水晶公司(Biocryst Pharmaceuticals)(7,388,002;7,429,571;7,514,410;7,560,434;7,994,139;8,133,870; 8,163,703;8,242,085和8,440,813)、Alla Chem,LLC(8,889,701和WO 2015/053662)、 Inhibitex(8,759,318和WO/2012/092484)、杨森(Janssen Products)(8,399,429;8,431,588, 8,481,510,8,552,021,8,933,052;9,006,29和9,012,428)、the University of Georgia Foundation (6,348,587;7,307,065;7,662,938;8,168,583;8,673,926,8,816,074;8,921,384和8,946,244)、RFS Pharma,LLC(8,895,531;8,859,595;8,815,829;8,609,627;7,560,550;US 2014/0066395; US 2014/0235566;US 2010/0279969;WO/2010/091386和WO 2012/158811)、University CollegeCardiff Consultants Limited(WO/2014/076490、WO 2010/081082;WO/2008/062206)、Achillion Pharmaceuticals,Inc.(WO/2014/169278和WO 2014/169280)、CocrystalPharma, Inc.(US 9,173,893)、Katholieke Universiteit Leuven(WO 2015/158913)、Catabasis(WO 2013/090420)和明尼苏达大学董事会(the Regents of the Universityof Minnesota)(WO 2006/004637)。
尽管如此,仍然存在开发安全、有效且良好耐受的抗-HCV治疗剂的强烈医学需要。预期重点需求是抗药性。对于感染所有HCV基因型的患者,更有效的直接作用抗病毒药可以显著地缩短治疗持续时间和提高顺应性和SVR速率。.
因此,本发明的一个目的是提供用于治疗和/或预防HCV感染的化合物、药物组合物、以及方法和用途。
发明简述
已经发现式I、式II、式III、式IV、式V、式VI、式VII的化合物,包括β-D-2′- 脱氧-2′-α-氟-2′-β-C-取代的-N
公开的核苷酸包括具有体外抗HCV的纳摩尔活性且治疗指数范围达25,000或更多的那些。
令人惊奇地,在本发明之前,所公开化合物的母体N
然而,现在发现本发明的化合物被合成代谢为N
特别地,已经发现β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
化合物5-2(表7)
同样,母体核苷β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
在另一个实施例中,在复制子试验中,化合物((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N- 甲基-N-环丙基-氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯显示出EC
表7中的化合物27和下述结构。.
如上所述,呈氨基磷酸酯的β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
因此,在一个实施方案中,本发明为:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
在一个实施方案中,本发明为:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
R
R
R
R
R
例如,包括但不限于下述实施方式:氯、溴、氟、氰基、叠氮基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基和正戊基、1,1-二甲基丙基、2,2-二甲基丙基、3-甲基丁基、1-甲基丁基、1-乙基丙基、乙烯基、烯丙基、1-丁炔基、2-丁炔基、乙炔基、环丙基、环丁基、环戊基、环己基、-(CH
R
R
R
R
R
R
R
R
R
R
x为1、2或3。
β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
2’-脱氧-2’-α-氟-2’-β-C-取代的-N
尽管有大量的抗病毒核苷文献和专利申请,但是还没有公开如本文所述的2’-脱氧 -2’-α-氟-2’-β-甲基-N
除非另有说明,否则本文所述的化合物以β-D-构型提供。同样,当以磷酰胺或硫代氨基磷酸酯形式时,氨基酸部分可以为L-或D-构型。在另一个实施方案中,所述化合物可以以β-L构型提供。同样,显示出手性的任何取代基团都可以以外消旋的、对映异构的、非对映异构的形式或其任何混合物提供。当其中磷显示出手性的氨基磷酸酯、硫代氨基磷酸酯或其它稳定的磷前药用作R
因此,本发明包括式I-VII的化合物或其可药用组合物、盐或前药,如本文所述:
在一个特定的实施方案中,母体核苷,即其中R
化合物、方法和组合物通过给药有效量的所述化合物或其可药用盐而被提供用于治疗感染HCV病毒的宿主。
所述化合物和组合物也可以用于治疗相关病症,例如抗-HCV抗体阳性和抗原阳性病症、基于病毒的慢性肝炎、由晚期丙型肝炎导致的肝癌、肝硬化、慢性或急性丙型肝炎、暴发性丙型肝炎、慢性持续性丙型肝炎和基于抗-HCV的疲劳。所述化合物或包括该化合物的制剂也可以预防性用于预防或限制抗-HCV抗体或抗原阳性或已经暴露于丙型肝炎的个体的临床病症的进展。
在另一个实施方案中,公开了式Ia的化合物:
其中:
Y、R
在式Ia的一个实施方案中,R
在式Ia的一个实施方案中,当Y为NR
在式Ia的一个实施方案中,当Y为NR
在式Ia的一个实施方案中,当Y为NR
在另一个实施方案中,公开了式Ib的化合物
其中:
Y、R
在式Ib的一个实施方案中,R
在式Ib的一个实施方案中,当Y为NR
在式Ib的一个实施方案中,当Y为NR
在一个实施方案中,公开了式II的化合物:
其中:
Y、R
在另一个实施方案中,公开了式IIa的化合物:
其中:
Y、R
在另一个实施方案中,公开了式IIb的化合物
其中:
Y、R
在一个实施方案中,公开了式III的化合物:
其中变量Y、R
在一个实施方案中,公开了式IV的化合物:
其中变量Y、R
在一个实施方案中,公开了式V的化合物:
其中变量Y、R
在一个实施方案中,公开了式VI的化合物::
其中:
R
变量Y、R
在一个实施方案中,公开了式VII的化合物::
其中变量Y、R
任一个上述结构式中的磷都可以是手性的,因此可以提供为R或S对映异构体或其混合物(包括外消旋混合物)。
将化合物5分离成对映异构体化合物5-1和5-2。化合物5-2还通过手性合成制备,指定为化合物24。
在一个实施方案中,化合物、方法和组合物提供用于治疗感染或暴露于本文所述丙型肝炎的宿主。本发明的化合物可以以有效量单独或与另外的抗-HCV药物组合给药以治疗感染的宿主。在一些实施方案中,给药调节相同或不同途径或抑制病毒中的不同靶点的药物组合是有用的。由于所公开的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
本发明的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
附图简述
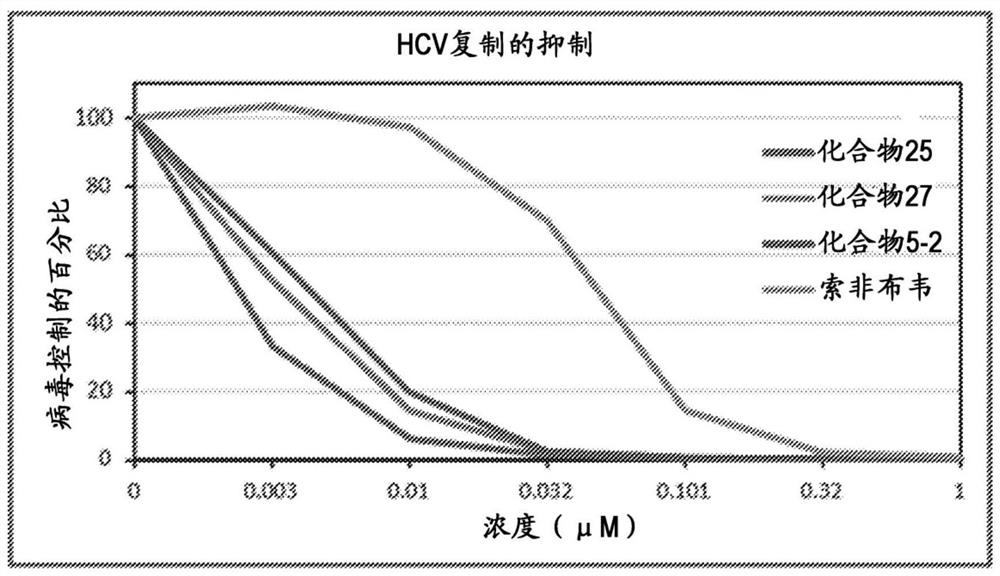
图1为示出如实施例9公开的使用Phenominex Luna柱分离化合物5的立体异构体的半制备运行的样品色谱图。y轴以mAU显示,x轴以分钟测量。图2为化合物5-2(表 7)和索非布韦的HCV复制抑制曲线图。化合物5-2具有EC
图3为化合物25(表7)和索非布韦的HCV复制抑制曲线图。如实施例27所述,化合物25具有EC
图4为化合物5-2、25、27(表7)和索非布韦的抗-HCV活性的批内(intra-assay)试验比较。y-轴为病毒控制的百分比,x-轴为药物浓度(μM)。参见,实施例27。
图5为显示化合物5-2;化合物5-2的N
图6为显示在人肝S9级分的存在下,2’-脱氧-2’-α-氟-2’-β-甲基-N
图7为显示在人肝S9级分的存在下,化合物5-2;化合物5-2的N
图8显示在人肝细胞中产生的主要化合物25的代谢产物。x轴为培养时间(小时)。y轴为细胞内浓度(pmol/10
图9显示在人肝细胞中产生的主要化合物27的代谢产物。x轴为培养时间(小时)。y轴为细胞内浓度(pmol/106细胞)。参见实施例33。
图10显示在人肝细胞中产生的主要化合物5-2的代谢产物。x轴为培养时间(小时)。 y轴为细胞内浓度(pmol/10
图11为显示化合物25、27和5-2的活化途径的图。如可以看到的,化合物25、27 和5-2被转化成其相应的单磷酸酯类似物,接着被代谢成常见MP类似物;β-D-2’-脱氧-2’-α-氟-2’-β-甲基-鸟嘌呤单磷酸酯。然后,该单磷酸酯逐步磷酸化成活性三磷酸酯:β-D-2’-脱氧-2’-α-氟-2’-β-甲基-鸟嘌呤三磷酸酯。参见实施例33。
发明详述
本文公开的本发明为用于治疗感染HCV病毒或暴露于HCV病毒的人和其它宿主动物的化合物、方法和组合物,其包括给药任选在可药用载体中的有效量的如本文所述的式I-VII的化合物或其可药用盐或前药。本发明的化合物具有抗病毒活性,或者代谢为显示出这种活性的化合物。
所述化合物和组合物也可以用于治疗涉及HCV病毒暴露后果的病症或作为该后果存在的病症。例如,所述活性化合物可用于治疗HCV抗体阳性和HCV抗原阳性病症、基于病毒的慢性肝炎、由晚期丙型肝炎导致的肝癌、肝硬化、急性丙型肝炎、暴发性丙型肝炎、慢性持续性丙型肝炎和基于抗-HCV的疲劳。在一个实施方案中,所述化合物或包括该化合物的制剂也可以预防性用于预防或阻止HCV抗体或HCV抗原阳性或已经暴露于丙型肝炎的个体的临床病症的进展。
特别地,已经发现β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
尽管有大量的抗病毒核苷文献和专利申请,但是没有特别地公开2’-脱氧-2’-α-氟 -2’-β-甲基-N
除非另有说明,否则本文所述的化合物以β-D-构型提供。在另一个实施方案中,所述化合物可以以β-L构型提供。同样,显示出手性的任何取代基团都可以以外消旋的、对映异构体的、非对映异构体的形式或其任何混合物提供。当其中磷显示出手性的氨基磷酸酯、硫代氨基磷酸酯或其它稳定的磷前药用作R
本发明包括下述特征:
(a)如本文所述的式I-VII的化合物,及其可药用盐和前药;
(b)如本文所述的式I-VII,及其可药用盐和前药,用于治疗或预防丙型肝炎病毒感染;
(c)式I-VII及其可药用盐和前药在制备用于治疗丙型肝炎病毒感染的药物中的用途;
(d)一种制备预期用于治疗丙型肝炎病毒感染的治疗用途的药物的方法,特征在于在该制备方法中使用如本文所述的式I-VII;
(e)药物制剂,包括有效的宿主治疗量的式I-VII或其可药用盐或前药与可药用载体或稀释剂;
(f)如本文所述的式I-VII,基本上不存在所述化合物的立体异构体或基本上分离了其它化学实体;和
