胎儿浓度的确定方法及装置
文献发布时间:2024-04-18 19:58:21
技术领域
本申请涉及生物信息技术领域,更具体的说是涉及一种胎儿浓度的确定方法及装置。
背景技术
在无创产前基因检测(Non-Invasive Prenatal Test,NIPT)中,胎儿浓度指的是孕妇的外周血游离脱氧核糖核酸(Cell-free DNA,cfDNA)中,来源于胎儿的脱氧核糖核酸(DeoxyriboNucleic Acid,DNA)的比例。胎儿浓度是NIPT中的重要参数。
针对第二代测序技术,目前已有评估胎儿浓度的方法,例如,利用cfDNA长度的分布特征、利用甲基化特征、利用SNP(单核苷酸多态性)特征、利用男胎的XY染色体含量等确定胎儿浓度。第三代测序技术是单分子测序技术,其测序原理与第二代测序技术有所不同,测序产生的数据特征和第二代测序技术有较大差异,因此,上述用于第二代测序的胎儿浓度评估方法不适用于第三代测序平台。
发明内容
基于第三代测序技术的测序原理与第二代测序不同,第三代测序技术测序产生的数据特征也有不同于第二代测序技术得到的测序数据的特征,这些不同使得两者在应用上也呈现了一定的差异性。如第三代测序平台无法测量插入序列长度,因此无法通过cfDNA长度的分布特征来确定胎儿浓度;又如,第三代测序平台无法测量甲基化信息,因此不适用通过提取甲基化特征的方式来确定胎儿浓度;再如,第三代测序平台的测序原理的差异性,导致其测序过程中产生的错误特征不同与第二代测序产生的错误特征,导致适用于第二代测序的SNP法不能适用于第三代测序平台胎儿浓度的确定。利用XY染色体含量确定胎儿浓度,只能基于胎儿为男胎的前提下进行,而对于性别为女胎的胎儿浓度的确定同样不适应。因此,基于上述情况,需要开发能兼顾各种测序平台、且能够适用于不同性别胎儿的胎儿浓度评估方法。
有鉴于此,本申请提供如下技术方案:
一种胎儿浓度的确定方法,包括:
获取待测cfDNA样本的测序序列;
将所述测序序列与参考基因组进行比对,得到比对结果;
基于所述比对结果,确定胎儿浓度。
作为本申请一种可能的实现方式,所述将所述测序序列与参考基因组进行比对,得到比对结果,包括:
对参考基因组进行分段,得到多个分段区间;
确定每一所述测序序列落入各个所述分段区间的第一比对数,将所述第一比对数确定为比对结果。
作为本申请一种可能的实现方式,所述对参考基因组进行分段,得到多个分段区间,包括:
基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;
剔除所述初始分段区间中特定染色体对应的分段区间,得到多个分段区间;
或者,
剔除参考基因组中的特定染色体,得到剔除后的参考基因组;
基于预设的分割长度对所述剔除后的参考基因组进行分段,得到多个分段区间。
作为本申请一种可能的实现方式,所述特定染色体包括X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体中的至少一种。
作为本申请一种可能的实现方式,所述方法还包括:
对所述第一比对数进行校正,得到校正后的第一比对数,将所述校正后的第一比对数确定为比对结果。
作为本申请一种可能的实现方式,所述对所述第一比对数进行校正,得到校正后的第一比对数,包括:
基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;
对每一分段区间所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述对每一分段区间所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线;
基于所述第一关系曲线对所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,在所述基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线的步骤之前,还包括:
基于GC含量对所述分段区间进行过滤,使得基于GC过滤后的每个分段区间的GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线;
或者,
所述基于所述第一关系曲线对所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
基于GC含量对所述分段区间进行过滤,并基于所述第一关系曲线对GC过滤后的归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述GC校正为根据所述归一化第一比对数以及与所述归一化第一比对数对应的分段区间的GC含量的减法关系式或者除法关系式,确定GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述对所述第一比对数进行校正,得到校正后的第一比对数,还包括:
基于特定滑窗长度对参考基因组进行截取,将截取到的序列与所述参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将所述第一比对数确定为滑窗比对数;
基于所述滑窗比对数,确定每一分段区间归一化后的滑窗比对数;
基于每一所述分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于每一所述分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数,包括:
基于每一所述分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线;
基于所述第二关系曲线对所述归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于每一所述分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线的步骤之前,还包括:
基于归一化后的滑窗比对数对分段区间进行过滤,获得归一化后的滑窗对比数不小于第一目标阈值的分段区间,以使得所述第二关系曲线在归一化后的滑窗比对数不小于所述第一目标阈值的分段区间生成;
或者,
所述基于所述第二关系曲线对所述归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数,包括:
基于归一化后的滑窗比对数,对所述分段区间进行过滤,保留所述第二关系曲线中归一化后的滑窗比对数不小于所述第一目标阈值的第二关系曲线区间,以得到比对概率过滤后的第二关系曲线;
基于所述比对概率过滤后的第二关系曲线对所述滑窗归一化比对数进行比对概率校正,得到比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于所述比对结果,确定胎儿浓度,包括:
获得第一训练样本数据,所述第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,所述第一特征值为样本比对数,所述第一目标值为样本实际胎儿浓度;
基于特定模型结构对所述第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
将所述比对结果输入至所述第一胎儿浓度定量模型,得到第一胎儿浓度,并将所述第一胎儿浓度确定为所述胎儿浓度。
作为本申请一种可能的实现方式,所述基于特定模型结构对所述第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型,包括:
将所述第一训练样本数据划分为训练集和测试集;
基于所述训练集进行机器学习建模,得到初始模型;
基于所述初始模型对所述测试集进行处理,得到与所述测试集中每一测试样本的预估胎儿浓度;
基于所述预估胎儿浓度与所述测试集中每一测试样本的实际胎儿浓度进行比较,得到比较结果;
基于所述比较结果对所述初始模型的模型参数进行调整,得到第一胎儿浓度定量模型。
作为本申请一种可能的实现方式,所述基于所述比对结果,确定胎儿浓度,包括:
将所述比对结果输入至第一预设模型,得到初始胎儿浓度,其中,所述第一预设模型为基于第二样本数据中的对比结果以及与所述比对结果对应的初始胎儿浓度建立的线性关系模式,所述第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;
根据第二预设模型对所述初始胎儿浓度进行校正,获得胎儿浓度,其中,所述第二预设模型是基于线性拟合确定的常数对所述第一预设模型进行处理后得到的模型。
作为本申请一种可能的实现方式,所述将所述测序序列与参考基因组进行比对,得到比对结果,包括:
将参考基因组的各碱基位点为所述测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于所述第二比对数,计算所述参考基因组的每个碱基位点对应的核小体中心分数;
基于所述核小体中心分数以及中心分数筛选阈值,确定核小体中心位置;
基于所述核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数;
将所述加和处理后的第二比对数进行降维处理,得到降维后的归一化第二比对数,并将所述降维后的归一化第二比对数确定为比对结果。
作为本申请一种可能的实现方式,所述计算所述参考基因组的每个碱基位点对应的核小体中心分数,包括:
计算所述参考基因组的每个碱基位点左右分别对应的第一特定数量的碱基范围的第一平均计数;
计算所述参考基因组的每个碱基位点左右分别对应的第二特定数量的碱基范围的第二平均计数;
根据所述第一平均计数和所述第二平均计数,确定每个位点对应的核小体中心分数。
作为本申请一种可能的实现方式,所述核小体中心分数的计算公式为:
式中,x为碱基位点,[x-93,x-74-n]表示从距离x一侧93个核苷酸到同侧距离x74-n个核苷酸的区间,[x+93,x+74-n]表示从距离x另一侧93个核苷酸到同侧距离x 74-n个核苷酸的区间,[x-73-n,x+73-n]表示从距离x另一侧73-n个核苷酸到距离x另一侧73-n个核苷酸的区间,n为小于或等于5的自然数。
作为本申请一种可能的实现方式,所述基于所述核小体中心分数以及中心分数筛选阈值,确定核小体中心分数,包括:
基于所述核小体中心分数的最大值,确定所述最大值在所述参考基因组上的位置,并将所述位置确定为第一位置;
将所述第一位置两侧特定数据的碱基的核小体中心分数清零,并基于清零后剩余的核小体中心分数再次确定最大值,并执行确定最大值在所述参考基因组上的位置,直至核小体中心分数小于第二目标阈值,将筛选出的位置确定为候选核小体中心位置;
基中心分数筛选阈值以及与所述候选核小体中心位置对应的核小体中心分数,确定核小体中心位置。
作为本申请一种可能的实现方式,所述基于所述核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理的步骤中,所述核小体区域内对应位置包括:
以所述核小体中心位置为中心,左右各特定数量的碱基的范围。
作为本申请一种可能的实现方式,所述基于所述比对结果,确定胎儿浓度,包括:
获取第二训练样本数据,所述第二训练样本数据中的每一样本均标注有特征值和目标值,所述特征值为所述降维后的归一化第二比对数,所述目标值为实际胎儿浓度;
基于所述第二训练样本数据创建第二胎儿浓度定量模型;
将所述比对结果输入至所述第二胎儿浓度定量模型,得到胎儿浓度。
一种胎儿浓度的确定方法,包括:
获取待测cfDNA样本的测序序列;
对参考基因组进行分段,得到多个分段区间,确定每一所述测序序列落入各个所述分段区间的第一比对数;
基于所述第一比对数,确定第一胎儿浓度;
基于所述参考基因组的各碱基位点是所述测序序列比对起始位置的第二比对数,确定核小体中心位置;
基于所述核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数;
基于所述第一胎儿浓度和所述加和处理后的第二比对数,确定比对结果;
基于所述比对结果,确定胎儿浓度。
作为本申请一种可能的实现方式,所述对参考基因组进行分段,得到多个分段区间,包括:
基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;
剔除所述初始分段区间中特定染色体对应的分段区间,得到多个分段区间;
或者,
剔除参考基因组中的特定染色体,得到剔除后的参考基因组;
基于预设的分割长度对所述剔除后的参考基因组进行分段,得到多个分段区间。
作为本申请一种可能的实现方式,所述特定染色体包括X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体中的至少一种。
作为本申请一种可能的实现方式,所述基于所述第一比对数,确定第一胎儿浓度的步骤之前,还包括:
对所述第一比对数进行校正,得到校正后的第一比对数,将所述校正后的第一比对数确定第一胎儿浓度。
作为本申请一种可能的实现方式,所述对所述第一比对数进行校正,得到校正后的第一比对数,包括:
基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;
对每一分段区间所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述对每一分段区间所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