(g)用于制备包含有效量的如本文所述的式I-VII的治疗产品的方法。
I.本发明的2’-脱氧-2’-α-氟-2’-β-C-取代的-2-改性的-N
本发明的活性化合物为式I中所述的那些,其可以以可药用组合物、其盐或前药提供:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
一种稳定的磷酸酯前药为可以递送单、二或三磷酸酯的任何部分。
在另一个实施方案中,公开了式Ia的化合物:
其中:
Y、R
在另一个实施方案中,公开了式Ib的化合物
其中:
Y、R
在另一个实施方案中,所述化合物为根据式Ic的化合物:
其中:
R
R
R
R
R
Y、R
在一个实施方案中,公开了式II的化合物:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
R
R
R
R
R
例如,包括但不限于下述实施方式:氯、溴、氟、氰基、叠氮基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基和正戊基、1,1-二甲基丙基、2,2-二甲基丙基、3-甲基丁基、1-甲基丁基、1-乙基丙基、乙烯基、烯丙基、1-丁炔基、2-丁炔基、乙炔基、环丙基、环丁基、环戊基、环己基、-(CH
R
R
R
R
R
R
R
R
R
R
x为1、2或3。
在另一个实施方案中,公开了式IIa的化合物:
其中:
Y、R
在另一个实施方案中,公开了式IIb的化合物:
其中:
Y、R
在一个典型的实施方案中,所述化合物为相对于相应核苷(即,天然存在构型)的β-D 异构体。在一个可替代的构型中,该化合物提供为β-L异构体。所述化合物通常为至少90%不含相反的对映异构体,并且可以为至少98%、99%或甚至100%不含相反对映异构体。除非另有描述,否则所述化合物为至少90%不含相反对映异构体。
在另一个实施方案中,所述化合物为根据式III的化合物:
其中:
R
R
R
R
R
Y、R
在一个实施方案中,公开了式IV的化合物:
其中变量Y、R
在一个实施方案中,公开了式V的化合物:
其中变量Y、R
在另一个实施方案中,化合物、方法和组合物提供用于治疗感染或暴露于丙型肝炎的宿主。
在一个实施方案中,公开了式VI的化合物:
其中:
R
变量Y、R
在一个实施方案中,公开了式VII的化合物:
其中变量Y、R
β-D-2’-脱氧-2’-α-氟-2’-β-C-取代的-N
β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
方案1
β-D-2’-脱氧-2’-α-氟-2’-β-甲基-N
方案2
方案3
稳定的磷酸酯前药
稳定的磷酸酯前药是可以体内递送单磷酸酯、二磷酸酯或三磷酸酯的部分。例如,McGuigan在美国专利Nos.:8,933,053;8,759,318;8,658,616;8,263,575;8,119,779; 7,951,787和7,115,590中公开了氨基磷酸酯。Alios在US 8,895,723和8,871,737(通过引用并入本文)中公开了硫代氨基磷酸酯。Alios还在美国专利No.8,772,474(通过参考引入本文)中公开了环核苷酸。Idenix在WO 2013/177219(通过引用并入本文)中公开了环状氨基磷酸酯和氨基磷酸酯/SATE衍生物。Idenix也在WO 2013/039920(通过引用并入本文)中公开了取代的羰氧基甲基氨基磷酸酯化合物。Hostetler公开了脂质磷酸酯前药,参见例如US7,517,858。Hostetler还公开了膦酸酯前药的共轭物,参见例如US 8,889,658; 8,846,643;8,710,030;8,309,565;8,008,308;和7,790,703。Emory University在WO 2014/124430中公开了核苷酸鞘氨基醇和脂质衍生物。RFS Pharma在WO 2010/091386中公开了嘌呤核苷单磷酸酯前药。Cocrystal Pharma Inc.也在美国专利No.:9,173,893(通过引用并入本文)中公开了嘌呤核苷单磷酸酯前药。HepDirect
在一个可替代的实施方案中,稳定的磷酸酯前药包括,但不限于在美国专利No.9,173,893和美国专利No.8,609,627(通过引用并入本文)中所述的那些,包括其制备方法。例如,式I-V的5’-前药可以由下述基团表示:
在一个可替代的实施方案中,式I-V的3’,5’-前药可以由下述基团表示:
其中:
当手性存在于亚磷(phosphorous)中心时,其可以是完全或部分的R
Z为O或S;
R
其中R
R
(a)OR
R
(b)
(c)D-氨基酸或L-氨基酸的酯
其中R
R
(d)R
其中R
(e)R
其中R
R
所述化合物可以例如通过制备5’-OH类似物,然后将这些化合物转化成单磷酸酯类似物来制备。
实施方案
特别的实施方案:
(i)在式Ia中,Y为NR
(ii)在式Ia中,Y为NR
(iii)在式Ia中,Y为NR
(iv)在式Ia中,Y为NR
(v)在式Ia中,Y为NR
(vi)在式Ia中,Y为NR
(vii)在式Ia中,Y为NR
(viii)在式Ia中,Y为NR
(ix)在式Ia中,Y为NR
(x)在式Ia中,Y为NR
(xi)在式Ia中,Y为NR
(xii)在式Ia中,Y为NR
(xiii)在式Ia中,Y为NR
(xiv)在式Ia中,Y为NR
(xv)在式Ia中,Y为NR
(xvi)在式Ia中,Y为NR
(xvii)在式Ia中,Y为NR
(xviii)在式Ia中,Y为NR
(xix)在式Ia中,Y为NR
(xx)在式Ia中,Y为NR
(xxi)在式Ia中,Y为NR
(xxii)在式Ia中,Y为NR
(xxiii)在式Ia中,Y为NR
(xxiv)在式Ia中,Y为NR
(xxv)在式Ia中,Y为NR
(xxvi)在式Ia中,Y为NR
(xxix)在式Ia中,Y为NR
(xxx)在式Ia中,Y为NR
(xxxi)在式Ia中,Y为NR
(xxxii)在式Ia中,Y为NR
(xxxiii)在式Ia中,Y为NR
(xxxiv)在式Ia中,Y为NR
(xxxv)在式Ia中,Y为NR
(xxxvi)在式Ib中,Y为NR
(xxxvii)在式Ib中,Y为NR
(xxxviii)在式Ib中,Y为NR
(xxxix)在式Ib中,Y为NR
(xl)在式Ib中,Y为NR
(xli)在式Ib中,Y为NR
(xlii)在式Ib中,Y为NR
(xliii)在式Ib中,Y为NR
(xliv)在式Ib中,Y为NR
(xlv)在式Ib中,Y为NR
(xlvi)在式Ib中,Y为NR
(xlvii)在式Ib中,Y为NR
(xlviii)在式Ib中,Y为NR
(xlix)在式Ib中,Y为NR
(l)在式Ib中,Y为NR
(li)在式Ib中,Y为NR
(lii)在式Ib中,Y为NR
(liii)在式Ib中,Y为NR
(liv)在式Ib中,Y为NR
(lv)在式Ib中,Y为NR
(lvi)在式Ia中,Y为NR
在任一个上述的可替代实施方案中,所述化合物具有R
在化合物(i)至(lvi)的可替代实施方案中,在式I-VII中使用L-核苷。
在一个可替代的实施方案中,式I中R
在一个可替代的实施方案中,式I中R
在一个可替代的实施方案中,式I中R
在一个实施方案中,提供式Ia的化合物。式Ia的化合物的非限制性实例包括:
在一个实施方案中,提供式Ia的硫代氨基磷酸酯。式Ia的硫代氨基磷酸酯的非限制性实例包括,但不限于:
在一个实施方案中,提供式Ia的稳定的磷酸酯前药。式Ia的稳定的磷酸酯前药的非限制性实例示例如下:
在另一个实施方案中,提供式Ia的化合物。式Ia的化合物的非限制性实例包括:
在一个实施方案中,提供式Ia的硫代氨基磷酸酯。式Ia的硫代氨基磷酸酯的非限制性实例包括,但不限于:
在一个实施方案中,提供式Ia的稳定的磷酸酯前药。式Ia的稳定的磷酸酯前药的非限制性实例示例如下::
在一个实施方案中,提供式II的化合物。式II的化合物的非限制性实例包括:
在一个实施方案中,提供式I的化合物。式I的化合物的非限制性实例包括:
在一个实施方案中,提供式II的化合物。式II的化合物的非限制性实例包括:
在一个实施方案中,R
在一个实施方案中,提供式II的化合物。式II的化合物的非限制性实例包括:
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在一个实施方案中,提供式II的化合物。式II的化合物的非限制性实例包括:
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
在某些实施方案中,R
II.定义
使用下述术语描述本发明。当术语在本文没有特别地定义的情况下,该术语被在上下文中应用该术语描述本发明的普通技术人员赋予本领域公认的含义。
术语“烷基”在其上下文中应当指直链或支链完全饱和的烃基或烷基,其可以任选地被取代(例如,被卤素(包括F)取代)。例如,烷基可以具有1、2、3、4、5、6、7或8 个碳原子(即,C
术语“烯基”指在相邻碳原子之间包含至少一个双键的非芳香烃基和如本文以其它方式所述的类似于烷基的结构。例如,烯基基团可以具有2至8个碳原子(即,C
术语“炔基”指在相邻碳原子之间包含至少一个三键的非芳香烃基和如本文以其它方式所述的类似于烷基的结构。例如,炔基基团可以具有2至8个碳原子(即,C2-C8炔),或2至4个碳原子(即,C
术语“酰基“指–C(O)R部分,其中所述羰基部分键合R,例如-C(O)烷基。R可以选自烷氧基、烷基、环烷基、低级烷基(即,C
术语“低级酰基”指其中羰基部分为低级烷基的酰基基团(即,C
术语“烷氧基“指基团–OR’,其中–OR’为-O-烷基、-O-烯基、-O-炔基、-O-(C
术语“氨基”指基团–NH
术语“氨基酸"或“氨基酸残基"指D-或L-天然或非天然存在的氨基酸。代表性的氨基酸包括,但不限于丙氨酸、β-丙氨酸、精氨酸、天门冬酰胺、天门冬氨酸、半胱氨酸、胱氨酸、谷氨酸、谷氨酰胺、甘氨酸、苯丙氨酸、组氨酸、异亮氨酸、赖氨酸、亮氨酸、甲硫氨酸、脯氨酸、丝氨酸、苏氨酸、缬氨酸、色氨酸或酪氨酸等。
术语“叠氮基"指基团–N
术语“芳基”或“芳香族”在上下文中指取代的(如本文在别处所述的)或未取代的具有单环(例如,苯基或苄基)或稠环(例如,萘基、蒽基、菲基等)的一价芳香族基团,并且可以在环上任何可获得的稳定的位点或如所呈现的化学结构中以其它方式指示的位点结合到根据本发明的化合物。芳基基团可以任选地被取代,如本文所述。
“环烷基”、“碳环”或“碳环基”指呈单环的具有3至7个碳原子的饱和的(即,环烷基) 或部分不饱和的(例如,环烯基、环二烯基等)环。单环碳环具有3至7个环原子,仍然更通常5或6个环原子。环烷基基团的非限制性实例包括环丙基、环丁基、环戊基、1-环戊 -1-烯基、1-环戊-2-烯基、1-环戊-3-烯基、环己基、l-环己-1-烯基、l-环己-2-烯基和1- 环己-3-烯基。
术语“氰基”指基团–CN。
术语“卤素”或“卤代”指氯、溴、氟或碘。
杂芳基环系统为环(单环)中具有一个或多个氮、氧或硫原子的饱和的或不饱和的环,包括但不限于咪唑、呋喃基(furyl)、吡咯、呋喃基(furanyl)、噻吩、噻唑、吡啶、嘧啶、嘌呤、吡嗪、三唑、噁唑或稠环系统例如吲哚、喹啉等,等等,其可任选地被取代,如上所述。杂芳基基团包括含氮杂芳基基团,例如吡咯、吡啶、吡啶酮、哒嗪、嘧啶、吡嗪、吡唑、咪唑、三唑、三嗪、四唑、吲哚、异吲哚、吲嗪、嘌呤、吲唑、喹啉、异喹啉、喹嗪、酞嗪、萘啶、喹喔啉、喹唑啉、噌啉、蝶啶、咪唑并吡啶、咪唑并三嗪、吡嗪并哒嗪、吖啶、菲啶、咔唑、咔唑啉(carbazoline)、萘嵌间二氮杂苯、菲咯啉、phenacene、噁二唑、苯并咪唑、吡咯并吡啶、吡咯并嘧啶和吡啶并嘧啶;含硫芳香族杂环例如噻吩和苯并噻吩;含氧芳香族杂环例如呋喃、吡喃、环五吡喃、苯并呋喃和异苯并呋喃;包括两个或多个选自氮、硫和氧杂原子的芳香族杂环,例如噻唑、噻二唑、异噻唑、苯并噁唑、苯并噻唑、苯并噻二唑、吩噻嗪、异噁唑、呋咱、吩噁嗪、吡唑并噁唑、咪唑并噻唑、噻吩并呋喃、呋喃并吡咯、吡啶并噁嗪、呋喃并吡啶、呋喃并嘧啶、噻吩并嘧啶和噁唑等,其都可以任选地被取代。
术语“杂环(heterocycle)”或“杂环(heterocyclo)”指包含至少一个杂原子(即O、N 或S)的环基团,并且可以是芳香族(杂芳基)或非芳香族的。本发明中使用的示例性的非芳香族杂环基团包括,例如吡咯烷基、哌啶基、哌嗪基、N-甲基哌嗪基、咪唑啉基、吡唑烷基、咪唑烷基、吗啉基、四氢吡喃基、氮杂环丁烷基、氧杂环丁烷基、氧硫杂环戊基、吡啶酮、2-吡咯烷酮、乙烯脲(ethyleneurea)、1,3-二氧戊环、1,3-二噁烷、1,4-二噁烷、邻苯二甲酰亚胺和琥珀酰亚胺等,其全部都可以任选地被取代。
术语“羟基"指基团–OH。
术语“硝基"指基团–NO
术语"可药用盐"或"前药"在整个说明书中描述β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
“可药用前药”指在宿主中代谢(例如水解或氧化)形成本发明的化合物的化合物。前药的典型的实例包括在活性化合物的功能部分上具有生物学不稳定的保护基的化合物。前药包括可以氧化、还原、胺化、脱氨基化、羟基化、脱羟基化、水解、脱水、烷基化、脱烷烃化、酰化、脱酰化、磷酸化、脱磷酸化、硫代氨基磷酸化、脱硫代氨基磷酸化、氨基磷酸化或脱氨基磷酸化产生活性化合物的化合物。本发明的化合物具有针对HCV的抗病毒活性,或者代谢为显示出这样的活性的化合物。β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
术语“膦酸”指基团–P(O)(OH)
在一个实施方案中,术语嘌呤或嘧啶碱基包括,但不限于腺嘌呤、N
术语“取代的”或“任选取代的”指可以具有至少一个另外的取代基的部分,所述取代基包括但不限于卤素(F、Cl、Br、I)、OH、苯基、苄基、N
术语“磺酸酯”由包括R
术语“磺酸”指基团–SO
术语“硫醇”指基团–SH。
如本文使用的术语“氮-保护基”指共价连接氮的部分,当适合时其可以被除去且通常用氢替代。例如,氮保护基可以是在给药至宿主之后在体内除去、在体外由细胞除去、或其可以在制备过程期间被除去的基团。用于本发明的合适的氮保护基为由Greene和Wuts在Protective Groups in Organic Synthesis(1991)New York,John Wiley and Sons,Inc中描述的。
如本文使用的术语“氧-保护基”指共价连接氧的部分,当适合时其可以被除去且通常用氢替代。例如,氧保护基可以是在给药至宿主之后在体内除去、在体外由细胞除去、或其可以在制备过程期间被除去的基团。用于本发明的合适的氧保护基为由Greene和Wuts在Protective Groups in Organic Synthesis(1991)New York,John Wiley and Sons,Inc中描述的。
“磷酸酯(phosphate)”指基团–OP(O)(OH)
除非另有说明,否则“磷酸酯(phosphate ester)”指单、二和三磷酸酯。
术语“氨基磷酸酯(phosphoamidate)”、“氨基磷酸酯(phosphoramidate)”或“氨基磷酸酯 (phosphoroamidate)”为具有结合三个氧基和一个胺(其可以任选地被取代)的磷的部分。用于本发明的合适的氨基磷酸酯为由Madela,Karolina和McGuigan在2012,“Progress in the development of anti-hepatitis C virus nucleoside andnucleotide prodrugs”,Future Medicinal Chemistry 4(5),第625-650页10:1021/jm300074y,以及Dominique,McGuigan和Balzarini 在2004,“Aryloxy PhosphoramidateTriesters as Pro-Tides”,Mini Reviews in Medicinal Chemistry 4(4),第371-381页中描述的。用于本发明的另外的氨基磷酸酯为在美国专利号: 5,233,031、7,115,590、7,547,704、7,879,815、7,888,330、7,902,202、7,951,789、7,964,580、 8,071,568;8,148,349、8,263,575、8,324,179、8,334,270、8,552,021、8,563,530、8,580,765、 8,735,372、8,759,318;EP 2120565;EP 1143995;6,455,513;和8,334,270中描述的。其它氨基磷酸酯为在本发明的背景中描述的核苷专利中描述的。
用于本发明的氨基磷酸酯基团包括下述结构的那些:
用于本发明的其它氨基磷酸酯基团包括下述结构的那些:
其中:
R
R
其中:
R
R
B’为
其中:
R
R
R
R
R
R
B’为
其中:
R
R
R
优选的R
术语氨基磷酸酯在整个说明书中用于描述存在于核苷化合物的呋喃糖环的5’或3’位并形成核苷化合物的前药形式的基团。在一个实施方案中,氨基磷酸酯可以同时存在于核苷化合物的呋喃糖环的5’和3’位,并形成该核苷化合物的前药形式。在另一个实施方案中,存在于核苷的呋喃糖环的5’位的氨基磷酸酯可以通过与该核苷化合物的呋喃糖环的3’位的3’-羟基取代基形成键而形成环状氨基磷酸酯化合物,并形成该核苷化合物的前药形式。
术语“硫代氨基磷酸酯(thiophosphoamidate)”、“硫代氨基磷酸酯(phosphoramidate)”或"硫代氨基磷酸酯(phosphoroamidate)"为具有结合硫、两个氧基和一个胺(其可以任选地被取代)的磷的部分。用于本发明的硫代氨基磷酸酯描述在US专利No.8,772,474和WO 2012/040124中。用于本发明的硫代氨基磷酸酯基团包括下述结构的那些:
其它硫代氨基磷酸酯包括下述结构的那些:
其中:
R
R
其中:
R
R
B’为
其中:
R
R
R
R
R
R
B’为
R
优选的R
硫代氨基甲酸酯可以存在于核苷化合物的呋喃糖环的5’或3’位,形成该核苷化合物的前药形式。在一个实施方案中,硫代氨基甲酸酯可以同时存在于核苷化合物的呋喃糖环的5′和3′位,并形成该核苷化合物的前药形式。在另一个实施方案中,存在于核苷的呋喃糖环的5’位的硫代氨基磷酸酯可以通过与该核苷化合物的呋喃糖环的3’位的3’-羟基取代基形成键而形成环状硫代氨基磷酸酯化合物,并形成该核苷化合物的前药形式。
如在本发明的上下文中使用的术语“D-构型”指与非天然存在核苷或"L"构型相反的、模拟糖部分的天然结构的主要构型。术语“β”或“β端基异构体”用于说明核苷类似物,其中核苷碱被装配(配置)在该核苷类似物中呋喃糖部分的平面之上。
术语“共给药”和“共同给药”或组合治疗用于描述给药至少一种根据本发明的2′-脱氧-2′-α-氟-2′-β-C-核苷化合物与至少一种其它活性剂的组合,例如当合适时至少一种另外的抗-HCV剂,包括本文公开的其它2’-脱氧-2’-α-氟-2’-β-C-核苷试剂。共同给药的时机最好由治疗患者的医学专家确定。有时优选同时给药所述试剂。可选地,选择用于组合治疗的药物可以在不同时间给药予患者。当然,当存在多于一种病毒或其它感染或其它病症时,根据需要,本发明的化合物可以组合其它试剂治疗其它感染或病症。
如本文使用的术语“宿主”指其中HCV病毒可以复制的单细胞或多细胞生物,包括细胞系和动物,通常为人类。术语宿主特别地指受感染细胞、转染全部或部分HCV基因组的细胞、以及动物,特别是灵长类动物(包括黑猩猩)和人类。在本发明的大多数动物应用中,宿主为人类患者。然而,在某些情形中,本发明显然预期兽医应用(例如黑猩猩)。宿主可以为例如牛类、马类、鸟类、犬科动物、猫科动物等。
同位素取代
本发明包括具有原子的期望同位素取代的化合物和化合物的用途,所述同位素的量高于同位素的天然丰度,即富集。同位素是具有相同原子序数但不同质量数,即质子数相同但中子数不同的原子。作为一般实例而非限制,氢的同位素,例如氘(2H)和氚(3H)可以在所述结构中的任何地方使用。可选地或另外地,可以使用碳的同位素例如
用同位素例如氘取代可以得到由更大的代谢稳定性产生的某些治疗优势,例如增大的半衰期或降低的剂量要求。在代谢分解位点用氘取代氢可以降低所述键处的代谢速率或消除所述键处的代谢。在可以存在氢原子的化合物的任何位点,氢原子都可以是氢的任何同位素,包括氕(
术语“同位素标记的”类似物指其为“氘化类似物”、“
III.治疗或预防的方法
如本文使用的治疗指向感染HCV病毒的宿主给药活性化合物。
术语“预防性的”或预防,当使用时,指给药活性化合物以预防或减少病毒性病症发生的可能性。本发明同时包括治疗和预防或预防性治疗。在一个实施方案中,向已经暴露于且因此处于丙型肝炎病毒感染的感染风险中的宿主给药活性化合物。
本发明涉及一种治疗或预防下述疾病的方法:丙型肝炎病毒(包括HCV的耐药性和多药耐药性形式和相关疾病状态、病症)、或HCV感染的并发症(包括肝硬化和相关肝毒性),以及HCV感染继发的其它病症(例如虚弱、食欲不振、体重减轻、乳房增大(特别是在男性中)、皮疹(特别是在手掌上)、血凝固困难、皮肤上的蜘蛛样血管、意识错乱、昏迷(脑病)、腹腔体液蓄积(腹水)、食管静脉曲张、门静脉高压、肾衰竭、脾肿大、血细胞减少、贫血、血小板减少症、黄疸病和肝细胞性癌症等)。所述方法包括向有需要的宿主给药有效量的至少一种如本文所述β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
在另一个方面,本发明是一种预防或防止下述疾病的方法:HCV感染或疾病状态或相关或继发疾病状态、病症或HCV感染的并发症,包括肝硬化和相关肝毒性、虚弱、食欲不振、体重减轻、乳房增大(特别是在男性中)、皮疹(特别是在手掌上)、血凝固困难、皮肤上的蜘蛛样血管、意识错乱、昏迷(脑病)、腹腔体液蓄积(腹水)、食管静脉曲张、门静脉高压、肾衰竭、脾肿大、血细胞减少、贫血、血小板减少症、黄疸病和肝细胞性癌症等,所述方法包括向处于风险中的患者给药有效量的至少一种如上所述根据本发明的化合物与可药用载体、添加剂或赋形剂的组合,任选地与另一种抗-HCV试剂的组合。在另一个实施方案中,本发明的活性化合物可以给药至肝炎-相关肝移植之后的患者以保护新器官。
如果期望,5’-稳定的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
IV.药物组合物
在本发明的一个方面,根据本发明的药物组合物包括抗-HCV病毒有效量的至少一种如本文所述的5’-稳定的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