基于GC含量和每个分段区间对应的归一化第一比对数,生成与特定坐标系相匹配的第一关系曲线;
基于所述第一关系曲线对所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,在所述基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线的步骤之前,还包括:
基于GC含量对所述分段区间进行过滤,使得基于GC过滤后的每个分段区间的GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线;
或者,
所述基于所述第一关系曲线对所述归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
基于GC含量对所述分段区间进行过滤,并基于所述第一关系曲线对GC过滤后的归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述GC校正为根据所述归一化第一比对数以及与所述归一化第一比对数对应的分段区间的GC含量的减法关系式或者除法关系式,确定GC校正后的第一比对数。
作为本申请一种可能的实现方式,所述对所述第一比对数进行校正,得到校正后的第一比对数,还包括:
基于特定滑窗长度对参考基因组进行截取,将截取到的序列与所述参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将所述第一比对数确定为滑窗比对数;
基于所述滑窗比对数,确定每一分段区间归一化后的滑窗比对数;
基于每一所述分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于每一所述分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数,包括:
基于每一所述分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线;
基于所述第二关系曲线对所述归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于每一所述分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线的步骤之前,还包括:
基于归一化后的滑窗比对数对分段区间进行过滤,获得归一化后的滑窗对比数不小于第一目标阈值的分段区间,以使得所述第二关系曲线在归一化后的滑窗比对数不小于所述第一目标阈值的分段区间生成;
或者,
所述基于所述第二关系曲线对所述归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数,包括:
基于归一化后的滑窗比对数,对所述分段区间进行过滤,保留所述第二关系曲线中归一化后的滑窗比对数不小于所述第一目标阈值的第二关系曲线区间,以得到比对概率过滤后的第二关系曲线;
基于所述比对概率过滤后的第二关系曲线对所述滑窗归一化比对数进行比对概率校正,得到比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,所述基于所述第一比对结果,确定第一胎儿浓度,包括:
获得第一训练样本数据,所述第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,所述第一特征值为比对数,所述第一目标值为实际胎儿浓度;
基于特定模型结构对所述第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
将所述第一比对结果数输入至所述第一胎儿浓度定量模型,得到第一胎儿浓度。
作为本申请一种可能的实现方式,所述基于特定模型结构对所述第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型,包括:
将所述第一训练样本数据划分为训练集和测试集;
基于所述训练集进行机器学习建模,得到初始模型;
基于所述初始模型对所述测试集进行处理,得到与所述测试集中每一测试样本的预估胎儿浓度;
基于所述预估胎儿浓度与所述测试集中每一测试样本的实际胎儿浓度进行比较,得到比较结果;
基于所述比较结果对所述初始模型的模型参数进行调整,得到第一胎儿浓度定量模型。
作为本申请一种可能的实现方式,所述基于所述第一比对数,确定第一胎儿浓度,包括:
将所述第一比对数输入至第一预设模型,得到初始胎儿浓度,其中,所述第一预设模型为基于第二样本数据中的对比结果以及与所述比对结果对应的初始胎儿浓度建立的线性关系模式,所述第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;
根据第二预设模型对所述初始胎儿浓度进行校正,获得第一胎儿浓度,其中,所述第二预设模型是基于线性拟合确定的常数对所述第一预设模型进行处理后得到的模型。
作为本申请一种可能的实现方式,所述基于所述参考基因组的各碱基位点是所述测序序列比对起始位置的第二比对数,确定核小体中心位置,包括:
将参考基因组的各碱基位点为所述测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于所述第二比对数,计算所述参考基因组的每个碱基位点对应的核小体中心分数;
基于所述核小体中心分数以及中心分数筛选阈值,确定核小体中心位置。
作为本申请一种可能的实现方式,所述基于所述第一胎儿浓度和所述加和处理后的第二比对数,确定比对结果,包括:
将核小体区域内各碱基位点对应的所述加和处理后的第二比对数进行降维处理,得到降维后的归一化第二比对数;
基于所述第一胎儿浓度和所述降维后的归一化第二比对数确定为比对结果;
或者,
基于所述第一胎儿浓度和所述加和处理后的第二比对数,确定初始比对结果;
对所述初始比对结果进行降维处理,将降维后的初始比对结果确定为比对结果。
作为本申请一种可能的实现方式,所述计算所述参考基因组的每个碱基位点对应的核小体中心分数,包括:
计算所述参考基因组的每个碱基位点左右分别对应的第一特定数量的碱基范围的第一平均计数;
计算所述参考基因组的每个碱基位点左右分别对应的第二特定数量的碱基范围的第二平均计数;
根据所述第一平均计数和所述第二平均计数,确定每个位点对应的核小体中心分数。
作为本申请一种可能的实现方式,所述核小体中心分数的计算公式为:
式中,x为碱基位点,[x-93,x-74-n]表示从距离x一侧93个核苷酸到同侧距离x74-n个核苷酸的区间,[x+93,x+74-n]表示从距离x另一侧93个核苷酸到同侧距离x 74-n个核苷酸的区间,[x-73-n,x+73-n]表示从距离x另一侧73-n个核苷酸到距离x另一侧73-n个核苷酸的区间,n为小于或等于5的自然数。
作为本申请一种可能的实现方式,所述基于所述核小体中心分数以及中心分数筛选阈值,确定核小体中心分数,包括:
基于所述核小体中心分数的最大值,确定所述最大值在所述参考基因组上的位置,并将所述位置确定为第一位置;
将所述第一位置两侧特定数据的碱基的核小体中心分数清零,并基于清零后剩余的核小体中心分数再次确定最大值,并执行确定最大值在所述参考基因组上的位置,直至核小体中心分数小于第二目标阈值,将筛选出的位置确定为候选核小体中心位置;
基中心分数筛选阈值以及与所述候选核小体中心位置对应的核小体中心分数,确定核小体中心位置。
作为本申请一种可能的实现方式,所述基于所述核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理的步骤中,所述核小体区域内对应位置包括:
以所述核小体中心位置为中心,左右各特定数量的碱基的范围。
作为本申请一种可能的实现方式,所述基于所述比对结果,确定胎儿浓度,包括:
获取第二训练样本数据,所述第二训练样本数据中的每一样本均标注有特征值和目标值,所述特征值为所述降维后的归一化第二比对数,所述目标值为实际胎儿浓度;
基于所述第二训练样本数据创建第二胎儿浓度定量模型;
将所述比对结果输入至所述第二胎儿浓度定量模型,得到胎儿浓度。
一种胎儿浓度的确定装置,包括:
第一获取单元,用于获取待测cfDNA样本的测序序列;
第一比对单元,用于将所述测序序列与参考基因组进行比对,得到比对结果;
第一确定单元,用于基于所述比对结果,确定胎儿浓度。
经由上述的技术方案可知,本申请公开了一种胎儿浓度的确定方法及装置,包括:获取待测cfDNA样本的测序序列;将测序序列与参考基因组进行比对,得到比对结果;基于比对结果,确定胎儿浓度。本申请能够基于测序序列与参考基因组的比对结果确定胎儿浓度,可以满足包括单分子测序平台在内的各种测序平台的应用,提升了胎儿浓度确定的准确性。此外,本申请提供的胎儿浓度的确定方法,不依赖于对性别具有依赖性的特征进行识别和确认,因此,能够同时适用于男胎和女胎的胎儿浓度的确定,弥补第三代测序平台不能有效评估女胎胎儿浓度的不足。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1为本申请实施例提供的一种胎儿浓度的确定方法的流程示意图;
图2为本申请实施例提供的一种Enet模型计算的胎儿浓度与参考胎儿浓度关系的示意图;
图3为本申请实施例提供的一种WRSC模型计算的胎儿浓度与参考胎儿浓度关系的示意图;
图4为本申请实施例提供的一种gmFF_V7原始参数计算出的胎儿浓度与参考胎儿浓度关系的示意图;
图5为本申请实施例提供的一种种gmFF_V7经线性校正后计算出的胎儿浓度与参考胎儿浓度关系的示意图;
图6为本申请实施例提供的一种一组数据按照核小体区域对齐并加和之后获得的分布的示意图;
图7为本申请实施例提供的一种随机拆分测试集训练集后的模型预测效果的示意图;
图8为本申请实施例提供的另一种胎儿浓度的确定方法的流程示意图;
图9为本申请实施例提供的一种胎儿浓度的确定装置的结构示意图;
图10为本申请实施例提供的另一种胎儿浓度的确定装置的结构示意图。
具体实施方式
下面将结合本申请实施例及其附图,对本申请的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本申请一部分实施例,而不是全部的实施例。基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本申请保护的范围。
本申请的说明书和权利要求书及上述附图中的术语“第一”和“第二”等是用于区别不同的对象,而不是用于描述特定的顺序,也不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。例如,在不脱离本申请实施例范围的情况下,本申请中的“第一对比数”和“第二比对数”的表述可以对换。
术语“包括”和“具有”以及他们任何变形,意图在于覆盖不排他的包含。例如包含了一系列步骤或单元的过程、方法、系统、产品或设备,并不限定于已列出的步骤或单元,而是可包括没有列出的步骤或单元。
术语“测序”又可称为“核酸测序”或“基因测序”,即三者在表述上可以互换,指核酸序列中碱基类型和排列顺序的测定;包括合成测序(边合成边测序,SBS)和/或连接测序(边连接边测序,SBL),包括DNA测序和/或RNA测序,包括长片段测序和/或短片段测序,所称的长片段和短片段是相对的,如长于1Kb、2Kb、5Kb或者10Kb的核酸分子可称为长片段,短于1Kb或者800bp的可称为短片段。
术语“单分子测序技术”是指基于碱基信号采集单元单拷贝的测序技术,即无需使用特定的扩增技术对待测的核酸分子进行信号放大,可直接进行核酸分子序列的读取的技术。术语“单分子测序平台”是指基于单分子测序技术的测序平台,包括以单分子测序技术为基础的各种测序仪,包括但不限于真迈生物的Genocare测序平台。
在本申请实施例提供了一种胎儿浓度的确定方法,该方法可以应用于各种测序平台对胎儿浓度进行评估,特别是通过该方法,可以在单分子测序平台的基础上,建立胎儿浓度评估方法,特别是适用于女胎的胎儿浓度评估方法。
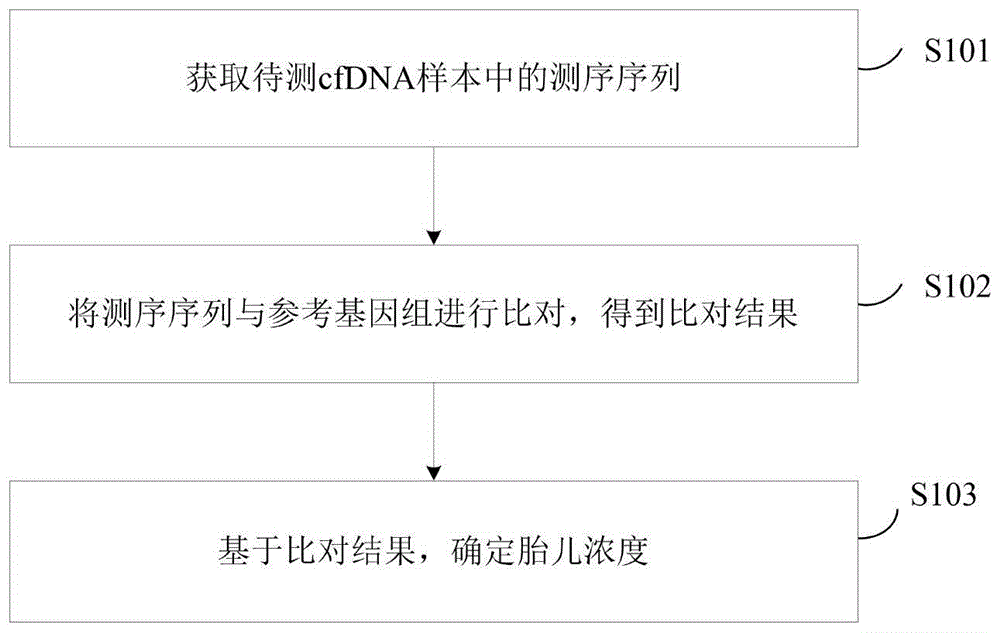
参见图1,为本申请实施例提供的一种胎儿浓度的确定方法的流程示意图,该方法可以包括以下步骤:
S101.获取待测cfDNA样本的测序序列。
其中,cfDNA指存在于人体血液循环中的游离于细胞外的高度片段化DNA。在本申请实施例中,待测cfDNA样本是指需要确定胎儿浓度的cfDNA样本。示例性的,cfDNA样本可以为含有游离脱氧核糖核酸(或游离DNA)的孕妇外周血。
本申请实施例中,待测cfDNA样本是指有待测定胎儿浓度的含有cfDNA的样本,待测cfDNA样本的测序序列是指待测样本中cfDNA经测序获得的序列,即待测样本中片段化DNA的测序序列。应当理解的是,待测cfDNA样本的测序序列对测序仪器和测序平台没有要求,即待测cfDNA样本的测序序列可以来源于任何测序平台所得到的的测序数据。在一些实施例中,待测cfDNA样本的测序序列可以通过单分子测序平台(真迈生物的Genocare测序平台、纳米孔测序平台等)获得,也可以用第二测序平台(如二代测序的illumina平台、华大平台)获得。示例性的,待测cfDNA样本的测序序列为基于单分子测序平台对待测cfDNA样本进行DNA测序得到的数据。由此,可通过本申请实施例提供的方法实现来源于单分子测序平台的待测cfDNA样本的测序序列,实现待测cfDNA样本胎儿浓度的分析。
S102.将测序序列与参考基因组进行比对,得到比对结果。
该步骤中,参考基因组是指人类参考基因组,示例性的,如人类参考基因组hg19,但不限于此。
S103.基于比对结果,确定胎儿浓度。