在本发明的一个方面,根据本发明的药物组合物包括抗-HCV病毒有效量的至少一种如本文所述的活性β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
本发明包括药物组合物,其包括在可药用载体或赋形剂中的治疗丙型肝炎病毒感染有效量的一种本发明的β-D-2′-d-2′-α-氟-2′-β-C-取代的-2-改性的-N
本领域普通技术人员应当认识到治疗有效量将随待治疗的感染或病症、其严重程度、施用的治疗方案、所使用试剂的药代动力学、以及待治疗的患者或受试者(动物或人类)而变化,并且这种治疗量可以由主治医师或专业医师确定。
根据本发明的5’-稳定的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
根据本发明的治疗活性制剂之内包括的化合物的量为用于治疗HCV感染、减少HCV感染的可能性或抑制、减少和/或消除HCV或其继发效应(包括疾病状态、病症和/ 或HCV继发的并发症)的有效量。一般而言,本发明的化合物在药物剂型中的治疗有效量通常范围为每日约0.001mg/kg患者至约100mg/kg患者或以上,更通常地,稍小于约 0.1mg/kg至约25mg/kg患者或显著更多,取决于使用的化合物、治疗的病症或感染和给药途径。根据本发明的活性核苷化合物通常的给药量范围为每日约0.1mg/kg至约15mg/kg 患者,取决于试剂在患者中的药代动力学。该剂量范围通常产生活性化合物的有效血液水平浓度,其范围可以为在患者中约0.001至约100、约0.05至约100微克/cc血液。
通常,为了治疗、预防或延迟这些感染的发病和/或减少HCV病毒感染的可能性或HCV的继发疾病状态、病症或并发症,所述组合物应当以口服剂型形式的总量为约250 微克至至多约500mg或更多、至少一天一次的量给药(例如至少25、50、100、150、250 或500毫克,至多一天四次)。本发明的化合物通常口服给药,但是可以以非肠道、局部或栓剂形式以及鼻内给药,呈鼻腔喷雾剂或如本文在别处所述的形式。
在共同给药本发明的化合物与如本文在别处所述另外的抗-HCV化合物的组合的情况下,根据本发明的化合物的给药量范围为约0.01mg/kg患者至约500mg/kg患者或显著更多,取决于共同给药的第二种试剂及其抗病毒效力、患者的病症和待治疗的疾病或感染的严重程度和给药途径。其它抗-HCV试剂的给药量范围可以例如为约0.01mg/kg至约500mg/kg。在某些优选的实施方案中,这些化合物的给药量通常可以为约0.5mg/kg到约50 mg/kg或更多(一般至多约100mg/kg),通常取决于两种试剂在患者中的药代动力学。这些剂量范围通常在患者中产生活性化合物的有效血液水平浓度。
用于本发明的目的,预防性或预防有效量的根据本发明的组合物落入与上述治疗有效量相同的浓度范围之内,并且一般与治疗有效量相同。.
活性化合物的给药包括连续的(静脉滴注)到每日数次口服或鼻内给药(例如Q.I.D.) 或透皮给药,并且可以包括口服、局部、非肠道、肌内、静脉内、皮下、透皮(其可以包括渗透增强剂)、颊内和栓剂给药,以及其它给药途径。为了增强口服途径化合物的生物利用度,也可以使用肠溶包衣口服片剂。最有效的剂型将取决于选择的具体药剂的生物利用度/药代动力学以及患者中疾病的严重程度。口服剂型是特别优选的,因为易于给药和预期有利的患者顺应性。
为了制备根据本发明的药物组合物,通常根据常规药物混合技术,将治疗有效量的根据本发明的一种或多种化合物与可药用载体充分混合以产生一个剂量。载体可以采取多种形式,取决于期望给药的制剂形式,例如口服给药或非肠道给药。在制备口服剂型的药物组合物中,可以使用任一种常用药物介质。因此,对于液体口服制剂,例如悬浮液、酏剂和溶液,可以使用包括水、二醇、油、乙醇、调味剂、防腐剂、着色剂等的合适载体和添加剂。对于固体口服制剂,例如散剂、片剂、胶囊,和对于固体制剂例如栓剂,可以使用包括淀粉、糖载体例如葡萄糖、甘露糖醇(manifold)、乳糖和相关载体、稀释剂、造粒剂、润滑剂、粘合剂、崩解剂等的合适载体和添加剂。如果期望,片剂或胶囊可以是肠溶包衣的或通过标准技术缓释释放。这些剂型的使用可以显著地增加所述化合物在患者中的生物利用度。
对于非肠道制剂,载体一般应当包括无菌水或氯化钠水溶液,但是也可以包括其它成分,包括有助于分散的那些成分。当然,当使用无菌水且保持为无菌时,所述组合物和载体也必须是灭菌的。也可以制备可注射的悬浮液,在这样的情况下,可以使用合适的液体载体、助悬剂等。
脂质体悬浮液(包括靶向病毒抗原的脂质体)也可以通过产生可药用载体的常规方法制备。这可以适于递送根据本发明的核苷化合物的游离核苷、酰基/烷基核苷或磷酸酯前药形式。
在根据本发明的典型的实施方案中,使用所述化合物和组合物治疗、预防或延迟HCV感染或继发疾病状态、病症或HCV的并发症。
V.组合和交替治疗
已经认识到,在延长用抗病毒剂治疗之后,可出现病毒的耐药突变体。耐药性最通常因为编码病毒复制中使用的酶的基因突变而出现。可以通过所述化合物与诱导与主要药物诱导突变不同的突变或经由与主要化合物途径不同的途径起作用的另一种、和或许甚至两种或三种其它抗病毒化合物的组合或交替给药,来延长、增强或恢复药物抗HCV感染的功效。可选地,可以通过这样的组合治疗(其可以包括交替治疗,如果认为一致)改变药物的药代动力学、生物分布、半衰期或其它参数。由于所公开的β-D-2′-D-2′-α-氟-2′-β-C- 取代的-2-改性的-N
(1)蛋白酶抑制剂,例如NS3/4A蛋白酶抑制剂;
(2)NS5A抑制剂;
(3)另一种NS5B聚合酶抑制剂;
(4)NS5B非底物抑制剂;
(5)干扰素α-2a,其可以被PEG化或以其它方式改性,和/或利巴韦林;
(6)非底物-基抑制剂;
(7)解螺旋酶抑制剂;
(8)反义寡脱氧核苷酸(S-ODN);
(9)适体;
(10)核酸酶-耐药性核酶;
(11)iRNA,包括microRNA和SiRNA;
(12)病毒的抗体、部分抗体或结构域抗体,或
(13)诱导宿主抗体反应的病毒抗原或部分抗原。
可以与本发明的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
(i)蛋白酶抑制剂,例如替拉瑞韦
(ii)NS5A抑制剂例如ACH-2928、ACH-3102、IDX-719、达卡他韦(daclatasvir)、ledispasvir和Ombitasvir(ABT-267);
(iii)NS5B抑制剂例如ACH-3422;AZD-7295;Clemizole;ITX-5061;PPI-461; PPI-688、
(iv)NS5B抑制剂,例如ABT-333、MBX-700;和
(v)抗体,例如GS-6624。
如果β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
目前批准用于流行性感冒的药物为金刚烷胺、金刚烷乙胺和奥塞米韦。任一种这些药物都可以与本文提供的活性化合物组合或交替使用,用于治疗对其敏感的病毒感染。利巴韦林用于治疗麻疹、流行性感冒A、流行性感冒B、副流行性感冒、重度RSV细支气管炎和SARS以及其它病毒感染,因此特别地与本发明的化合物组合用于治疗感染单链 RNA病毒的宿主。帕丽珠单抗批准用于处于RSV感染高风险的婴儿。
目前,对于西方尼罗河病毒仍然没有任何批准的药物。医师推荐提供强化的支持治疗,其可以包括住院治疗、静脉内输液、使用呼吸器协助呼吸、使用药物控制发作、脑肿胀、恶心和呕吐,以及使用抗生素预防使该疾病变得更糟的细菌感染。这强化了本发明的化合物用于病毒医学治疗的重要性。
VI.本发明的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
用于提供本发明的化合物的一般方法是本领域已知或本文描述的。2’-氯核苷酸的合成描述在US 20150366888、WO 2014058801;WO 2015/066370和WO 2015200219中。
在合成方案中使用下述缩写。
CBr
DBU:1,8-二氮杂二环[5.4.0]十一碳-7-烯
DCM:二氯甲烷
THF:四氢呋喃(THF),无水
EtOAc:乙酸乙酯
EtOH:乙醇
Li(OtBu)
Na
MeCN:乙腈
MeNH
MeOH:甲醇
Na
NaHCO
NH
NH
PE:石油醚
Ph
硅胶(230至400目,吸附剂)
t-BuMgCl:叔丁基氯化镁
t-BuOK:叔丁醇钠
t-BuOH:叔丁醇
实施例
通用方法
i)Li(OtBu)
实施例1.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备
步骤1.((2R,3R,4R,5R)-3-(苯甲酰氧基)-5-溴-4-氟-4-甲基四氢呋喃-2-基)甲基苯甲酸酯(2) 的制备
向在氮气氛和冷却至-30℃下的(2R)-3,5-二-O-苯甲酰基-2-氟-2-C-甲基-D-核糖酸-γ- 内酯(24.8g,66.6mmol)在无水THF(333mL)中的溶液中,滴加三叔丁氧基氢化铝锂(在 THF中1.0M,22.6mL,22.6mmol)。在加入完成之后,经90分钟,使该反应混合物慢慢地升温至-15℃,然后加入EtOAc(300mL),并用饱和的NH
步骤2.(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(苯甲酰氧基甲基)-4-氟-4-甲基四氢呋喃-3-基苯甲酸酯(3)的制备
在氮气氛下,将2-氨基-6-氯嘌呤(2.63g,15.5mmol)悬浮在t-BuOH(54mL)中。将该反应混合物加热至30℃,然后加入叔丁醇钾(1.69g,15.1mmol)。在45分钟之后,加入溴呋喃糖苷2(2.24g,5.12mmol)溶于无水MeCN(6mL)中的溶液,并将该反应混合物加热至65℃16小时,然后冷却至室温。加入饱和的NH
步骤3.(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃-3-醇(4)的制备
向化合物3(575mg,1.09mmol)在MeOH(9mL)中的溶液中加入甲胺(在无水EtOH 中33%,1.7mL,1.81mmol)。在密封管中,将该反应混合物加热至85℃16小时,冷却至室温并浓缩。先通过柱色谱(梯度DCM/MeOH 100∶0至85∶15),然后通过反相柱色谱(梯度 H
步骤4.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(5)的制备
经10分钟,向在氮气氛和冷却至0℃下的化合物4(114mg,365μmol)在无水THF(4mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,0.66mL,660μmol)。在0℃下,搅拌该反应混合物15分钟,然后在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,然后经10分钟滴加((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(Ross,B.S.,Reddy, P.G.,Zhang,H.R.,Rachakonda,S.,和Sofia,M.J.,J.Org,Chem.,(2011),(253mg,558 μmol)溶于无水THF(1mL)中的溶液。在0℃下,搅拌反应混合物30分钟,接着在室温下搅拌反应混合物18小时,然后用饱和的NH