在本申请实施例中为了能够准确获得最终的胎儿浓度,通过测序序列与参考基因组进行比对,得到的比对结果可以是母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率,可以是母体与婴儿的cfDNA的两端在核小体上位置的分布差异对应的比对结果,也可以是母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率和母体与婴儿的cfDNA的两端在核小体上位置的分布差异对应的比对结果,以及还可以是对上述两种比对结果进行处理后新形成的比对结果。
下面对本申请实施例提供的胎儿浓度的确定方法进行详细说明。
利用母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率差异来确定胎儿浓度。
在母体的外周血获得的游离DNA(cfDNA)中,一部分来源于母体,一部分来源于胎儿,来源于母体和胎儿的DNA与参考基因组进行比对时,分别在参考基因组不同区域的覆盖深度会有一定的差异,因此,可以根据该差异对应的比对结果来确定胎儿浓度。
在本申请实施例的一种实施方式中,利用母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率差异来确定胎儿浓度。具体的,胎儿浓度的确定方法包括:
S111.获取待测cfDNA样本的测序序列。
该步骤中,待测cfDNA样本为需要确定胎儿浓度的cfDNA样本。本申请实施提供的方法,对待测cfDNA样本的来源以及待测cfDNA样本的特征参数等没有明确要求。
S112.将测序序列与参考基因组进行比对,得到比对结果。
该步骤包括:
S1121.对参考基因组进行分段,得到多个分段区间。
S1122.确定每一测序序列落入各个所述分段区间的第一比对数,以使得将所述第一比对数确定为比对结果。
需要说明的是,该第一比对数中的“第一”仅是与后续出现的比对数进行区分,“第一”和“第二”并不存在先后顺序关系。该方法将参考基因组按照固定的长度分段,然后对落入每一分段区间的测序序列的数量进行统计,即计算有多少条测序序列落入了到对应的分段区间内,每一分段区间的测序序列的落入数量即为第一比对数,也为该场景下的比对结果。对应的,在某些场景中也可以将每个分段区间内落入的测序序列的数量称为“覆盖数”,为了便于描述在本申请后续实施例中称为比对数,并且与其他类型的比对结果进行区分,称为“第一比对数”。
应当理解的是,本申请实施例所述的“第一比对数”,可以是落入对应的分段区间内种的测序序列的绝对数量,也可以是落入对应的分段区间内种的测序序列的中位数。另外,在统计过程中,对落入每一分段区间的测序序列的数量进行统计,允许一定的容错比例出现,即允许一定长度范围内存在预设数量碱基错误的测序序列,作为比对上的测序序列。
为了能够准确获得比对结果,可以按照固定的长度对参考基因组进行分段,如根据测序序列的特征来确定参考基因组的分割长度。
参考基因组中,存在一些特殊的染色体,这些染色体可能在通过参考基因组比对方式确定胎儿浓度的过程中存在一定的偏向性或其他特殊性。在一种实施情形中,通过去除参考基因组中特殊的染色体(下文称为特定染色体)带来的分段区间,来减少这些染色体带来的偏向性,从而降低其特殊性对测试结果的影响。在一些实施例中,特定染色体包括X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体中的至少一种。其中,通过去除X染色体和Y染色体对应的分段区间,避免男胎和女胎上述染色体的差异,对后续浓度定量模型的结果造成的影响;去除13号染色体、18号染色体和21号染色体,是因为这几条染色体本身存在较大的重复缺失概率。而NIPT的筛查目标之一是对13、18、21号染色体的重复缺失情况进行筛查,当以13号染色体、18号染色体和21号染色体作为参考染色体时,对数据进行归一化统计时可能出现偏差,如影响归一化比对数,进而对计算结果造成影响,影响NIPT筛查结果的准确性。
本申请实施例可以根据胎儿浓度检测预期的需要,调整特定染色体的范围,如剔除X染色体和Y染色体,或剔除13号染色体、18号染色体和21号染色体,或剔除线粒体染色体。当然,可以对上述情形中的多种情形同时去除,如将X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体或其对应的分段片段全部去除,以使参考基因组分段后得到的分段区间中不含有上述染色体对应的分段区间。
本申请可以通过多种方式去除特定染色体进入分段区间。可以对分段后得到的分段区间基于特定染色体进行筛选,也可以先将这些特定染色体进行剔除后再分段,具体的,可以基于实际的应用需求选取对应的处理方式,本申请对此不进行限制。
在一种实施方式中,对参考基因组进行分段,得到多个分段区间,包括:基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;剔除初始分段区间中特定染色体对应的分段区间,得到多个分段区间。即在将参考基因组分段为多个分段区间后,对参考基因组中特定染色体对应的分段区间进行剔除,不参与计算。应当理解的是,所指的“特定染色体对应的分段区间”,是指特定染色体按照预设的分割长度进行分段后,形成的多个分段区间。
在另一种实施方式中,对参考基因组进行分段,得到多个分段区间,包括:剔除参考基因组中的特定染色体,得到剔除后的参考基因组;基于预设的分割长度对所述剔除后的参考基因组进行分段,得到多个分段区间。即在将参考基因组分段之前,先剔除特定染色体。
其中,预设的分割长度可以根据数据处理的数据量以及预期的相对准确度等调整,例如可以按照每50k(5000)碱基数量的长度对参考基因组进行分段。为了能够使得获得的第一比对数更加准确,还可以对第一比对数进行校正,得到校正后的第一比对数,以使得将校正后的第一比对数确定为比对结果。
在一些实施例中,校正包括GC校正。GC校正是生物信息学分析过程中的操作,之所以进行GC校正是基因测序仪以及对应的测序流程会造成一定的GC偏好,例如,某些测序仪测出高GC序列的概率更高,而某些测序仪测出低测出低GC序列的概率更高。在该实施方式中,确定胎儿浓度的处理方式是基于参考基因组上不同区域的比对概率进行的,而测序仪的GC偏好将会影响对真实的不同区域的比对概率的判断,为了提升准确性,因而需要进行GC校正。
此时,对第一比对数进行校正,得到校正后的第一比对数,包括:基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数。其中,所有分段区间的平均第一比对数是指,各分段区间的第一比对数之和与分段区间的总数量的比值。
在获得了所有分段区间的第一比对数之后,为了便于计算和处理,会对所有分段区间的第一比对数进行归一化处理,对应的公式为:
每个分段区间归一化第一比对数=每个分段区间的第一比对数/所有分段区间的平均第一比对数。
获得了每一分段区间的第一比对数之后,将进行GC校正,得到GC校正后的第一比对数。
在一种实施方式中,对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
(1)基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线。
该实施方式中,GC含量可以通过多种参数来表示,如每个分段区间的GC含量或GC含量中位数,还可以是能够反馈分段区间GC含量的其他参数。
本申请实施例中,可以通过对形成第一关系曲线的分段区间进行GC过滤,如根据GC含量对分段区间进行过滤,以过滤掉一些异常或者不具备统计意义的区段,优化第一关系曲线,降低了在数据处理过程中数据处理资源的占用。
在一种实施方式中,GC过滤通过下述方式实现:在基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线的步骤之前,进行如下处理:基于GC含量对分段区间进行过滤,去除GC含量不满足预设要求的分段区间,使得基于GC过滤后的每个分段区间的GC含量和每个分段区间的归一化第一比对数,生成第一关系曲线。
在另一种实施方式中,GC过滤通过下述方式实现:基于第一关系曲线进行GC校正对归一化第一比对数的过程中,基于GC含量对分段区间进行过滤,并基于第一关系曲线对GC过滤后的归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
上述实施例中,作为一种示例,基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线,通过通过下述方式实现:以x轴表示每个分段区间的GC含量(即参考基因组上该分段区间对应的GC碱基的占比),y轴为每个分段区间对应的归一化第一比对数。整合每个分段区间及其区间内的GC含量的数据,即根据每个分段区间的GC含量以及对应的归一化第一比对数,生成散点图。对散点图中的散点数据进行平滑,获得平滑曲线。其中,对散点图中的散点数据进行平滑可以利用如LOWESS算法进行,获得平滑曲线,即获得第一关系曲线,如y
具体的,作为一种示例,第一关系曲线可以通过下述方式生成:将横坐标分为不同的区段,然后计算同一区段内散点的GC含量中位数,利用这个GC含量中位数来代表这一区段对应的数值,再将所有这些GC含量中位数画到图上进行平滑,获得第一关系曲线。在另一种示例中,可以基于所有的数据直接进行平滑处理,即将所有数据点均纳入考虑,共同平滑获得平滑曲线,这样测试集获得的斯皮尔曼相关系数更高。需要说明的是,这种平滑方法同样适用于后续描述的对比概率的平滑和校正。
(2)基于第一曲线对归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
在一些实施方式中,根据归一化第一比对数以及归一化第一比对数对应的分段区间的GC含量的减法关系式或者除法关系式进行GC校正,确定GC校正后的第一比对数。
在一种示例中,GC校正的方法为“减法”,对应的校正公式为:y
上述两个校正公式中,x为所述分段区间的GC含量,y为该分段区间对应的归一化第一比对数,y
在一些实施例中,校正还包括比对概率校正,以提升获得的第一比对数的准确率。即在一些实施例中,校正同时包括GC校正和比对概率校正。
在本申请实施例的一种实施方式中,对第一比对数进行校正,得到校正后的第一比对数,还包括:基于特定滑窗长度对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将第一比对数确定为滑窗比对数;基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;基于每一分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
本申请实施例采用“比对概率校正”方法对第一比对数进行校正,基于以下考虑:参考基因组对应的参考序列的不同区域,如果按照特定长度截取下来当作一条测序序列,然后再比对回参考基因组时,不同的分段区间被比对上的次数会有差异,因为对于一些区域的序列,在参考基因组的参考序列上的多处都存在着与其类似的序列,因而这些区域更容易被比对上。例如,将人类基因组参考序列hg19按照2个碱基的步长、37个碱基的窗口进行滑窗截取序列,将这些序列与人类参考基因组hg19进行比对,然后统计在人类参考基因组的不同分段区域上的比对数,可以把这个比对数称为“滑窗比对数”。具体的,特定滑窗长度可以基于实际的应用场景考虑,如基于采用的参考基因组的特征,或者待测样本的特征等确定。在确定了特定滑窗长度后对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组上的每一分段区间上的第一比对数,将此时的第一比对数确定为滑窗比对数。在获得了滑窗比对数之后,为了便于计算和处理,基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;对归一化后的滑窗比对数进行比对概率校正。比对概率校正的方法,可以参考前文GC校正的方法。
在一种实施方式中,基于每一分段区间归一化后的滑窗比对数和归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数,包括:
(1)基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线。
其中,每一分段区间归一化后的滑窗比对数可以通过多种参数来表示,如每一分段区间的归一化后的滑窗比对数或滑窗比对数的中位数,还可以是能够反馈每一分段区间归一化后的滑窗比对数的其他参数。
本申请实施例中,可以通过对形成第二关系曲线的归一化的滑窗比对数进行过滤,以过滤掉一些异常或者不具备统计意义的区段,优化第二关系曲线,提升基于第二关系曲线进行相关数据处理时的处理准确性。
在一种实施方式中,对形成第二关系曲线的归一化的滑窗比对数进行过滤,通过下述方式实现:在基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线的步骤前,进行如下处理:基于归一化的滑窗比对数对分段区间进行过滤,获得归一化后的滑窗比对数不小于第一目标阈值的分段区间,以使得第二关系曲线在归一化后的滑窗比对数不小于第一目标阈值的分段区间生成。
在一种实施方式中,对形成第二关系曲线的归一化的滑窗比对数进行过滤,通过下述方式实现:基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数的过程中,基于归一化后的滑窗比对数,对分段区间进行过滤,保留第二关系曲线中归一化后的滑窗比对数不小于第一目标阈值的第二关系曲线区间,以得到比对概率过滤后的第二关系曲线;基于比对概率过滤后的第二关系曲线对每一分段区间对应的滑窗归一化比对数进行比对概率校正,得到比对概率校正后的第一比对数。
上述实施例中,基于反复验证,第一目标阈值可以选取0.8,即去掉归一化后的滑窗比对数小于0.8的分段区间。
上述实施例中,作为一种示例,基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线,通过通过下述方式实现:以x轴表示每个分段区间归一化后的滑窗比对数,y轴为每个分段区间对应的归一化第一比对数,整合每个分段区间及其对应的GC含量的数据,即根据每个分段区间的GC含量以及对应的归一化第一比对数,生成散点图;对散点图中的散点数据进行平滑,获得平滑曲线。示例性的,对散点图中的散点数据进行平滑可以利用LOWESS算法实现。
(2)基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数。
在一些实施方式中,根据归一化第一比对数以及归一化第一比对数对应的分段区间的滑窗比对数的减法关系式或者除法关系式进行比对概率校正,确定比对概率校正后的第一比对数。
在一种示例中,比对概率校正的方法为“减法”,对应的校正公式为:y
上述两个校正公式中,x为每个分段区间归一化后的滑窗比对数,y对应的就是每个分段区间的归一化第一比对数,y
S113、基于比对结果,确定胎儿浓度。
在本申请实施例中可以结合神经网络的机器学习方法确定胎儿浓度。在本申请实施例的一种实施方式中,基于对比对结果,确定胎儿浓度,包括:
获得第一训练样本数据,所述第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,第一特征值为样本比对数,第一目标值为样本实际胎儿浓度;
基于特定模型结果对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
将比对结果输入至第一胎儿浓度定量模型,得到第一胎儿浓度,并将所述第一胎儿浓度确定为胎儿浓度。
在一些实施例中,基于特定模型结构对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型,包括:
将第一训练样本数据划分为训练集和测试集;基于训练集进行机器学习建模,得到初始模型;
基于初始模型对测试集进行处理,得到与测试集中每一测试样本的预估胎儿浓度;
基于预估胎儿浓度与测试集中每一测试样本的实际胎儿浓度进行比较,得到比较结果;
基于比较结果对初始模型的模型参数进行调整,得到第一胎儿浓度定量模型。
其中,第一训练样本数据可以是基于包括单分子测序平台在内的各种测序平台获得的已知胎儿浓度的孕妇外周血cfDNA的数据库中的数据,例如,采用一定数含量的已知胎儿浓度的样本,对参考基因组染色体参考序列进行区间分段(如,可通过5k碱基、50k碱基、100k碱基、300k碱基或者800k碱基等分段),并分别统计每个样本的比对数据在不同分段区间的比对数,并利用LOWESS法对这些比对数进行归一化、GC和比对概率的校正,将最终归一化和校正后不同分段区间的比对数作为特征值,将每个样本对应的已知的实际胎儿浓度作为目标值,用特定的模型进行学习和建模,其中,该特定的模型可以是分类模型、卷积神经网络模型等本申请实施例对特定模型结构不进行限制,只要能够对训练样本数据进行学习,并能预测出胎儿浓度即可。模型学习和训练的过程是一个反复迭代的过程,因此可以将训练样本分为训练集和测试集,通过测试集测试当前训练得到的模型的准确度,若准确度低于设定的准确度阈值,则通过测试集对当前的模型的模型结构中的模型参数进行调整,直至获得的模型输出的胎儿浓度与实际的胎儿浓度的误差满足对应的误差范围即可。
当用于机器学习的样本数量本身较少,而特征值较多(例如约三千到六十万个特征,具体取决于染色体参考序列区间分段的大小)时,无论是降维还是学习的过程中,都无法避免过拟合的影响。最终建立的胎儿浓度定量模型中,对测试集胎儿浓度的预测效果会与实际的胎儿浓度存在一定偏差。如采用615例已知胎儿浓度的样本建立的胎儿浓度定量模型中,模型对应测试集R
在另一种实施方式中,基于比对结果,确定胎儿浓度,包括:
将比对结果输入至第一预设模型,得到初始胎儿浓度;
根据第二预设模型对初始胎儿浓度进行校正,获得胎儿浓度。
其中,第一预设模型为基于第二样本数据中的比对结果以及与比对结果对应的初始胎儿浓度建立的线性关系模式,第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;第二预设模型是基于现象拟合确定的常数对第一预设模型处理后得到的模型。
示例性的,第一预设模型可以为
示例性的,利用样本在参考基因组参考序列不同区间的比对概率差异来建立模型并估算胎儿浓度的模型为SeqFF。SeqFF法中用到了两种机器学习模型,一种是权重排序选择标准(WRSC),另一种是弹性网络(Enet),其最终输出的胎儿浓度结果是两个模型预测值的平均值。当利用测序平台特别是单分子测序平台的数据测试SeqFF模型,其中WRSC模型的效果优于Enet,即WRSC估算的胎儿浓度与实际胎儿浓度的线性相关系数比Enet或者SeqFF要高,因而优先考虑WRSC算法涉及的模型参数。具体的,可以参见图2和图3,图2为本申请实施例提供的一种Enet模型计算的胎儿浓度与参考胎儿浓度关系的示意图,图3为本申请实施例提供的一种WRSC模型计算的胎儿浓度与参考胎儿浓度关系的示意图。由图可见,WRSC模型计算的胎儿浓度与参考胎儿浓度之间正相关性优于Enet模型计算的胎儿浓度与参考胎儿浓度之间的正相关性。
示例性的,WRSC模型中,按照特定数量碱基如50k碱基的区间对染色体参考序列进行划分。通过WRSC算法训练,找出了不同参考序列区间上的覆盖密度与胎儿浓度之间的线性关系,如下列公式所展示。下列公式中,FF为胎儿浓度,
由于SeqFF是基于二代测序数据训练处的模型,其测序原理与单分子测序有较差差异,因而若直接采用SeqFF的上述模型参数计算包括单分子测序平台在内的其他测序平台得到的数据的胎儿浓度,计算出的胎儿浓度与实际胎儿浓度差异较大。仍利用上述实施例中的615例已知胎儿浓度的NIPT临床样本为例进行分析和测试。直接使用SeqFF的算法和参数对包括单分子测序平台在内的其他测序平台原始的比对文件数据(如,sam文件)进行处理,获得的中间参数与真实胎儿浓度之间的皮尔森相关系数为0.714。
SeqFF模型计算出的胎儿浓度的计算值与实际胎儿浓度值线性关系较好,因而可以采用上述公式中的
仍利用上述实施例中的615例已知胎儿浓度的NIPT临床样本为例进行分析和测试。改为仅使用测序序列落入各个分段区间的第一比对数进行分析;剔除参考基因组中X、Y、M、13、18、21以及线粒体染色体上比对概率异常的区域进行分析;对校正方法(GC校正,或GC和比对概率校正)进行优化等。使用优化后的参数重新对数据进行分析,中间参数与胎儿浓度之间的皮尔森相关系数为0.755。随机进行100次测试集(30%)和训练集(70%)的拆分,用于建立线性模型、确定上述常数参数c和d。100次建模获得的平均测试集预测R
需要说明的是,真正建立线性模型时,测试集和训练集的拆分是为了在有限的数据中评估我们目前随机拆分的训练集建立的模型的可信程度。因此随机进行100次测试集(30%)和训练集(70%)的拆分,100次建模获得的平均测试集预测R2值为0.561。
下表1展示9个基于利用母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率差异来确定胎儿浓度的示例。
表1中,示例1是原始的seqFF WRSC方法用到的条件。表格中“Bin筛选”的“Bin”就是基因组参考序列上的分段区间,“Mappability校正”是指“比对概率的校正”,“Mappability”指的是上述滑窗法归一化比对数”,“groupby”指seqFF法中一种GC校正方法。groupby相当于做lowess平滑时,对GC含量进行了区间分段,每一个分段用一个保留了三位小数的GC含量数值指代其GC含量,并用这个GC含量范围内所有分段区间对应的归一化统计数的中位数来指代该GC含量下的归一化统计数。在表格中提到了,这样的处理方式我们标记为“median”。相对应的,表格中还有标记为“all”的归一化方式。“All”指的就是,把所有分段区间的GC含量以及它对应的归一化比对数一并考虑进去进行Lowess平滑,并不对GC含量进行区间化分组,即平滑的时候用到了所有分段区间的数据。“median”是指中位数,“Frac”是指Lowess平滑所用参数,“所有mapped”是指所有比对上的序列,“unique”是指唯一比对上的序列(即仅比对上了染色体的一处的序列),“XYM”或“XYM染色体”是指X染色体、Y染色体和线粒体染色体,“flag筛选”是指seqFF法原始论文中,对不同的分段区间做了一些筛选,并提供了筛选文件。“seqFF的bin flag筛选”就是按照原始文献中标记的标签进行筛选。我们最终选定的方案并没有按照这个原始文献中的标签进行筛选。
表1
/>
皮尔相关系数评估的是算法直接计算出的数值与参考胎儿浓度之间的线性相关程度。参见图4,虽然二者线性相关程度高,但是二者绝对数值差异很大。因此,需要一个线性模型对算法的计算结果进行校正,使其达到准确估算胎儿浓度的目的。可以利用上述的
表1通过预测出的胎儿浓度和参考胎儿浓度(其他平台提供的胎儿浓度)之间的皮尔森相关系数,评价不同的处理参数、条件的优劣。
由表1可见,相较于示例1(皮尔森相关系数为0.71425)的算法,其他各算法的皮尔森相关系数均大于0.74,示例6、示例8和示例9的算法的皮尔森相关系数大于0.75。相较其他算法,采用示例8的算法处理可以获得最高的皮尔森相关系数,为0.75471。
校正后的示例8计算的胎儿浓度与参考胎儿浓度的关系图如图5所示。由图5可见,计算的胎儿浓度与参考胎儿浓度之间呈现高度正相关关系。
可以利用母体与婴儿的cfDNA的两端在核小体上位置的分布差异计算胎儿浓度。
核小体是组成人类染色质的基本单位,当DNA受到酶切作用,在核小体内部进行切割比在核小体之间的连接外进行切割要困难,因而DNA在被消化的过程中,更有可能被切断在核小体之间的连接处。而由于在细胞凋亡过程中,母亲和胎儿的DNA中核小体的结构特征上的差异,使核小体的捆绑强度也形成差异,因而母亲和胎儿的DNA在被消化的过程中剪切在核小体内部与核小体之间连接处的概率也会有差异。具体的,胎儿的DNA在核小体内部被剪切的概率相比高于母亲的DNA在核小体内部被剪切的概率,因此,通过评估待测cfDNA样本中cfDNA的端点相对于核小体的位置分布,能够获得胎儿浓度的信息。
在本申请的另一种实施方式中,提供一种利用母体与婴儿的cfDNA的两端在核小体上位置的分布差异确定胎儿浓度的方法。具体的,胎儿浓度的确定方法包括:
S121.获取待测cfDNA样本的测序序列。
该步骤中,待测cfDNA样本为需要确定胎儿浓度的cfDNA样本。本申请实施提供的方法,对待测cfDNA样本的来源以及待测cfDNA样本的特征参数等没有明确要求。
S122.将测序序列与参考基因组进行比对,得到比对结果。
该步骤包括:
S1221.将参考基因组的各碱基位点为测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于第二比对数,计算参考基因组的每个碱基位点对应的核小体中心分数;
S1222.基于核小体中心分数及中心分数筛选阈值,确定核小体中心位置;
S1223.基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数;
S1224.将加和处理后的第二对比数进行降维处理,得到降维后的归一化第二比对数,并将降维后的归一化第二比对数确定为比对结果。
需要说明的是,在该实施方式中的第二比对数是为了与上述实施例中的第一比对数进行区分,二者并不存在先后顺序。将参考基因组的各碱基位点为所述测序序列比对起始位置的碱基位点的计数确定为第二比对数,对应的,该第二比对数也可以称为“计数”,即统计不同序列的比对起始位置,然后获得参考基因组上不同位点正好是序列比对起始位置的次数。
由于核小体内部区域作为cfDNA的切口(亦即测序序列的起始端)的概率小于核小体连接区作为cfDNA切口的概率,因此,需要查找确认核小体的区域范围。
步骤S1221中,统计参考基因组中测序序列比对起始位置的碱基位点的计数,是指分析所有的测序序列的比对起始位置,然后统计参考基因组上每个位点作为这些序列的比对起始位置出现的计数。本申请实施例将该计数确定为第二比对数。应当理解的是,第二比对数可以是统计参考基因组上每个位点作为这些序列的比对起始位置的绝对次数,也可以是统计参考基因组上每个位点作为这些序列的比对起始位置的次数中位数。另外,在统计过程中,对参考基因组上每个位点作为这些序列的比对起始位置进行统计,允许一定的容错比例出现,即允许一定长度范围内存在预设数量碱基错误的测序序列,作为比对上的测序序列。
在将该计数确定为第二比对数后,基于第二比对数,计算参考基因组的每个碱基位点对应的核小体中心分数。“核小体中心分数”可以理解为每个位点是核小体中心位置的概率评分。“核小体中心位置”是指这个位点在核小体区域的最中心。
在一些实施例中,计算参考基因组的每个碱基位点对应的核小体中心分数,包括:
(1)计算参考基因组的每个碱基位点左右分别对应的第一特定数量的碱基范围的第一平均计数;