i)Me
-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
实施例2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(7)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃-3-醇(6)的制备
向来自实施例1的化合物3(500mg,0.95mmol)在MeOH(6mL)中的溶液中加入二甲胺盐酸盐(783mg,9.6mmol)和1,8-二氮杂二环[5.4.0]十一碳-7-烯(1.43mL,9.6mmol)。在密封管中,在85℃下,加热反应混合物6小时,冷却至室温并浓缩。先通过柱色谱(梯度DCM/MeOH 100∶0至85∶15),然后通过反相柱色谱(梯度H
步骤2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(7)的制备
经10分钟,向在氮气氛和冷却至0℃下的化合物6(80mg,245μmol)在无水THF(4mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,0.64mL,640μmol)。在0℃下,搅拌该反应混合物15分钟,然后在室温下再搅拌15分钟。将反应混合物冷却至0℃,然后经 10分钟,滴加((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(167mg,367μmol)溶于无水THF(4mL)中的溶液。在0℃下,搅拌该反应混合物30分钟,并在室温下搅拌 18小时。用饱和的NH
i)a)N-甲基环丙基胺盐酸盐,Et
((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
实施例3.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丙基氨基)-9H-嘌呤-9-基)-4-氟 -3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(9)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丙基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4- 甲基四氢呋喃-3-醇(8)的制备
向化合物3(600mg,1.14mmol)在MeOH(10mL)中的溶液中加入N-甲基环丙基胺盐酸盐(366mg,3.40mmol)和三乙胺(470μL,3.40mmol)。在密封管中,在100℃下加热该反应混合物15小时,并冷却至室温。加入包含30%NH
步骤2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丙基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(9)的制备
经10分钟,向在0℃下化合物8(200mg,0.57mmol)在无水THF(15mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,680μL,0.68mmol)。在0℃下,搅拌该反应混合物 15分钟,然后在室温下再搅拌15分钟。将反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(283mg,0.62mmol)溶于无水THF(4 mL)中的溶液。在0℃下,搅拌该反应混合物30分钟,并在室温下搅拌18小时。用饱和的NH
i)2,6-二氯嘌呤,tBuOK,tBuOH/MeCN,65℃;ii)MeNH
((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃ toRT
实施例4.((((R,S)-(2R,3R,4R,5R)-5-(2,6-二甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(12)的制备
步骤1.(2R,3R,4R,5R)-5-(2,6-二氯-9H-嘌呤-9-基)-2-(苯甲酰氧基甲基)-4-氟-4-甲基四氢呋喃-3-基苯甲酸酯(10)的制备.
在氮气氛下,将化合物2,6-二氯嘌呤(1.30g,6.86mmol)悬浮在t-BuOH(25mL)中。分批加入叔丁醇钾(778mg,6.92mmol),然后在室温下搅拌反应混合物。在1小时之后,加入溴呋喃糖苷2(1.0g,2.29mmol)溶于无水MeCN(20mL)中的溶液,并在65℃下加热该反应混合物过夜,然后冷却至室温。加入饱和的NH
步骤2.(2R,3R,4R,5R)-5-(2,6-二甲基氨基-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃 -3-醇(11)的制备
在密封管中,在130℃下,加热化合物10(148mg,0.27mmol)在甲胺(在EtOH中33%,30mL)中的溶液4天,冷却至室温并浓缩。先通过柱色谱(梯度DCM/MeOH 100∶0 至50∶50),然后通过反相柱色谱(梯度H2O/MeOH 100∶0至0∶100)纯化残余物,得到呈白色固体的产物11(33mg,0.10mmol,37%)。
步骤3.((((R,S)-(2R,3R,4R,5R)-5-(2,6-二甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(12)的制备
经10分钟,向在0℃下的化合物11(55mg,0.17mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1M,304mL,0.30mmol)。在0℃下,搅拌该反应混合物15分钟,然后在室温下搅拌15分钟。将该溶液冷却至0℃,并经10分钟滴加((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(115mg,0.25mmol)溶于无水THF(1mL)中的溶液。使该混合物慢慢地升温至室温,并搅拌4天。用饱和的NH
i)TIPDSCl
((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
实施例5.((((R,S)-(2R,3R,4R,5R)-5-(2-异丁酰氨基-6-甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(16)的制备
步骤1.化合物13的制备
向在0℃下的化合物4(286mg,0.92mmol)和咪唑(370mg,5.43mmol)在无水DMF(6mL)中的溶液中加入1,3-二氯-1,1,3,3-四异丙基二硅氧烷(300μL,0.94mmol)。在室温下,搅拌该反应混合物2小时,用EtOAc(50mL)稀释,并用饱和的NH
步骤2.化合物14的制备
向在0℃下,化合物13(200mg,0.36mmol)在无水吡啶(3mL)中溶液中加入异丁酰氯(38μL,0.36mmol)。在室温下,搅拌反应混合物2小时。通过加入水(500μL)淬灭该反应。浓缩该混合物,并与甲苯(3×10mL)共同蒸发。通过柱色谱(梯度PE/EtOAc 1∶0至1∶1) 纯化残余物,得到呈白色固体的产物14(99mg,0.16mmol,44%)。MS(ESI)m/z C
步骤3.(2R,3R,4R,5R)-5-(2-异丁酰氨基6-甲基氨基-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃-3-醇(15)的制备
向化合物14(90mg,0.14mmol)在无水THF(2mL)中的溶液中加入氟化四丁铵(在THF中1M,38μL,0.38mmol)。在室温下,搅拌该混合物2小时并浓缩。先通过柱色谱 (梯度DCM/MeOH 10∶0至9∶1),接着通过反相柱色谱(梯度H2O/MeOH 100∶0至0∶100)纯化残余物,得到呈白色固体的产物15(42mg,0.11mmol,77%)。
步骤4.((((R,S)-(2R,3R,4R,5R)-5-(2-异丁酰氨基6-甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(16)的制备
经10分钟,向在0℃下化合物15(27mg,0.07mmol)在无水THF(1mL)中的溶液中滴加丁基氯化镁(在THF中1.0M,130μL,0.13mmol)。在0℃下,搅拌反应混合物15 分钟,然后在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(50mg,0.11mmol)溶于无水THF(1 mL)中的溶液。在0℃下,搅拌反应混合物30分钟,接着在室温下,搅拌18小时,然后用饱和的NH
i)N-甲基乙胺,MeOH,100℃;ii)((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯, tBuMgCl,THF,0℃.
实施例6.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-乙基氨基)-9H-嘌呤-9-基)-4-氟-3- 羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(18)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-乙基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4- 甲基四氢呋喃-3-醇(17)的制备
向化合物3(150mg,0.29mmol)在MeOH(4mL)中的溶液中加入N-甲基乙胺 (245μL,2.90mmol)。在密封管中,在100℃下加热该反应混合物15小时,冷却至室温并浓缩。通过柱色谱(梯度DCM/MeOH 100∶0至90∶10)纯化残余物,得到呈白色固体的产物 31(89mg,0.26mmol,89%)。
步骤2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-乙基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基 -4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(18)的制备
经10分钟,向在0℃下,化合物17(30mg,0.09mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,110μL,0.11mmol)。在0℃下,搅拌反应混合物15分钟,然后在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(48mg,0.11mmol)溶于无水 THF(1mL)中的溶液。在0℃下,搅拌反应混合物30分钟,并在室温下,搅拌18小时。用饱和的NH
i)N-甲基丙胺,MeOH,100℃;ii)((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯, tBuMgCl,THF,0℃.