(2)计算参考基因组的每个碱基位点左右分别对应的第二特定数量的碱基的第二平均计数;
(3)根据第一平均计数和第二平均计数,确定每个位点对应的核小体中心分数。
其中,第一特定数量和第二特定数量基于核小体区域的碱基数量以及核小体连接区域的碱基数量确定。以x表示一个碱基位点,第一特定数量=(核小体区域的碱基数量-1)/2,第二特定数量=(核小体区域的碱基数量-1)/2+核小体连接区域的碱基数量/2。
示例性的,结合文献报道,核小体区域长度为147个碱基,核小体两端连接区各约20个碱基,此时,第一特定数量为(147-1)/2=73,第二特定数量为(147-1)/2+20=93。即假定x为核小体中心位置,这个位置的左右各73个碱基的范围为核小体区域,这个位置左右分别对应的第74至第93个碱基的范围为核小体的连接区。
上述公式计算了待考察位置左右分别对应的74-93个碱基范围的平均计数之和,与待考察位置左右各73个碱基范围的平均计数的比值。显然,对于一个特定的核小体区域附近,待考察位置越接近于核小体中心位置,该数值就越大。
进一步地,根据第一平均计数和第二平均计数,确定每个位点对应的核小体中心分数,核小体中心分数的计算公式可表示为:
式中,x为碱基位点,[x-93,x-74]表示从距离x一侧93个核苷酸到同侧距离x 74个核苷酸的区间,[x+93,x+74]表示从距离x另一侧93个核苷酸到同侧距离x 74个核苷酸的区间,[x-73,x+73]表示从距离x另一侧73个核苷酸到距离x另一侧73个核苷酸的区间。
在一些实施例中,考虑到单分子测序仪的测序错误率倾向于高于二代测序技术的错误率,因此,对核小体区域内各碱基作为核小体中心位置进行概率分析比对时,通过截去序列两端错误率相对较高的区域,可以提高确定核小体中心位置的准确率和效率。
在一些实施例中,将核小体区域左右两端各缩短n个碱基之后再计算“核小体中心分数”,以及后续的“核小体比值”。n为小于或等于5的自然数,示例性的,n为1、2、3、4或5。
此时,核小体中心分数的计算公式可表示为:
式中,x为碱基位点,[x-93,x-74-n]表示从距离x一侧93个核苷酸到同侧距离x74-n个核苷酸的区间,[x+93,x+74-n]表示从距离x另一侧93个核苷酸到同侧距离x 74-n个核苷酸的区间,[x-73-n,x+73-n]表示从距离x另一侧73-n个核苷酸到距离x另一侧73-n个核苷酸的区间。
示例性的,当n=5时,核小体中心分数的计算公式可表示为:
式中,x为碱基位点,[x-93,x-69]表示从距离x一侧93个核苷酸到同侧距离x 69个核苷酸的区间,[x+93,x+69]表示从距离x另一侧93个核苷酸到同侧距离x 69个核苷酸的区间,[x-68,x+68]表示从距离x另一侧68个核苷酸到距离x另一侧68个核苷酸的区间。
步骤S1223中,基于核小体中心分数以及中心分数筛选阈值,确定核小体中心分数,包括:
(1)基于核小体中心分数的最大值,确定最大值在参考基因组上的位置,并将位置确定为第一位置;
(2)将第一位置两侧特定数据的碱基的核小体中心分数清零,并基于清零后剩余的核小体中心分数再次确定最大值,并执行确定最大值在参考基因组上的位置,直至核小体中心分数小于第二目标阈值,将筛选出的位置确定为候选核小体中心位置;
(3)基中心分数筛选阈值以及与候选核小体中心位置对应的核小体中心分数,确定核小体中心位置。
其中,第一位置是基于核小体中心分数最大值在参考基因组上的位置,即找出上述计算的核小体中心分数的最大值,记录它们在参考基因组上的位置,并将该位置两侧特定数据的碱基(如,两侧各147碱基)的核小体中心分数清零,这是因为找出的位置被认为是核小体中心位置,而该核小体中心位置两侧至少147个碱基范围内不可能再有另一个核小体中心位置。然后再次找出当前的核小体中心分数的最大值对应的位置,继续执行两侧核小体中次年分数清零的步骤。如此循环直至最大的核小体中心分数值低于第二目标阈值停止,基于该执行过程筛选出的位置即为核小体中心位置。理论上,核小体连接区的平均技术应该高于核小体区域。基于上述公式,核小体中心分数应该是大于2。为了便于处理和提升准确度,在一个实施例中,第二目标阈值的取值可以设定为2.2。亦即,在筛选核小体中心区域时,循环持续到最大的核小体中心分数的值低于2.2就停止迭代处理了。
在获得了所有候选核小体中心位置后,但是由于在候选核小体中心位置中有些“核小体中心分数”异常偏高的位置,可能受到了比对概率偏好等各种位置因素的影响,其对应的第二比对数存在不可靠的情况,因此,使用这些候选核小体中心位置中的一部分作为最终选定的核小体中心位置,可以对核小体中心分数进行筛选。
下表2提供了几种Score筛选条件以及对应的条件下胎儿浓度与核小体比值的Pearson相关系数。
表2
在表2中,“score”是指“核小体中心分数”,“bp”是指碱基数,“linker”是指核小体连接区,“核小体ratio”是指“核小体比值”。通过“核小体比值”与参考胎儿浓度之间的皮尔森相关系数来初步筛选核小体中心位置以及核小体比值的计算方法。[10,)表示大于等于10的范围,右边圆括号是开区间。
从表2可见,相比不对核小体区域进行处理,将核小体区域左右两端各缩短5个碱基之后再计算“核小体中心分数”,以及后续的“核小体比值”,胎儿浓度与核小体ratio的Pearson相关系数较高。
步骤S1223中,基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数。
在一些实施例中,按照核小体区域进行对齐叠加时,取了“核小体中心位置”左右各200个碱基的范围进行对齐加和,即获得的是一个401个元素的向量,第201个元素为核小体中心位置。这样的操作对于计算“核小体比值”没有任何影响,但是由于后续会引入机器学习算法建立模型,因而提取了核小体区域周围更大范围内的信息,以提升模型潜在的信息量与预测能力。
在确定了核小体中心位置后,对于待分析的样本数据,可以先按照上述方法计算参考基因组上每个位置对应的序列起始位置计数,即获得第二比对数,然后基于核小体中心位置的处理过程,将核小体中心位置左右各147碱基的第二比对数取出。对于所有的核小体中心位置都进行该操作。在该操作完成后,将所有取出的数据对其加和,即基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,其中,核小体区域内对应位置包括:以核小体中心位置为中心,左右特定数量的碱基范围。经过反复验证左右各200个碱基范围的处理效果最佳。即每次去除的是一个295长度的向量,向量的第148是核小体中心位置的第二比对数,向量其他元素是核小体中心位置左右的位置对应的第二比对数,将这些向量加和,加和结果可以参见图6所示,其示出了其中一组数据按照核小体区域对齐并加和之后获得的分布。由图6可知,按照我们的分析处理方法获得的对齐后的核小体区域统计数据,确实能看到序列起始位点落入核小体中心区域的概率远远低于落入核小体连接区域的概率,与生化原理相符,侧面佐证了我们上述分析方法的可靠性。
计算加和后的向量对应的核小体区域147个碱基(也就是向量第148个元素左右各73个碱基的范围内)的计数均值,再计算该均值与左右核小体连接区的计数均值的比值(如,核小体中心位置左右分别对应的74-93个碱基的范围为核小体的连接区),可以将该比值称为“核小体比值”,即本申请实施例中的基于核小体相关信息确定的比对结果,对应的“核小体比值”越高,胎儿浓度理论上应该越高,通过核小体比值与参考胎儿浓度之间的关系建立线性模型,从而获得该比对结果与胎儿浓度之间的关系,因此实现基于对比结果确定胎儿浓度的目的。
具体的,也可以建立第二比对数与胎儿浓度之间的线性模型,可以通过机器学习的处理手段实现,其中,所述基于比对结果,确定胎儿浓度,包括:
(1)获取第二训练样本数据,第二训练样本数据中每一样本均标注有特征值和目标值,特征值为降维后的归一化第二比对数,目标值为实际胎儿浓度;
(2)基于第二训练样本创建第二胎儿浓度定量模型;将比对结果输入至第二胎儿浓度定量模型,得到胎儿浓度。
其中,生成该第二胎儿浓度定量模型的处理过程与生成第第一胎儿浓度定量模型的处理过程相似,只是二者学习的特征值不同,因此,可以参考第一胎儿浓度定量模型的创建过程,此处不再详述。
例如,以核小体区域的平均计数与核小体连接区域的平均技术的比值作为参数,与已知的胎儿浓度建立线性模型。由于单分子测序数据,起始位点准确率偏低,在不截去核小体区域两端错误率高的碱基时,皮尔森相关系数仅0.3左右(参见表2)。因此在一实施例将上述图6中所有401个位置的计数全部作为原始特征,利用机器学习建立模型(主成分分析降维+弹性网络建模)估算胎儿浓度。示例性的,对于所有一定数量如615组已有参考胎儿浓度的数据,计算其按照核小体中心位置对齐加和的计数向量。每一组实现对应于一个包含401元素的向量,这401个元素就是原始输入的特征值。随机进行100次测试集(30%)和训练集(70%)的拆分,其测试集估算的胎儿浓度与真实胎儿浓度的皮尔森相关系数为0.58,R
在一些实施例中,通过对原始特征数进行处理,可以提高基于各测序平台特别是单分子测序平台获得的测序数据的胎儿浓度的计算准确性。
在一种实施例中,对原始特征进行归一化处理。具体的,以中间位置为中心,左右各200个计数(第二比对数),这些计数分别除以中心位置的计数,从而得到归一化的401个特征,这种归一化处理可以减少噪声对模型的影响,从而提高模型的性能。
在另一种实施例中,基于原始特征创建新特征。示例性的,基于上文所说的401个原始特征,创建了两个新的特征,第一个新特征为最坐标20个位置的计数均值与中心147个位置的计数均值的比值,第二个新特征为最右边20个位置的计数均值与中心147个位置的计数均值的比值。由此,形成403个特征。
在再一种实施例中,可以对原始特征进行主成分降维(在训练集上得到主成分变换模型并用在测试集上进行相同的变换),以将各变量之间互相关联的复杂关系进行简化,使模型更简洁,减少模型的过拟合。
上述三种实施例可以单独用于对原始特征进行处理,也可以组合用于对原始特征进行处理。
示例性的,基于原始特征创建新特征形成403个特征后,对403个特征进行主成分分析降维(在训练集上得到主成分变换模型并用在测试集上进行相同的变换),权衡主成分个数和保留的信息,最终在保留90%有效信息下将403个特征降维到33个。
机器学习建立模型,为避免模型过拟合,对模型进行L1和L2正则化处理(L1和L2正则化是一种正则化方式,是为了防止模型过拟合在损失函数后面加入附加项的处理方法),并通过迭代选择最佳的L1和L2正则化参数组合。在训练模型时,为了更准确的评价模型的预测效果,随机进行100次测试集(30%)和训练集(70%)的拆分,进行模型训练,最终模型测试集估算的胎儿浓度与真实胎儿浓度的皮尔森相关系数为0.58,R
在本申请的另一实施例中还提供了另一种胎儿浓度的确定方法,参见图8,该方法可以包括以下步骤:
S201、获取待测cfDNA样本的测序序列。
测序序列如前所述,为了节约篇幅,此处不再赘述。
S202、对参考基因组进行分段,得到多个分段区间,确定每一测序序列落入各个分段区间的第一比对数。
该步骤中,为了能够准确获得比对结果,可以按照固定的长度对参考基因组进行分段,如根据测序序列的特征来确定参考基因组的分割长度。
参考基因组中,存在一些特殊的染色体,这些染色体可能在通过参考基因组比对方式确定胎儿浓度的过程中存在一定的偏向性或其他特殊性。在一种实施情形中,通过去除参考基因组中特殊的染色体(下文称为特定染色体)带来的分段区间,来减少这些染色体带来的偏向性,从而降低其特殊性对测试结果的影响。在一些实施例中,特定染色体包括X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体中的至少一种。其中,通过去除X染色体和Y染色体对应的分段区间,避免男胎和女胎上述染色体的差异,对后续浓度定量模型的结果造成的影响;去除13号染色体、18号染色体和21号染色体,是因为这几条染色体本身存在较大的重复缺失概率。而NIPT的筛查目标之一是对13、18、21号染色体的重复缺失情况进行筛查,当以13号染色体、18号染色体和21号染色体作为参考染色体时,对数据进行归一化统计时可能出现偏差,如影响归一化比对数,进而对计算结果造成影响,影响NIPT筛查结果的准确性。
本申请实施例可以根据胎儿浓度检测预期的需要,调整特定染色体的范围,如剔除X染色体和Y染色体,或剔除13号染色体、18号染色体和21号染色体,或剔除线粒体染色体。当然,可以对上述情形中的多种情形同时去除,如将X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体或其对应的分段片段全部去除,以使参考基因组分段后得到的分段区间中不含有上述染色体对应的分段区间。
本申请可以通过多种方式去除特定染色体进入分段区间。可以对分段后得到的分段区间基于特定染色体进行筛选,也可以先将这些特定染色体进行剔除后再分段,具体的,可以基于实际的应用需求选取对应的处理方式,本申请对此不进行限制。
在一种实施方式中,对参考基因组进行分段,得到多个分段区间,包括:基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;剔除初始分段区间中特定染色体对应的分段区间,得到多个分段区间。即在将参考基因组分段为多个分段区间后,对参考基因组中特定染色体对应的分段区间进行剔除,不参与计算。应当理解的是,所指的“特定染色体对应的分段区间”,是指特定染色体按照预设的分割长度进行分段后,形成的多个分段区间。