实施例7.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-丙基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(20)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-丙基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4- 甲基四氢呋喃-3-醇(19)的制备
向化合物3(150mg,0.29mmol)在MeOH(4mL)中的溶液中加入N-甲基丙胺 (295μL,2.90mmol)。在密封管中,在100℃下,加热该反应混合物15小时,冷却至室温并浓缩。先通过柱色谱(梯度DCM/MeOH 100∶0至90∶10),然后通过反相柱色谱(梯度 H2O/MeOH 100∶0至0∶100)纯化残余物,得到呈白色固体的产物19(80mg,0.23mmol, 78%)。
步骤2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-丙基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基 -4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(20)的制备
经10分钟,向在0℃下,化合物19(30mg,0.09mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,110μL,0.11mmol)。在0℃下,搅拌反应混合物15分钟,然后在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(46mg,0.11mmol)溶于无水 THF(1mL)中的溶液。在0℃下,搅拌该反应混合物30分钟,并在室温下搅拌18小时。用饱和的NH
i)a)N-甲基环丁胺盐酸盐,Et
((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
实施例8.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丁基氨基)-9H-嘌呤-9-基)-4-氟 -3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(22)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丁基氨基)-9H-嘌呤-9-基)-4-氟-2-(羟甲基)-4-甲基四氢呋喃-3-醇(21)的制备
向化合物3(150mg,0.29mmol)在MeOH(4mL)中的溶液中加入N-甲基环丁胺盐酸盐(105mg,0.90mmol)和三乙胺(190μL,1.00mmol)。在密封管中,在100℃下加热该反应混合物15小时,并冷却至室温。加入包含30%NH
步骤2.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基-环丁基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基 -4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(22)的制备
经10分钟,向在0℃下化合物21(50mg,0.14mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,210μL,0.21mmol)。在0℃下,搅拌该反应混合物 15分钟,然后在室温下再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(74mg,0.16mmol)溶于无水THF(2 mL)中的溶液。在0℃下,搅拌该反应混合物30分钟,并在室温下搅拌18小时。用饱和的NH
活性化合物中2-氨基部分的改性
本领域普通技术人员可以通过本领域技术人员熟知的方法向2-氨基嘌呤部分增加取代基。此处提供一种非限制性的方法,并且可以容易地采用其它方法。在升高的温度下,用市售可获得的2,6-二氯嘌呤、碱和有机溶剂的混合物处理((2R,3R,4R,5R)-3-(苯甲酰氧基)-5-溴-4-氟-4-甲基四氢呋喃-2-基)苯甲酸甲酯,得到(2R,3R,4R,5R)-5-(2,6-二氯-9H-嘌呤 -9-基)-2-(苯甲酰氧基甲基)-4-氟-4-甲基四氢呋喃-3-基苯甲酸酯。在一个实施方案中,所所述碱为叔丁醇钾。在一个实施方案中,有机溶剂的混合物包括叔丁醇和乙腈。在环境温度下,用胺、碱和有机溶剂处理化合物(2R,3R,4R,5R)-5-(2,6-二氯-9H-嘌呤-9-基)-2-(苯甲酰氧基甲基)-4-氟-4-甲基四氢呋喃-3-基苯甲酸酯,得到2-氯-N
立体特异性的磷对映异构体的制备
一些本文所述活性化合物具有手性磷部分。任一种本文所述活性化合物都可以使用本领域技术人员已知的方法提供为分离的磷对映异构体形式,例如,至少80、90、95或98%的R或S对映异构体。例如,存在描述如何获得这样的化合物的大量出版物,包括但不限于柱色谱,例如如下实施例17和Ross等人的美国专利No.8,859,756;8,642,756和 8,333,309中所述的。
实施例9.化合物5立体异构体的分离
在Phenominex Luna柱上,使用下述条件分离化合物5的立体异构体:柱:Phenominex Luna 5micron C18(2)250x 10mm part#OOG-4252-BO
样品浓度:在乙腈中约50mg/mL
注射体积:50μl
流动相A:HPLC级水
流动相B:HPLC级乙腈。
流速:5ml/min
UV:283nm
梯度:
运行时间:45分钟
柱温:40℃
半制备运行的样品色谱图示于图1中。
使用用下述条件的分析柱分析合并的级分:
柱:Phenominex Luna 5micron C18(2)250x 2mm part#OOG-4252-BO
注射体积:10μl
流动相A:HPLC级水
流动相B:HPLC级乙腈。
流速:0.2ml/min
UV:283nm
梯度:
运行时间:45分钟
柱温:40℃
使用旋转蒸发器(rotovap)将每种立体异构体的合并级分蒸干,浴温30℃。将得到的固体溶于1ml乙腈中,转移到1.7ml微量离心管中,并在30℃的温度下,在真空离心机上蒸发溶剂。.
关于最终样品的数据如下:
1.第一洗脱峰:化合物5#1(5-1)(21.7mgs–97.8%ee).
2.第二洗脱峰:化合物5#2(5-2)(13.2mgs–95.9%ee).
第一峰和第二峰的最终重量完全对应于它们在原混合物中的百分比(分别为62.2%和37.8%)。
式I-VII的化合物的立体特异性合成
实施例10.(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(羟甲基)-4-氟-4-甲基四氢呋喃-3-醇(23)的制备
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(羟甲基)-4-氟-4-甲基四氢呋喃-3-醇 (23)的制备
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(苯甲酰氧基甲基)-4-氟-4- 甲基四氢呋喃-3-基苯甲酸酯3(80g,140mmol)加入到三甲胺在甲醇(7M,800mL)中的溶液中,并在室温下搅拌过夜。浓缩该混合物,然后通过柱色谱(DCM∶MeOH=100∶1)纯化,得到(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(羟甲基)-4-氟-4-甲基-四氢呋喃-3-醇 (23)(40g,90%).
实施例11.((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸酯的制备.
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃 -3-醇(4)的制备
向(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(羟甲基)-4-氟-4-甲基-四氢呋喃-3- 醇(2.0g,1.0eq)在二噁烷(15mL)中的溶液中加入MeNH
[M+H]
步骤2.((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸酯的制备.
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-(甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇(1.47g,1.0eq)和PPAL-S(2.35g,1.1eq)溶于无水THF(29mL)中。在将该混合物冷却至-10℃之后,在N
实施例12.((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(25)的制备.
步骤1.(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇的制备
向(2R,3R,4R,5R)-5-(2-氨基-6-氯-9H-嘌呤-9-基)-2-(羟甲基)-4-氟-4-甲基-四氢呋喃 -3-醇(2.8g,8mmol)在二噁烷(20mL)中的溶液中加入二甲胺水溶液(5mL)。在室温下搅拌 3小时之后,TLC显示给予起始物质耗尽。浓缩该混合物,并通过柱色谱(DCM∶MeOH=60∶1) 纯化,得到(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇(2.2g)。
步骤2.((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(25)的制备
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇(8g,1.0eq)和PPAL-S(11.1g,1eq)溶于无水THF(100mL)中。将该混合物冷却至-5-0℃,并在N
实施例13.((((R)-(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(26)的制备
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-(二甲基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇(3g,1.0eq)和PPAL-R(4.17g,1eq)溶于无水THF(60mL)中。将该混合物冷却至-5-0℃,并在N
实施例14.((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备
步骤1:(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-3-醇(8)的制备
将K
步骤2:((((S)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-3-醇(8g,1.0eq)和PPAL-S(10.3g,1eq)溶于无水THF(100mL)中。在将该混合物冷却至-5-0℃之后,在N
实施例15.((((R)-(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基 -4-甲基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
将化合物(2R,3R,4R,5R)-5-(2-氨基-6-(甲基环丙氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4- 甲基四氢呋喃-3-醇(3g,1.0eq)和PPAL-R(2.8g,1eq)溶于无水THF(60mL)中。在将该混合物冷却至-5-0℃之后,在N
实施例16.化合物32的制备
步骤1.化合物29的制备.
在0℃下,向6(3.0g,1.0eq)在吡啶(30mL)中的溶液中加入TIPDSCl
步骤2.化合物30的制备。
在0℃下,向化合物29(800mg,1.0eq)、DMAP(16mg,0.1eq)、吡啶(1.6mL)和 DCM(10mL)的混合物中加入异丁酰氯(209mg,1.5eq)。在室温下搅拌2小时之后,TLC 显示起始物质耗尽。将该混合物用水淬灭,用1M HCl水溶液、饱和的NaHCO
步骤3.化合物31的制备。
在室温下,向30(560mg,1.0eq)在THF(10mL)中的混合物中加入E
步骤4.化合物32的制备。
将化合物31(280mg,1.0eq)和PPAL-S(320mg,1eq)溶于无水THF(10mL)中。在将该混合物冷却至-5℃之后,在N
实施例17.化合物35的制备。
步骤1.化合物33的制备。
在0℃下,向29(2.0g,1.0eq)、DMAP(0.04g,0.1eq)、吡啶(4mL)和DCM(20mL) 的混合物中加入AcCl(0.414g,1.5eq)。在室温下,搅拌2小时之后,TLC显示起始物质耗尽。将该混合物用水淬灭,用1M HCl水溶液、饱和的NaHCO
步骤2.化合物34的制备。
在室温下,向33(1.58g,1.0eq)在THF(20mL)中的混合物中加入Et
[M+H]+=369.6.
步骤3.化合物35的制备。
将化合物34(136mg,1.0eq)和PPAL-S(184mg,1.1eq)溶于无水THF(3mL)中。在将该混合物冷却至-5℃之后,在N
β-D-2’-脱氧-2’-α-氟-2’-β-乙炔基-N
实施例18.β-D-2’-脱氧-2’-α-氟-2’-β-乙炔基-N
步骤1.化合物36的制备。
在N
在0-5℃下滴加异丁酰氯。在该温度下搅拌2小时之后,TLC显示中间体耗尽。加入水,并用DCM萃取内含物。然后,用0.5N HCl洗涤有机相以除去吡啶。.
在将内容物的pH洗涤至5~6之后,在0-5℃下加入pTSA·H
步骤2.化合物37的制备。
在室温下,向36(10.0g,16.3mmol)在DCM(100mL)中的溶液中加入Dess-Martin 过碘烷,并搅拌该反应12小时。TLC显示起始物质耗尽。然后,用DCM(200mL)稀释反应混合物,并用饱和的Na
步骤3.化合物38的制备。
在N
步骤4.化合物39的制备。
在N
步骤5.化合物40的制备。
在室温下,向39(3.8g,5.3mmol)在THF(120mL)中的溶液中加入AcOH(1.3g,22mmol) 和TBAF(4.2g,15.9mmol)。在室温下,搅拌反应30分钟。TLC显示起始物质耗尽。在真空干燥之后,用柱色谱(EA)纯化残余物,得到呈白色固体的产物(2.0g,95%)。
用于氨基取代和脱保护的通用方法:
在室温下,向40(350mg,0.88mmol)在二噁烷(20mL)中的溶液中加入甲醇或相应胺(游离碱或盐如盐酸盐+DIEA)的水溶液。在室温下,搅拌内容物1-12小时。TLC显示起始物质耗尽。在真空浓缩之后,将残余物直接用于下一步而无需纯化。将上述残余物溶于甲醇(10mL)中。加入NaOH水溶液(2.5N,10mL)。在室温下搅拌过夜之后,TLC显示起始物质耗尽。用1N HCl将内容物的pH调节至7-8。浓缩该溶液,并用柱色谱 (DCM/MeOH=100->20/1)纯化,得到呈黄白色固体的产物(经2步的产率:40-80%)。表 1图解了化合物57-63的结构,以及各个化合物的相应质谱和1H NMR。
表1.
实施例19.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-二甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4- 乙炔基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备
i)((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
步骤1.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-二甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-乙炔基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
经10分钟,向在0℃下的化合物41(30mg,0.09mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,125μL,0.13mmol)。在0℃下,搅拌该反应混合物 15分钟,并在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(49mg,0.11mmol)溶于无水THF(2 mL)中的溶液。在0℃下,搅拌反应混合物30分钟,并在室温下,搅拌18小时。用饱和的NH
实施例20.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-乙炔基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
i)((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸并丙酯,tBuMgCl,THF,0℃.
步骤1.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-甲基氨基-9H-嘌呤-9-基)-4-氟-3-羟基-4-乙炔基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
经10分钟,向在0℃下的42(30mg,0.09mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M 125μL,0.13mmol)。在0℃下,搅拌反应混合物15 分钟,然后在室温下再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(49mg,0.11mmol)溶于无水THF(2 mL)中的溶液。在0℃下,搅拌该反应混合物30分钟,并在室温下搅拌18小时。用饱和的NH
实施例21.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基环丙基氨基)-9H-嘌呤-9-基)-4-氟 -3-羟基-4-乙炔基四氢呋喃-2-基)甲氧基)-苯氧.基-磷酰基)-L-丙氨酸异丙酯的制备
i)((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯,tBuMgCl,THF,0℃.