在另一种实施方式中,对参考基因组进行分段,得到多个分段区间,包括:剔除参考基因组中的特定染色体,得到剔除后的参考基因组;基于预设的分割长度对所述剔除后的参考基因组进行分段,得到多个分段区间。即在将参考基因组分段之前,先剔除特定染色体。
其中,预设的分割长度可以根据数据处理的数据量以及预期的相对准确度等调整,例如可以按照每50k(5000)碱基数量的长度对参考基因组进行分段。为了能够使得获得的第一比对数更加准确,还可以对第一比对数进行校正,得到校正后的第一比对数,以使得将校正后的第一比对数确定为比对结果。
在一些实施例中,校正包括GC校正。GC校正是生物信息学分析过程中的操作,之所以进行GC校正是基因测序仪以及对应的测序流程会造成一定的GC偏好,例如,某些测序仪测出高GC序列的概率更高,而某些测序仪测出低测出低GC序列的概率更高。在该实施方式中,确定胎儿浓度的处理方式是基于参考基因组上不同区域的比对概率进行的,而测序仪的GC偏好将会影响对真实的不同区域的比对概率的判断,为了提升准确性,因而需要进行GC校正。
此时,对第一比对数进行校正,得到校正后的第一比对数,包括:基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数。其中,所有分段区间的平均第一比对数是指,各分段区间的第一比对数之和与分段区间的总数量的比值。
在获得了所有分段区间的第一比对数之后,为了便于计算和处理,会对所有分段区间的第一比对数进行归一化处理,对应的公式为:
每个分段区间归一化第一比对数=每个分段区间的第一比对数/所有分段区间的平均第一比对数。
获得了每一分段区间的第一比对数之后,将进行GC校正,得到GC校正后的第一比对数。
在一种实施方式中,对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
(1)基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线。
该实施方式中,GC含量可以通过多种参数来表示,如每个分段区间的GC含量或GC含量中位数,还可以是能够反馈分段区间GC含量的其他参数。
本申请实施例中,可以通过对形成第一关系曲线的分段区间进行GC过滤,如根据GC含量对分段区间进行过滤,以过滤掉一些异常或者不具备统计意义的区段,优化第一关系曲线,降低了在数据处理过程中数据处理资源的占用。
在一种实施方式中,GC过滤通过下述方式实现:在基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线的步骤之前,进行如下处理:基于GC含量对分段区间进行过滤,去除GC含量不满足预设要求的分段区间,使得基于GC过滤后的每个分段区间的GC含量和每个分段区间的归一化第一比对数,生成第一关系曲线。
在另一种实施方式中,GC过滤通过下述方式实现:基于第一关系曲线进行GC校正对归一化第一比对数的过程中,基于GC含量对分段区间进行过滤,并基于第一关系曲线对GC过滤后的归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
上述实施例中,作为一种示例,基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线,通过通过下述方式实现:以x轴表示每个分段区间的GC含量(即参考基因组上该分段区间对应的GC碱基的占比),y轴为每个分段区间对应的归一化第一比对数。整合每个分段区间及其区间内的GC含量的数据,即根据每个分段区间的GC含量以及对应的归一化第一比对数,生成散点图。对散点图中的散点数据进行平滑,获得平滑曲线。其中,对散点图中的散点数据进行平滑可以利用如LOWESS算法进行,获得平滑曲线,即获得第一关系曲线,如y
具体的,作为一种示例,第一关系曲线可以通过下述方式生成:将横坐标分为不同的区段,然后计算同一区段内散点的GC含量中位数,利用这个GC含量中位数来代表这一区段对应的数值,再将所有这些GC含量中位数画到图上进行平滑,获得第一关系曲线。在另一种示例中,可以基于所有的数据直接进行平滑处理,即将所有数据点均纳入考虑,共同平滑获得平滑曲线,这样测试集获得的斯皮尔曼相关系数更高。需要说明的是,这种平滑方法同样适用于后续描述的对比概率的平滑和校正。
(2)基于第一曲线对归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
在一些实施方式中,根据归一化第一比对数以及归一化第一比对数对应的分段区间的GC含量的减法关系式或者除法关系式进行GC校正,确定GC校正后的第一比对数。
在一种示例中,GC校正的方法为“减法”,对应的校正公式为:y
上述两个校正公式中,x为所述分段区间的GC含量,y为该分段区间对应的归一化第一比对数,y
在一些实施例中,校正还包括比对概率校正,以提升获得的第一比对数的准确率。即在一些实施例中,校正同时包括GC校正和比对概率校正。
在本申请实施例的一种实施方式中,对第一比对数进行校正,得到校正后的第一比对数,还包括:基于特定滑窗长度对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将第一比对数确定为滑窗比对数;基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;基于每一分段区间归一化后的滑窗比对数和所述归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
本申请实施例采用“比对概率校正”方法对第一比对数进行校正,基于以下考虑:参考基因组对应的参考序列的不同区域,如果按照特定长度截取下来当作一条测序序列,然后再比对回参考基因组时,不同的分段区间被比对上的次数会有差异,因为对于一些区域的序列,在参考基因组的参考序列上的多处都存在着与其类似的序列,因而这些区域更容易被比对上。例如,将人类基因组参考序列hg19按照2个碱基的步长、37个碱基的窗口进行滑窗截取序列,将这些序列与人类参考基因组hg19进行比对,然后统计在人类参考基因组的不同分段区域上的比对数,可以把这个比对数称为“滑窗比对数”。具体的,特定滑窗长度可以基于实际的应用场景考虑,如基于采用的参考基因组的特征,或者待测样本的特征等确定。在确定了特定滑窗长度后对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组上的每一分段区间上的第一比对数,将此时的第一比对数确定为滑窗比对数。在获得了滑窗比对数之后,为了便于计算和处理,基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;对归一化后的滑窗比对数进行比对概率校正。比对概率校正的方法,可以参考前文GC校正的方法。
在一种实施方式中,基于每一分段区间归一化后的滑窗比对数和归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数,包括:
(1)基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线。
其中,每一分段区间归一化后的滑窗比对数可以通过多种参数来表示,如每一分段区间的归一化后的滑窗比对数或滑窗比对数的中位数,还可以是能够反馈每一分段区间归一化后的滑窗比对数的其他参数。
本申请实施例中,可以通过对形成第二关系曲线的归一化的滑窗比对数进行过滤,以过滤掉一些异常或者不具备统计意义的区段,优化第二关系曲线,提升基于第二关系曲线进行相关数据处理时的处理准确性。
在一种实施方式中,对形成第二关系曲线的归一化的滑窗比对数进行过滤,通过下述方式实现:在基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线的步骤前,进行如下处理:基于归一化的滑窗比对数对分段区间进行过滤,获得归一化后的滑窗比对数不小于第一目标阈值的分段区间,以使得第二关系曲线在归一化后的滑窗比对数不小于第一目标阈值的分段区间生成。
在一种实施方式中,对形成第二关系曲线的归一化的滑窗比对数进行过滤,通过下述方式实现:基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数的过程中,基于归一化后的滑窗比对数,对分段区间进行过滤,保留第二关系曲线中归一化后的滑窗比对数不小于第一目标阈值的第二关系曲线区间,以得到比对概率过滤后的第二关系曲线;基于比对概率过滤后的第二关系曲线对每一分段区间对应的滑窗归一化比对数进行比对概率校正,得到比对概率校正后的第一比对数。
上述实施例中,基于反复验证,第一目标阈值可以选取0.8,即去掉归一化后的滑窗比对数小于0.8的分段区间。
上述实施例中,作为一种示例,基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线,通过通过下述方式实现:以x轴表示每个分段区间归一化后的滑窗比对数,y轴为每个分段区间对应的归一化第一比对数,整合每个分段区间及其对应的GC含量的数据,即根据每个分段区间的GC含量以及对应的归一化第一比对数,生成散点图;对散点图中的散点数据进行平滑,获得平滑曲线。示例性的,对散点图中的散点数据进行平滑可以利用LOWESS算法实现。
(2)基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数。
在一些实施方式中,根据归一化第一比对数以及归一化第一比对数对应的分段区间的滑窗比对数的减法关系式或者除法关系式进行比对概率校正,确定比对概率校正后的第一比对数。
在一种示例中,比对概率校正的方法为“减法”,对应的校正公式为:y
上述两个校正公式中,x为每个分段区间归一化后的滑窗比对数,y对应的就是每个分段区间的归一化第一比对数,y
S203、基于第一比对数,确定第一胎儿浓度。
在本申请实施例中可以结合神经网络的机器学习方法确定胎儿浓度。在本申请实施例的一种实施方式中,基于对比对结果,确定胎儿浓度,包括:
获得第一训练样本数据,所述第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,第一特征值为样本比对数,第一目标值为样本实际胎儿浓度;
基于特定模型结果对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
将比对结果输入至第一胎儿浓度定量模型,得到第一胎儿浓度,并将所述第一胎儿浓度确定为胎儿浓度。
在一些实施例中,基于特定模型结构对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型,包括:
将第一训练样本数据划分为训练集和测试集;基于训练集进行机器学习建模,得到初始模型;
基于初始模型对测试集进行处理,得到与测试集中每一测试样本的预估胎儿浓度;
基于预估胎儿浓度与测试集中每一测试样本的实际胎儿浓度进行比较,得到比较结果;
基于比较结果对初始模型的模型参数进行调整,得到第一胎儿浓度定量模型。
其中,第一训练样本数据可以是基于包括单分子测序平台在内的各种测序平台获得的已知胎儿浓度的孕妇外周血cfDNA的数据库种的数据,例如,采用一定数含量的已知胎儿浓度的样本,对参考基因组染色体参考序列进行区间分段(如,可通过5k碱基、50k碱基、100k碱基、300k碱基或者800k碱基等分段),并分别统计每个样本的比对数据在不同分段区间的比对数,并利用LOWESS法对这些比对数进行归一化、GC和比对概率的校正,将最终归一化和校正后不同分段区间的比对数作为特征值,将每个样本对应的已知的实际胎儿浓度作为目标值,用特定的模型进行学习和建模,其中,该特定的模型可以是分类模型、卷积神经网络模型等本申请实施例对特定模型结构不进行限制,只要能够对训练样本数据进行学习,并能预测出胎儿浓度即可。模型学习和训练的过程是一个反复迭代的过程,因此可以将训练样本分为训练集和测试集,通过测试集测试当前训练得到的模型的准确度,若准确度低于设定的准确度阈值,则通过测试集对当前的模型的模型结构中的模型参数进行调整,直至获得的模型输出的胎儿浓度与实际的胎儿浓度的误差满足对应的误差范围即可。
当用于机器学习的样本数量本身较少,而特征值较多(例如约三千到六十万个特征,具体取决于染色体参考序列区间分段的大小)时,无论是降维还是学习的过程中,都无法避免过拟合的影响。最终建立的胎儿浓度定量模型中,对测试集胎儿浓度的预测效果会与实际的胎儿浓度存在一定偏差。如采用615例已知胎儿浓度的样本建立的胎儿浓度定量模型中,模型对应测试集R
在另一种实施方式中,基于比对结果,确定胎儿浓度,包括:
将比对结果输入至第一预设模型,得到初始胎儿浓度;
根据第二预设模型对初始胎儿浓度进行校正,获得胎儿浓度。
其中,第一预设模型为基于第二样本数据中的比对结果以及与比对结果对应的初始胎儿浓度建立的线性关系模式,第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;第二预设模型是基于现象拟合确定的常数对第一预设模型处理后得到的模型。
示例性的,第一预设模型可以为
示例性的,利用样本在参考基因组参考序列不同区间的比对概率差异来建立模型并估算胎儿浓度的模型为SeqFF。SeqFF法中用到了两种机器学习模型,一种是权重排序选择标准(WRSC),另一种是弹性网络(Enet),其最终输出的胎儿浓度结果是两个模型预测值的平均值。当利用测序平台特别是单分子测序平台的数据测试SeqFF模型,其中WRSC模型的效果优于Enet,即WRSC估算的胎儿浓度与实际胎儿浓度的线性相关系数比Enet或者SeqFF要高,因而优先考虑WRSC算法涉及的模型参数。具体的,可以参见图2和图3,图2为本申请实施例提供的一种Enet模型计算的胎儿浓度与参考胎儿浓度关系的示意图,图3为本申请实施例提供的一种WRSC模型计算的胎儿浓度与参考胎儿浓度关系的示意图。由图可见,WRSC模型计算的胎儿浓度与参考胎儿浓度之间正相关性优于Enet模型计算的胎儿浓度与参考胎儿浓度之间的正相关性。
示例性的,WRSC模型中,按照特定数量碱基如50k碱基的区间对染色体参考序列进行划分。通过WRSC算法训练,找出了不同参考序列区间上的覆盖密度与胎儿浓度之间的线性关系,如下列公式所展示。下列公式中,FF为胎儿浓度,
由于SeqFF是基于二代测序数据训练处的模型,其测序原理与单分子测序有较差差异,因而若直接采用SeqFF的上述模型参数计算包括单分子测序平台在内的其他测序平台得到的数据的胎儿浓度,计算出的胎儿浓度与实际胎儿浓度差异较大。仍利用上述实施例中的615例已知胎儿浓度的NIPT临床样本为例进行分析和测试。直接使用SeqFF的算法和参数对包括单分子测序平台在内的其他测序平台原始的比对文件数据(如,sam文件)进行处理,获得的中间参数与真实胎儿浓度之间的皮尔森相关系数为0.714。
SeqFF模型计算出的胎儿浓度的计算值与实际胎儿浓度值线性关系较好,因而可以采用上述公式中的
仍利用上述实施例中的615例已知胎儿浓度的NIPT临床样本为例进行分析和测试。改为仅使用测序序列落入各个分段区间的第一比对数进行分析;剔除参考基因组中X、Y、M、13、18、21以及线粒体染色体上比对概率异常的区域进行分析;对校正方法(GC校正,或GC和比对概率校正)进行优化等。使用优化后的参数重新对数据进行分析,中间参数与胎儿浓度之间的皮尔森相关系数为0.755。随机进行100次测试集(30%)和训练集(70%)的拆分,用于建立线性模型、确定上述常数参数c和d。100次建模获得的平均测试集预测R
需要说明的是,真正建立线性模型时,测试集和训练集的拆分是为了在有限的数据中评估我们目前随机拆分的训练集建立的模型的可信程度。因此随机进行100次测试集(30%)和训练集(70%)的拆分,100次建模获得的平均测试集预测R2值为0.561。
上表1展示9个基于利用母体与婴儿的cfDNA在参考基因组上不同位置区间的比对概率差异来确定胎儿浓度的示例。
由表1可见,相较于示例1(皮尔森相关系数为0.71425)的算法,其他各算法的皮尔森相关系数均大于0.74,示例6、示例8和示例9的算法的皮尔森相关系数大于0.75。相较其他算法,采用示例8的算法处理可以获得最高的皮尔森相关系数,为0.75471。
校正后的示例8计算的胎儿浓度与参考胎儿浓度的关系图如图5所示。
S204、基于参考基因组的各碱基位点是测序序列比对起始位置的第二比对数,确定核小体中心位置。
该步骤包括:
将参考基因组的各碱基位点为测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于第二比对数,计算参考基因组的每个碱基位点对应的核小体中心分数;
基于核小体中心分数及中心分数筛选阈值,确定核小体中心位置。
在一些实施例中,计算参考基因组的每个碱基位点对应的核小体中心分数,包括:
(1)计算参考基因组的每个碱基位点左右分别对应的第一特定数量的碱基范围的第一平均计数;
(2)计算参考基因组的每个碱基位点左右分别对应的第二特定数量的碱基的第二平均计数;
(3)根据第一平均计数和第二平均计数,确定每个位点对应的核小体中心分数。
其中,第一特定数量和第二特定数量基于核小体区域的碱基数量以及核小体连接区域的碱基数量确定。以x表示一个碱基位点,第一特定数量=(核小体区域的碱基数量-1)/2,第二特定数量=(核小体区域的碱基数量-1)/2+核小体连接区域的碱基数量/2。
示例性的,结合文献报道,核小体区域长度为147个碱基,核小体两端连接区各约20个碱基,此时,第一特定数量为(147-1)/2=73,第二特定数量为(147-1)/2+20=93。即假定x为核小体中心位置,这个位置的左右各73个碱基的范围为核小体区域,这个位置左右分别对应的第74至第93个碱基的范围为核小体的连接区。
上述公式计算了待考察位置左右分别对应的74-93个碱基范围的平均计数之和,与待考察位置左右各73个碱基范围的平均计数的比值。显然,对于一个特定的核小体区域附近,待考察位置越接近于核小体中心位置,该数值就越大。
进一步地,根据第一平均计数和第二平均计数,确定每个位点对应的核小体中心分数,核小体中心分数的计算公式可表示为:
式中,x为碱基位点,[x-93,x-74]表示从距离x一侧93个核苷酸到同侧距离x 74个核苷酸的区间,[x+93,x+74]表示从距离x另一侧93个核苷酸到同侧距离x 74个核苷酸的区间,[x-73,x+73]表示从距离x另一侧73个核苷酸到距离x另一侧73个核苷酸的区间。
在一些实施例中,考虑到单分子测序仪的测序错误率倾向于高于二代测序技术的错误率,因此,对核小体区域内各碱基作为核小体中心位置进行概率分析比对时,通过截去序列两端错误率相对较高的区域,可以提高确定核小体中心位置的准确率和效率。
在一些实施例中,将核小体区域左右两端各缩短n个碱基之后再计算“核小体中心分数”,以及后续的“核小体比值”。n为小于或等于5的自然数,示例性的,n为1、2、3、4或5。
此时,核小体中心分数的计算公式可表示为:
/>
式中,x为碱基位点,[x-93,x-74-n]表示从距离x一侧93个核苷酸到同侧距离x74-n个核苷酸的区间,[x+93,x+74-n]表示从距离x另一侧93个核苷酸到同侧距离x 74-n个核苷酸的区间,[x-73-n,x+73-n]表示从距离x另一侧73-n个核苷酸到距离x另一侧73-n个核苷酸的区间。
示例性的,当n=5时,核小体中心分数的计算公式可表示为:
式中,x为碱基位点,[x-93,x-69]表示从距离x一侧93个核苷酸到同侧距离x 69个核苷酸的区间,[x+93,x+69]表示从距离x另一侧93个核苷酸到同侧距离x 69个核苷酸的区间,[x-68,x+68]表示从距离x另一侧68个核苷酸到距离x另一侧68个核苷酸的区间。
基于核小体中心分数以及中心分数筛选阈值,确定核小体中心分数,包括:
(1)基于核小体中心分数的最大值,确定最大值在参考基因组上的位置,并将位置确定为第一位置;
(2)将第一位置两侧特定数据的碱基的核小体中心分数清零,并基于清零后剩余的核小体中心分数再次确定最大值,并执行确定最大值在参考基因组上的位置,直至核小体中心分数小于第二目标阈值,将筛选出的位置确定为候选核小体中心位置;
(3)基中心分数筛选阈值以及与候选核小体中心位置对应的核小体中心分数,确定核小体中心位置。
其中,第一位置是基于核小体中心分数最大值在参考基因组上的位置,即找出上述计算的核小体中心分数的最大值,记录它们在参考基因组上的位置,并将该位置两侧特定数据的碱基(如,两侧各147碱基)的核小体中心分数清零,这是因为找出的位置被认为是核小体中心位置,而该核小体中心位置两侧至少147个碱基范围内不可能再有另一个核小体中心位置。然后再次找出当前的核小体中心分数的最大值对应的位置,继续执行两侧核小体中次年分数清零的步骤。如此循环直至最大的核小体中心分数值低于第二目标阈值停止,基于该执行过程筛选出的位置即为核小体中心位置。理论上,核小体连接区的平均技术应该高于核小体区域。基于上述公式,核小体中心分数应该是大于2。为了便于处理和提升准确度,在一个实施例中,第二目标阈值的取值可以设定为2.2。亦即,在筛选核小体中心区域时,循环持续到最大的核小体中心分数的值低于2.2就停止迭代处理了。
在获得了所有候选核小体中心位置后,但是由于在候选核小体中心位置中有些“核小体中心分数”异常偏高的位置,可能受到了比对概率偏好等各种位置因素的影响,其对应的第二比对数存在不可靠的情况,因此,使用这些候选核小体中心位置中的一部分作为最终选定的核小体中心位置,可以对核小体中心分数进行筛选。
上表2提供了几种Score筛选条件以及对应的条件下胎儿浓度与核小体比值的Pearson相关系数。
S205、基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数。
在一些实施例中,按照核小体区域进行对齐叠加时,取了“核小体中心位置”左右各200个碱基的范围进行对齐加和,即获得的是一个401个元素的向量,第201个元素为核小体中心位置。这样的操作对于计算“核小体比值”没有任何影响,但是由于后续会引入机器学习算法建立模型,因而提取了核小体区域周围更大范围内的信息,以提升模型潜在的信息量与预测能力。
在确定了核小体中心位置后,对于待分析的样本数据,可以先按照上述方法计算参考基因组上每个位置对应的序列起始位置计数,即获得第二比对数,然后基于核小体中心位置的处理过程,将核小体中心位置左右各147碱基的第二比对数取出。对于所有的核小体中心位置都进行该操作。
S206、基于第一胎儿浓度和加和处理后的第二比对数,确定比对结果。
在该实施例中对第二处理模式提供的特征值的处理方法与单独使用第二处理模式时一致,所不同的是加入了第一处理模式中所预测的胎儿浓度作为其中一个特征,一起进行了主成分分析降维处理。在模型训练时,为了保证模型的稳定性和模型评估结果的可靠性,同样随机进行100次测试集(30%)和训练集(70%)的拆分。弹性网络模型同样对模型进行L1和L2正则化处理,并通过迭代选择最佳的L1和L2正则化参数组合。不同模型具体效果详见表3和表4。下表中,r2_test_enst代表了用enst模型建模时,测试集的R
需要说明的是,“随机进行100次测试集(30%)和训练集(70%)的拆分”并计算100次拆分获得的R
表3
/>
表4
作为本申请一种可能的实现方式在一些实施例中,基于第一胎儿浓度和加和处理后的第二比对数,确定比对结果,包括:将核小体区域内各碱基位点对应的加和处理后的第二比对数进行降维处理,得到降维后的归一化第二比对数;基于第一胎儿浓度和降维后的归一化第二比对数确定为比对结果。或者,基于第一胎儿浓度和加和处理后的第二比对数,确定初始比对结果;对初始比对结果进行降维处理,将降维后的初始比对结果确定为比对结果。
示例性的,将第二处理模式中的401条原始特征(即按照核小体中心位置对齐加和获得的401个计数)与第一确定模式中WRSC法直接获得的胎儿浓度值合并成402条参数数据,作为原始输入的特征值,使用主成分分析降维,并用机器学习算法建模。机器学习模型可以为弹性网络、Lasso、线性回归、逐步回归、岭回归、Keras、lightGBM等多种模型,相较于其他模型,岭回归效果更好,最终测试集胎儿浓度估算的R
S207、基于比对结果,确定胎儿浓度。
在另一种实施方式中,基于比对结果,确定胎儿浓度,包括:
获取第二训练样本数据,第二训练样本数据中的每一样本均标注有特征值和目标值,特征值为降维后的归一化第二比对数,目标值为实际胎儿浓度;基于第二训练样本数据创建第二胎儿浓度定量模型;将比对结果输入至第二胎儿浓度定量模型,得到胎儿浓度。
该实施例基于前述实施例中的利用母体与婴儿的cfDNA在参考基因组序列上不同位置区间的比对概率差异,确定胎儿浓度的方式(为了便于后续描述简称为第一确定模式);以及利用母体与婴儿的cfDNA的两端在核小体上位置的分布差异,确定胎儿浓度的方式(简称为第二确定模式)共同实现的。由于这两种方式是使用不同原理确定胎儿浓度的,结合着两种处理方式的参数共同确定胎儿浓度,特征值的有效信息更多,胎儿浓度的估算能力也应该更强。例如,将第二处理模式中的401条原始特征(即按照核小体中心位置对齐加和获得的401个计数)与第一确定模式中WRSC法直接获得的胎儿浓度值合并成402条参数数据,作为原始输入的特征值,使用主成分分析降维,并用机器学习算法建模,有利于获得更准确的胎儿浓度数据。
基于前述实施例,在本申请的另一实施例中还提供了一种胎儿浓度的确定装置,参见图9,该装置可以包括:
第一获取单元10,用于获取待测cfDNA样本的测序序列;
第一比对单元11,用于将测序序列与参考基因组进行比对,得到比对结果;
第一确定单元12,用于基于比对结果,确定胎儿浓度。
作为本申请一种可能的实现方式,第一比对单元包括:
第一分段子单元,用于对参考基因组进行分段,得到多个分段区间;
第一确定子单元,用于确定每一测序序列落入各个分段区间的第一比对数,将第一比对数确定为比对结果。
作为本申请一种可能的实现方式,第一分段子单元具体用于:
基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;
剔除初始分段区间中特定染色体对应的分段区间,得到多个分段区间;
或者,
剔除参考基因组中的特定染色体,得到剔除后的参考基因组;
基于预设的分割长度对剔除后的参考基因组进行分段,得到多个分段区间。
其中,特定染色体包括X染色体、Y染色体、线粒体染色体、13号染色体、18号染色体和21号染色体中的至少一种。
作为本申请一种可能的实现方式,装置还包括:
第一校正单元,用于对第一比对数进行校正,得到校正后的第一比对数,以使得将校正后的第一比对数确定为比对结果。
作为本申请一种可能的实现方式,第一校正单元包括:
第一处理子单元,用于基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;