步骤1.((((R,S)-(2R,3R,4R,5R)-5-(2-氨基-6-(N-甲基环丙基氨基)-9H-嘌呤-9-基)-4-氟-3-羟基-4-乙炔基四氢呋喃-2-基)甲氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯的制备。
经10分钟,向在0℃下的化合物43(40mg,0.11mmol)在无水THF(2mL)中的溶液中滴加叔丁基氯化镁(在THF中1.0M,160μL,0.16mmol)。在0℃下,搅拌反应混合物 15分钟,然后在室温下,再搅拌15分钟。将该反应混合物冷却至0℃,并经10分钟滴加 ((R,S)-(五氟苯氧基)-苯氧基-磷酰基)-L-丙氨酸异丙酯(55mg,0.12mmol)溶于无水THF(2 mL)中的溶液。在0℃下,搅拌反应混合物30分钟,并在室温下,搅拌18小时。用饱和的NH
实施例22.PPAL-S的制备
步骤1.外消旋的PPAL的制备
在-10℃下,向苯基二氯磷酸酯(250g)在EtOAc(800mL)中的搅拌溶液中加入在三乙胺(120g)中的L-丙氨酸异丙酯(200g)。在-10℃下,搅拌该反应1小时。在-5℃下,加入在三乙胺(120g)和EtOAc(400mL)中的化合物2,3,4,5,6-五氟苯酚(220g),并在该温度下搅拌0.5小时。使该反应混合物升温至25℃,并在该温度下搅拌2小时。过滤溶液,并用 EtOAc(2×200mL)洗涤,在真空下蒸发合并的有机相,得到固体PPAL-RS(消旋物)。
步骤2.PPAL-RS的制备
向PPAL-RS在EtOAc(200mL)和正庚烷(1.4L)中的搅拌溶液中加入在三乙胺中的2,3,4,5,6-五氟苯酚(10.1g),并继续搅拌约4-8小时。在固体的R-异构体小于0.5%之后,过滤固体。将该固体溶于EtOAc(4L)中,用水(2×100mL)、盐水(1L)洗涤,经无水Na
实施例23.PPAL-R的制备
向装有机械搅拌器的三颈圆底烧瓶中加入苯基二氯磷酸酯(189.6g,0.90mol)和无水 EtOAc(750mL)。在氮气氛下,将该溶液冷却至-10℃。将L-丙氨酸异丙酯(118g,0.90mmol) 和三乙胺(100g,1.1eq)加入到上述溶液中。在-5℃下,经由加料漏斗将2,3,4,5,6-五氟苯酚(165g,1eq)和三乙胺(90.5g,1eq)在EtOAc(300mL)中的预冷却(低于10℃)混合物加入到所述混合物中,并在20-25℃下搅拌得到的混合物1小时。过滤出白色沉淀物(TEA.HCl),并用EtOAc洗涤。在减压下浓缩滤液,得到约280g呈白色固体的PPAL-RS(S/R =1/1)。在室温下,在300mL的庚烷/EtOAc(20∶1)中研磨PPAL-RS(280g)5分钟。过滤该白色悬浮液,并用庚烷/EtOAc(20∶1)的混合物洗涤固体。将滤液冷却至8℃,并通过过滤收集固体。得到粗PPAL-R(10g),95%手性纯度。按照上述步骤纯化粗产物。得到 PPAL-R(5g),NLT98%手性纯度。
实施例24:化合物52的制备:.
步骤1.化合物49的制备。
向48(1.81g,3.23mmol)在二噁烷(18mL)中的溶液中加入40%CH
步骤2.化合物50的制备.
在0℃下,向49(1.34g,2.42mmol)和1-甲基咪唑(794mg,9.68mmol)在DCM(14mL)中的溶液中慢慢地加入氯甲酸戊酯(547mg,3.63mmol)。在室温下,搅拌该反应过夜。浓缩该混合物,并通过柱色谱(PE∶EtOAc=5∶1-2∶1)纯化,得到呈白色固体的50(1.01g, 62%)。
步骤3.化合物51的制备
在0℃下,向50(1.00g,1.5mmol)在THF(11mL)中的溶液中加入Et
步骤4.化合物52的制备
在N
实施例25:化合物56的制备.
步骤1.化合物48的制备
在0℃下,向23(600mg,1eq)在吡啶(30mL)中的溶液中加入TIPDSCl
步骤2.化合物53的制备。
在室温下,搅拌48(800mg,1eq)、吡啶(3.2mL)、DMAP(34.9mg,0.2eq)在DCM(20 mL)中的混合物。在0℃下,滴加氯甲酸正戊酯(3.2mL),并在室温下搅拌该混合物1天。将有机层用1M HCl水溶液、饱和的碳酸氢钠水溶液和饱和的氯化钠水溶液洗涤,经无水硫酸钠干燥,并在真空中蒸发。通过硅胶色谱(MeOH∶CH2Cl2=1∶50)纯化残余物,得到呈白色固体泡沫状物的53(255mg,26%)。.
步骤3.化合物54的制备.
向53(270mg,1eq)在1,4-二噁烷(10mL)中的溶液中滴加40%CH
步骤4.化合物55的制备。
将三乙胺(1011.9mg,10eq)和Et
步骤5.化合物56的制备
在-10℃下,向55(113mg,1eq)和PPAL-S(120mg,1eq)在THF(4mL)中的混合物中滴加在THF(0.327mL,2.1eq)中的1.7M t-BuMgCl。在室温下,搅拌该混合物1小时,然后用饱和的NH
实施例26:化合物60的制备.
步骤1.化合物57的制备.
在5±5℃下,向6(20g,1eq)在CH
步骤2.化合物58的制备
在0-5℃下,向57(9.8g,1eq)在THF(4mL)中的溶液中滴加在THF中的1.7M t-BuMgCl(50mL,4.8eq)。在室温下,搅拌该混合物0.5小时,并慢慢地加入氯甲酸正戊酯(2.7g,1.05eq)。在0-5℃下,搅拌该混合物3-4小时。用饱和的NH
步骤3.化合物59的制备
将三乙胺(10.119g)和Et
步骤4.化合物60的制备。
在-5℃下,向59(2g,1eq)和PPAL-S(2.3g,1.1eq)在THF(40mL)中的混合物中滴加在THF(5.6mL,2.1eq)中的1.7M t-BuMgCl。在-20±5℃下,搅拌该混合物1小时,然后用饱和的NH
生物学数据
实施例27.试验方法和另外的生物学数据
将载有双顺反子(discistronic)HCV基因型1b荧光素酶受体复制子的Huh-7luc/neo ET细胞以7.5×10
使用包含野生型和抗性相关突变体的各种患者来源的HCV基因型测定它们对测试化合物的相对复制敏感性。使用分离自HCV患者血浆的病毒RNA制备包含NS5B基因组区域的复制子抗性测试载体(RTV)。将每个NS5B区域通过反转录聚合酶链反应扩增,并克隆到HCV复制子RTV中,然后通过电穿孔转移到Huh-7细胞中。在不存在和存在连续稀释的测试化合物下培养72-96小时之后,通过荧光素酶活性测量病毒复制,并测定50%抑制浓度(IC
表2示出了化合物25、27、5-2和索非布韦针对包含野生型和抗性相关突变体的各种临床分离物的IC
针对HCV复制,所有化合物都比索非布韦显著更有效,无论25、27还是5-2化合物都没有显示出对于L159F、L159F和S282T和C316N突变体的交叉抗性的任何证据。.
表2:在源自患者的HCV基因型中测试化合物的抗病毒活性
进行瞬时转染测试以测定HCV野生型S282T突变体对于所测试化合物的敏感性。在从野生型转录的RNA或来自T7启动子的S282T HCV复制子质粒的存在下,使Huh-7 细胞电穿孔。将转染的细胞以7.5×10
表3报道了化合物25、27、5-3和索非布韦针对HCV野生型和S282T复制子的IC
针对HCV复制,所有化合物都比索非布韦显著更有效,无论25、27还是5-2化合物都没有显示出对于S282T突变体的交叉抗性的任何证据。
表3:在HCV瞬时感染测定中测试化合物的抗病毒活性
在包含10μM测试化合物的培养物中,测定所选择化合物在新鲜人全血和人肝S9级分中的稳定性。在培养0、30、60分钟和至多120分钟之后,移出等分试样,立即用3 体积冰冷的甲醇/乙腈(1∶1,v/v)萃取。离心萃取物,并通过LC-MS/MS分析上清液中未改变的测试化合物和可能代谢物的浓度。
图5示出了化合物5-2和所有2-氨基衍生物在人血中的优良的稳定性。
有趣地是,图6示出了使用人肝S9级分的2′-脱氧-2′-α-氟-2′-β-甲基-N
实施例28.HCV(gt1b)NS5B聚合酶测定
通过在包含TA的连续稀释液、与HCV(-)链3’UTR区域互补的体外转录病毒RNA、聚合酶、放射标记的核糖核苷酸、250μM非竞争性rNTP和1μM竞争性rNTP的反应混合物中,测量重新聚合来测定HCV(gt1b)NS5B聚合酶的抑制,一式三份。从得到的抑制曲线确定产生50%抑制的TA浓度(IC
实施例29.人骨髓祖细胞测定
将悬浮在BFU-E或GM-CSF特异性培养基的新鲜人骨髓祖细胞(Invitrogen)以10
化合物25、27和5-2显示出在体外对于骨髓干细胞没有细胞毒性。
实施例30.iPS心肌细胞测定
将iPS心肌细胞(Cellular Dynamics)以1.5×10
化合物25、27和5-2显示出在体外对于iPS心肌细胞没有细胞毒性。
实施例31.人DNA聚合酶测定
在连续稀释的TA、0.05mM dCTP、dTTP和dATP、10μCi[
三磷酸酯,β-D-2′-脱氧-2′-α-氟-2′-β-甲基-鸟嘌呤三磷酸酯以及化合物25、27和5-2的三磷酸酯类似物都不会抑制人DNA聚合酶α、β或γ。.
实施例32.人肝细胞共培养物
通过测量微模板的(micro-patterned)人肝细胞共培养物(
当用微模板共培养的人肝细胞培养最多12天时,如通过ALT渗漏、白蛋白分泌、脲产生和细胞ATP含量所测量的,最多30μM浓度的化合物5-2没有显示出细胞毒性的迹象。采用延长暴露(培养最多21天)检测的较小细胞毒性指征显著地小于用索非布韦观察到的那些。参见下表4、5和6。
INX-189对于人共培养的肝细胞是高度细胞毒性的,通过所有测量都显示出早在第2 天白蛋白分泌就减少且具有细胞毒性。在相同条件下,索非布韦显示比AT-511更大细胞毒性。
表4.测试制品对于细胞ATP浓度的影响
表5.测试制品对白蛋白分泌的影响
表6.测试制品对白蛋白分泌的影响
实施例33.代谢研究
在人类、狗和小鼠肝细胞的新鲜原代培养物中研究10μM浓度的化合物25、27和5-2的代谢。用10μM TA单次(singlet)培养在具有matrigel盖层的6孔板中铺板的来自人类(XenoTech,混合性别,由10个供体合并的)、雄性比格犬(BioreclamationIVT)和雄性 ICR/CD-1小鼠(BioreclamationIVT,8个供体)的肝细胞。在2、4、6、8或24小时之后,通过LC-MS/MS定量核苷酸前药及其可能的代谢物(前药、单磷酸酯、三磷酸酯和核苷)的细胞内水平。由标准曲线外推低于定量下限的浓度(前药、单磷酸酯和核苷为1.5pmol/10
化合物β-D-2′-脱氧-2′-α-氟-2′-β-甲基-鸟嘌呤三磷酸酯是在培养的人肝细胞中观察到的化合物25、27和5-2的主要代谢物,并且是HCV(gt1b)NS5B聚合酶的有效抑制剂,IC
图8显示在人肝细胞中主要的化合物25代谢物。
图9显示在人肝细胞中主要的化合物27代谢物。
图10显示在人肝细胞中主要的化合物5-2代谢物。
图11示出了化合物25、27和5-2的活化途径。如可以看到的,化合物25、27和5-2 被转化成其相应单磷酸酯类似物,其接着代谢成常见的MP类似物;β-D-2′-脱氧-2′-α-氟 -2′-β-甲基-鸟嘌呤单磷酸酯(化合物61)。然后,所述单磷酸酯逐步磷酸化成活性三磷酸酯:β-D-2′-脱氧-2′-α-氟-2′-β-甲基-鸟嘌呤三磷酸酯(化合物62)。.