GC校正子单元,用于对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,GC校正子单元具体用于:
基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线;
基于第一关系曲线对归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
其中,在基于GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线的步骤之前,还包括:
基于GC含量对分段区间进行过滤,使得基于GC过滤后的每个分段区间的GC含量和每个分段区间对应的归一化第一比对数,生成第一关系曲线;
或者,
基于第一关系曲线对归一化第一比对数进行GC校正,得到GC校正后的第一比对数,包括:
基于GC含量对分段区间进行过滤,并基于并基于第一关系曲线对GC过滤后的归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
具体的,GC校正为根据归一化第一比对数以及与归一化第一比对数对应的分段区间的GC含量的减法关系式或者除法关系式,确定GC校正后的第一比对数。
在另一种实施方式中,第一校正单元还包括:
截取子单元,用于基于特定滑窗长度对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将第一比对数确定为滑窗比对数;
第二确定子单元,用于基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;
第三确定子单元,用于基于每一分段区间归一化后的滑窗比对数和归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,第三确定子单元具体用于:
基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线;
基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数。
在一种实施方式中,基于每一分段区间归一化后的滑窗比对数和每个分段区间对应的归一化第一比对数,生成第二关系曲线的步骤之前,还包括:
基于归一化后的滑窗比对数对分段区间进行过滤,获得归一化后的滑窗对比数不小于第一目标阈值的分段区间,以使得第二关系曲线在归一化后的滑窗比对数不小于第一目标阈值的分段区间生成;
或者,
基于第二关系曲线对归一化第一比对数进行比对概率校正,得到比对概率校正后的第一比对数,包括:
基于归一化后的滑窗比对数,对分段区间进行过滤,保留第二关系曲线中归一化后的滑窗比对数不小于第一目标阈值的第二关系曲线区间,以得到比对概率过滤后的第二关系曲线;
基于比对概率过滤后的第二关系曲线对滑窗归一化比对数进行比对概率校正,得到比对概率校正后的第一比对数。
作为本申请一种可能的实现方式,第一确定单元包括:
第一样本获取子单元,用于获得第一训练样本数据,第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,第一特征值为比对数,第一目标值为实际胎儿浓度;
第一建模子单元,用于基于特定模型结构对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
模型处理子单元,用于将比对结果输入至第一胎儿浓度定量模型,得到第一胎儿浓度,并将第一胎儿浓度确定为胎儿浓度。
作为本申请一种可能的实现方式,第一建模子单元具体用于:
将第一训练样本数据划分为训练集和测试集;
基于训练集进行机器学习建模,得到初始模型;
基于初始模型对测试集进行处理,得到与测试集中每一测试样本的预估胎儿浓度;
基于预估胎儿浓度与测试集中每一测试样本的实际胎儿浓度进行比较,得到比较结果;
基于比较结果对初始模型的模型参数进行调整,得到第一胎儿浓度定量模型。
具体的,第一确定单元还用于:
将比对结果输入至第一预设模型,得到初始胎儿浓度,其中,第一预设模型为基于第二样本数据中的对比结果以及与比对结果对应的初始胎儿浓度建立的线性关系模式,第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;
根据第二预设模型对初始胎儿浓度进行校正,获得胎儿浓度,其中,第二预设模型是基于线性拟合确定的常数对第一预设模型进行处理后得到的模型。
在另一种实施方式中,第一比对单元具体用于:
将参考基因组的各碱基位点为测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于第二比对数,计算参考基因组的每个碱基位点对应的核小体中心分数;
基于核小体中心分数以及中心分数筛选阈值,确定核小体中心位置;
基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数;
将加和处理后的第二比对数进行降维处理,得到降维后的归一化第二比对数,并将降维后的归一化第二比对数确定为比对结果;
其中,计算参考基因组的每个碱基位点对应的核小体中心分数,包括:
计算参考基因组的每个碱基位点左右分别对应的第一特定数量的碱基范围的第一平均计数;
计算参考基因组的每个碱基位点左右分别对应的第二特定数量的碱基范围的第二平均计数;
根据第一平均计数和第二平均计数,确定每个位点对应的核小体中心分数。
作为本申请一种可能的实现方式,核小体中心分数的计算公式为:
式中,x为碱基位点,[x-93,x-74-n]表示从距离x一侧93个核苷酸到同侧距离x74-n个核苷酸的区间,[x+93,x+74-n]表示从距离x另一侧93个核苷酸到同侧距离x 74-n个核苷酸的区间,[x-73-n,x+73-n]表示从距离x另一侧73-n个核苷酸到距离x另一侧73-n个核苷酸的区间,n为小于或等于5的自然数。
作为本申请一种可能的实现方式,基于核小体中心分数以及中心分数筛选阈值,确定核小体中心分数,包括:
基于核小体中心分数的最大值,确定最大值在参考基因组上的位置,并将位置确定为第一位置;
将第一位置两侧特定数据的碱基的核小体中心分数清零,并基于清零后剩余的核小体中心分数再次确定最大值,并执行确定最大值在参考基因组上的位置,直至核小体中心分数小于第二目标阈值,将筛选出的位置确定为候选核小体中心位置;
基中心分数筛选阈值以及与候选核小体中心位置对应的核小体中心分数,确定核小体中心位置。
具体的,基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理的步骤中,核小体区域内对应位置包括:
以核小体中心位置为中心,左右各特定数量的碱基的范围。
在一种实施方式中,第一确定单元还用于:
获取第二训练样本数据,第二训练样本数据中的每一样本均标注有特征值和目标值,特征值为降维后的归一化第二比对数,目标值为实际胎儿浓度;
基于第二训练样本数据创建第二胎儿浓度定量模型;
将比对结果输入至第二胎儿浓度定量模型,得到胎儿浓度。
基于前述实施例,参见图10,在本申请的另一实施例中还提供了另一种胎儿浓度的确定装置,该装置可以包括:
第二获取单元20,用于获取待测cfDNA样本的测序序列;
第二确定单元21,用于对参考基因组进行分段,得到多个分段区间,确定每一测序序列落入各个分段区间的第一比对数;
第三确定单元22,用于基于第一比对数,确定第一胎儿浓度;
第四确定单元23,用于基于参考基因组的各碱基位点是测序序列比对起始位置的第二比对数,确定核小体中心位置;
处理单元24,用于基于核小体中心位置将所有核小体区域内对应位置的第二比对数进行对齐加和处理,得到加和处理后的第二比对数;
第五确定单元25,用于基于第一胎儿浓度和加和处理后的第二比对数,确定比对结果;
第六确定单元26,用于基于比对结果,确定胎儿浓度。
在一些实施例中,第二确定单元包括:
第二分段子单元,第二分段子单元具体用于:
基于预设的分割长度对参考基因组进行分段,得到多个初始分段区间;
剔除初始分段区间中特定染色体对应的分段区间,得到多个分段区间;
或者,
剔除参考基因组中的特定染色体,得到剔除后的参考基因组;
基于预设的分割长度对剔除后的参考基因组进行分段,得到多个分段区间。
在一些实施例中,装置还包括:
第二校正单元,用于对第一比对数进行校正,得到校正后的第一比对数,将校正后的第一比对数确定第一胎儿浓度。
在一些实施例中,第二校正单元包括:
第二处理子单元,用于基于每一分段区间的第一比对数以及所有分段区间的平均第一比对数,对每一分段区间的第一比对数进行归一化处理,得到每一分段区间归一化第一比对数;
GC校正子单元,用于对每一分段区间归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
作为本申请一种可能的实现方式,GC校正子单元具体用于:
基于GC含量和每个分段区间对应的归一化第一比对数,生成与特定坐标系相匹配的第一关系曲线;
基于第一关系曲线对归一化第一比对数进行GC校正,得到GC校正后的第一比对数。
在一些实施例中,第二校正子单元还用于:
基于特定滑窗长度对参考基因组进行截取,将截取到的序列与参考基因组进行比对,并统计在参考基因组的每一分段区间上的第一比对数,将第一比对数确定为滑窗比对数;
基于滑窗比对数,确定每一分段区间归一化后的滑窗比对数;
基于每一分段区间归一化后的滑窗比对数和归一化第一比对数进行比对概率校正,确定比对概率校正后的第一比对数。
在一些实施例中,第三确定单元具体用于:
获得第一训练样本数据,第一训练样本数据中的每一样本均标注有第一特征值和第一目标值,第一特征值为比对数,第一目标值为实际胎儿浓度;
基于特定模型结构对第一训练样本数据进行机器学习建模,得到第一胎儿浓度定量模型;
将第一比对结果输入至第一胎儿浓度定量模型,得到第一胎儿浓度。
作为本申请一种可能的实现方式,第三确定单元还用于:
将第一比对数输入至第一预设模型,得到初始胎儿浓度,其中,第一预设模型为基于第二样本数据中的对比结果以及与比对结果对应的初始胎儿浓度建立的线性关系模式,第二样本数据为cfDNA样本中的测序序列与参考基因组进行比对得到的比对结果,以及与比对结果对应的胎儿浓度;
根据第二预设模型对初始胎儿浓度进行校正,获得第一胎儿浓度,其中,第二预设模型是基于线性拟合确定的常数对第一预设模型进行处理后得到的模型。
在一些实施例中,第四确定单元包括:
分数计算子单元,用于将参考基因组的各碱基位点为测序序列比对起始位置的碱基位点的计数确定为第二比对数,并基于第二比对数,计算参考基因组的每个碱基位点对应的核小体中心分数;
中心位置确定子单元,用于基于核小体中心分数以及中心分数筛选阈值,确定核小体中心位置。
在一些实施例中,第五确定单元具体用于:
将核小体区域内各碱基位点对应的加和处理后的第二比对数进行降维处理,得到降维后的归一化第二比对数;
基于第一胎儿浓度和降维后的归一化第二比对数确定为比对结果;
或者,
基于第一胎儿浓度和加和处理后的第二比对数,确定初始比对结果;
对初始比对结果进行降维处理,将降维后的初始比对结果确定为比对结果。
需要说明的是,在该实施例的胎儿浓度的确定装置的各个单元的执行过程可以参见图8的胎儿浓度的确定方法,此处不再详述。
在本申请的另一实施例中,还提供了一种可读存储介质,其上存储有计算机程序,计算机程序被处理器执行时,实现如上任一项的胎儿浓度的确定方法各个步骤。
在本申请的另一实施例中,还提供了一种电子设备,电子设备可以包括:
存储器,用于存储应用程序和应用程序运行所产生的数据;
处理器,用于执行应用程序,以实现如上述中任一项胎儿浓度的确定方法。
需要说明的是,本实施例中处理器的具体实现可以参考前文中的相应内容,此处不再详述。
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。对于实施例公开的装置而言,由于其与实施例公开的方法相对应,所以描述的比较简单,相关之处参见方法部分说明即可。
专业人员还可以进一步意识到,结合本文中所公开的实施例描述的各示例的单元及算法步骤,能够以电子硬件、计算机软件或者二者的结合来实现,为了清楚地说明硬件和软件的可互换性,在上述说明中已经按照功能一般性地描述了各示例的组成及步骤。这些功能究竟以硬件还是软件方式来执行,取决于技术方案的特定应用和设计约束条件。专业技术人员可以对每个特定的应用来使用不同方法来实现所描述的功能,但是这种实现不应认为超出本申请的范围。
结合本文中所公开的实施例描述的方法或算法的步骤可以直接用硬件、处理器执行的软件模块,或者二者的结合来实施。软件模块可以置于随机存储器(RAM)、内存、只读存储器(ROM)、电可编程ROM、电可擦除可编程ROM、寄存器、硬盘、可移动磁盘、CD-ROM、或技术领域内所公知的任意其它形式的存储介质中。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本申请。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本申请的精神或范围的情况下,在其它实施例中实现。因此,本申请将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 胎儿游离DNA浓度获取方法和装置
- 答案质量确定模型训练方法、答案质量确定方法及装置
- 一种神经网络模型训练方法及装置、文本标签确定方法及装置
- 确定胎儿核酸浓度的方法以及胎儿基因分型方法
- 建立胎儿浓度校正模型的方法及装置与胎儿浓度定量的方法及装置