实施例34.对照
在上述实施例中,使用INX-189(INX-08189/BMS-986094)和索非布韦作为对照。
两种最有效的核苷酸前药化合物25和27都表现出优良的选择性,在Huh-7细胞、人骨髓干细胞和人心肌细胞中的CC
表7为说明在HCV复制子测定中测试的化合物与EC
表7.测试化合物的复制子测定结果.
本文所述的β-D-2′-D-2′-α-氟-2′-β-C-取代的-2-改性的-N
例如,可以以HCV亚基因组RNA复制子测定系统在Huh7 ET细胞中测量化合物的抗-HCV活性和细胞毒性。(参见Korba,et al.,Antiviral Research 2008,77,56)。可以与阳性对照2′-C-Me-胞嘧啶{2′-C-Me-C}进行比较来概述结果(Pierra,et al.,Journal ofMedicinal Chemistry 2006,49,6614)。
另一种抗-丙型肝炎病毒活性的体外试验描述在Stuyver等人且申请人为Pharmasset, Inc的美国专利No.7,718,790中。
已经参照本发明的实施方案描述了本说明书。考虑到本文的教导,本领域普通技术人员将能够根据期望的目的改变本发明,并且这些变化被认为在本发明的范围之内。
本申请还涉及以下项目。
1.式I的化合物或其可药用盐:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
R
R
2.项目1的化合物,其中:
R
3.项目2的化合物,其中:
R
4.项目2的化合物,其中:
Y为NR
R
5.项目2的化合物,其中:
Y为NR
R
6.项目2的化合物,其中:
Y为NR
R
7.项目1的化合物,其中R
8.项目1的化合物,其中R
9.下式的化合物或其可药用盐:
其中:
R
R
R
R
R
10.项目9的化合物,其中:
Y为NR
11.项目9的化合物,其中:
Y为NR
12.项目9的化合物,其中:
Y为NR
13.项目9的化合物,其中:
Y为NR
14.式II的化合物或其可药用盐:
其中:
Y为NR
R
R
其中R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
R
x为1、2或3。
15.项目14的化合物,其中:
R
16.项目15的化合物,其中:
R
17.项目15的化合物,其中:
Y为NR
18.项目15的化合物,其中R
19.项目15的化合物,其中R
20.项目15的化合物,其中:
R
21.项目15的化合物,其中:
R
22.项目15的化合物,其中:
R
23.项目15的化合物,其中:
R
R
24.式III的化合物或其可药用盐:
其中:
R
R
R
R
R
其中Y、R
25.项目24的化合物,其中:
Y为NR
26.项目24的化合物,其中:
Y为NR
27.项目24的化合物,其中:
Y为NR
28.项目24的化合物,其中:
Y为NR
29.式IV的化合物或其可药用盐:
其中变量Y、R
30.式V的化合物或其可药用盐:
其中变量Y、R
31.式VI的化合物或其可药用盐:
其中:
Y、R
R
32.式VII的化合物或其可药用盐:
其中:
Y、R
33.药物组合物,包括在可药用载体中的用于治疗宿主中HCV的有效量的项目1的化合物。
34.药物组合物,包括在可药用载体中的用于治疗宿主中HCV的有效量的项目9的化合物。
35.药物组合物,包括在可药用载体中的用于治疗宿主中HCV的有效量的项目14的化合物。
36.药物组合物,包括在可药用载体中的用于治疗宿主中HCV的有效量的项目24或29-32 中任一项的化合物。
37.项目33的药物组合物,其中所述组合物适于口服递送。
38.项目34的药物组合物,其中所述组合物适于口服递送。
39.项目35的药物组合物,其中所述组合物适于口服递送。
40.项目36的药物组合物,其中所述组合物适于口服递送。
41.一种用于在有需要的宿主中治疗丙型肝炎感染或由于丙型肝炎感染导致的病症的方法,包括给药任选地在可药用载体中的有效量的项目1的化合物。
42.一种用于在有需要的宿主中治疗丙型肝炎感染或由于丙型肝炎感染导致的病症的方法,包括给药任选地在可药用载体中的有效量的项目9的化合物。
43.一种用于在有需要的宿主中治疗丙型肝炎感染或由于丙型肝炎感染导致的病症的方法,包括给药任选地在可药用载体中的有效量的项目14的化合物。
44.一种用于在有需要的宿主中治疗丙型肝炎感染或由于丙型肝炎感染导致的病症的方法,包括给药任选地在可药用载体中的有效量的项目24或29-32中任一项的化合物。
45.项目41的方法,其中所述化合物是透皮给药的。
46.项目41的方法,其中所述化合物经由控制释放给药。
47.项目41的方法,其中所述化合物是静脉内给药的。
48.项目41的方法,其中所述由丙型肝炎导致的感染为抗体阳性和抗原阳性病症、基于病毒的慢性肝炎、由晚期丙型肝炎导致的肝癌、肝硬化或疲劳。
49.项目41的方法,进一步包括给药所述化合物与另一种抗-HCV试剂的组合。
50.项目49的方法,其中所述另外的抗-HCV试剂选自蛋白酶抑制剂;NS5A抑制剂;另外的NS5B聚合酶抑制剂;非底物(变构)抑制剂;干扰素α-2a,其可以是PEG基化的;利巴韦林;解螺旋酶抑制剂;反义寡脱氧核苷酸(S-ODN);适体;核酸酶-抗性核酶;iRNA; HCV的抗体;HCV的部分抗体;和HCV的结构域抗体。
51.项目50的方法,其中所述蛋白酶抑制剂选自替拉瑞韦、伯赛匹韦、西咪匹韦和paritaprevir。
52.项目41-51的方法,其中所述宿主为人类。
53.下式的化合物:
其中R
54.药物组合物,包括下述化合物:
其中R
55.下式的化合物:
其中R
56.药物组合物,包括下述化合物:
其中R
57.下述结构的化合物::
其中R
58.药物组合物,包括下述化合物:
其中R
59.选自下述结构的化合物或其可药用盐:
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体;
其中R
60.药物组合物,包括下述化合物或其可药用盐:
其中R
61.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
62.药物组合物,包括下述化合物或其可药用盐:
其中R
63.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
64.药物组合物,包括下述结构或其可药用盐:
其中R
65.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
66.药物组合物,包括下述结构或其可药用盐:
其中R
67.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
68.药物组合物,包括下述化合物或其可药用盐:
其中
R
R
69.选自下述结构的化合物或其可药用盐:
其中R
70.药物组合物,包括下述化合物或其可药用盐:
其中R
71.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
72.药物组合物,包括下述化合物或其可药用盐:
其中
R
R
73.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
74.药物组合物,包括下述化合物或其可药用盐:
其中R
75.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
76.药物组合物,包括下述结构或其可药用盐:
其中
R
R
77.选自下述结构的化合物或其可药用盐:
其中R
78.药物组合物,包括下述结构:
其中R
79.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
80.药物组合物,包括下述化合物或其可药用盐:
其中
R
R
81.选自下述结构的化合物:
其中R
82.药物组合物,包括下述化合物或其可药用盐:
其中R
83.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
84.药物组合物,包括下述化合物或其可药用盐:
其中
R
R
85.选自下述结构的化合物:
其中R
其为分离的磷S对映异构体的形式。
86.药物组合物,包括下述化合物或其可药用盐:
其中R
87.选自下述结构的化合物:
其中
R
R
其为分离的磷S对映异构体的形式,或其可药用盐。
88.药物组合物,包括下述结构或其可药用盐:
其中
R
R
89.选自下述结构的化合物或其可药用盐:
其中R
90.药物组合物,包括下述结构:
其中R
91.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
92.药物组合物,包括下述结构或其可药用盐:
其中
R
R
93.选自下述结构的化合物或其可药用盐:
其中R
94.药物组合物,包括下述化合物:
其中R
或其可药用盐。
95.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
96.药物组合物,包括下述化合物或其可药用盐:
其中
R
R
97.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
98.药物组合物,包括下述结构:
其中R
或其可药用盐。
99.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
100.药物组合物,包括下述结构或其可药用盐:
其中
R
R
101.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
102.药物组合物,包括下述结构或其可药用盐:
其中R
103.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
104.药物组合物,包括下述结构或其可药用盐:
其中
R
R
105.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
106.药物组合物,包括下述结构:
其中R
107.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
108.药物组合物,包括下述结构或其可药用盐:
其中
R
R
109.选自下述结构的化合物或其可药用盐:
其中R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
110.药物组合物,包括下述结构或其可药用盐:
其中R
111.选自下述结构的化合物或其可药用盐:
其中
R
R
其为分离的磷R或S对映异构体或其混合物的形式,所述分离的磷R或S对映异构体具有至少90%的指定对映异构体。
112.药物组合物,包括下述结构或其可药用盐:
其中
R
R
113.项目1、9、14、24或29-32中任一项的化合物,其中R
114.项目1、9、14、24或29-32中任一项的化合物,其中R
115.项目1、9、14、24或29-32中任一项的化合物,其中R
116.项目1、9、14、24或29-32中任一项的化合物,其中所述磷具有S-构型。
117.项目1、9、14、24或29-32中任一项的化合物,其中所述磷具有R-构型。
118.项目1、9、14、24或29-32中任一项的化合物,其中所述氨基酸具有D-构型。
119.项目1、9、14、24或29-32中任一项的化合物,其中所述氨基酸具有L-构型。
120.项目14、24、29、30或32中任一项的化合物,其中R
121.项目31的化合物,其中R
122.项目31的化合物,其中R
123.项目31的化合物,其中R
124.项目32的化合物,其中R
125.项目32的化合物,其中R
126.项目32的化合物,其中R
127.项目1、9、14、24或29-32中任一项的化合物及其可药用盐和前药,用于治疗或预防丙型肝炎病毒感染。.
128.项目1、9、14、24或29-32中任一项的化合物及其可药用盐和前药在制备用于治疗丙型肝炎病毒感染的药物中的用途。
129.一种用于制备预期用于治疗丙型肝炎病毒感染的治疗用途的药物的方法,特征在于在制备中使用项目1、9、14、24或29-32中任一项的化合物。
130.药物制剂,包括有效治疗宿主量的项目1、9、14、24或29-32中任一项的化合物或其可药用盐或前药连同可药用载体或稀释剂。
131.化合物,选自:
其中R
132.下式的化合物或其可药用盐:
其中R4为稳定的磷酸酯前药;和
R
133.化合物或其可药用盐,选自;
其中R4为稳定的磷酸酯前药;和
R
- 用于HCV治疗的β-D-2’-脱氧-2’α-氟-2’-β-C-取代的-2-改性的-N6-取代的嘌呤核苷酸
- 取代的嘌呤核苷酸