一种复方奥美拉唑干混悬剂中杂质的分离检测方法
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于医药检测技术领域,具体涉及一种复方奥美拉唑干混悬剂中杂质的分离检测方法。
背景技术
胃食管反流病及十二指肠溃疡易反复发作、较难治愈。胃粘膜分泌的胃酸有强烈的腐蚀作用,如果因为某种原因导致胃粘膜受损,无法及时补充粘膜液,或者胃酸分泌过多使粘膜出现缺口,胃粘膜就会受到胃酸腐蚀,造成胃溃疡、十二指肠溃疡等消化性溃疡病。抑制胃酸分泌是缓解消化性溃疡症状,帮助溃疡愈合的关键,胃酸分泌少会使灼痛减轻,胃粘膜有足够时间修复。
奥美拉唑是一种质子泵抑制剂。质子泵又称胃H
复方奥美拉唑干混悬剂用于十二指肠溃疡、活跃性良性胃溃疡、有烧心等症状的胃食管反流症(GERD)、糜烂性食管炎等的治疗,具备良好的疗效,值得在临床治疗中广泛使用。而复方奥美拉唑干混悬剂在生产、运输和贮藏过程中,可能产生降解产物、聚合物或晶型转变等多种杂质。杂质的存在会影响药物的意料效果和临床使用安全。国内外对药物的研究,可允许含有一定限量的无害或低毒性的有关物质,但对毒性较大,能危害人体健康的、无效的或能影响药物稳定性的有关物质或杂质则必须严格控制。因此,杂质的分离检测是控制药品质量的重要指标。
复方奥美拉唑干混悬剂中存在的杂质主要为工艺副产物和降解产物,需建立合适的分离、检测方法,对这些杂质进行有效的检测和监控。复方奥美拉唑干混悬剂中可能存在的主要杂质如下:
中国发明专利CN102735771B公开了一种用于奥美拉唑钠杂质的色谱检测方法的系统适用性检验方法,包括步骤:(1)采用高效液相色谱法,色谱条件为:用十八烷基硅烷键合硅胶为填充剂;以磷酸盐缓冲液、硫酸氢四丁胺缓冲液、乙腈按体积比64:6:30为流动相,检测波长为280nm;(2)系统适用性试验溶液的配制:取奥美拉唑10mL,置于50mL容量瓶内,按照1:1的体积比加入质量含量0.05%的高锰酸钾溶液与乙腈的混合溶液5mL,振摇使溶解,稀释至50mL,得到奥美拉唑与奥美拉唑磺酰化物的混合溶液,作为系统适用性试验溶液;(3)测定:取10-20μL所述系统适用性试验溶液注入色谱仪,记录色谱图,有效对杂质进行破坏,准确分析,系统稳定性好,平衡时间短,成本低。中国发明专利CN103592379B公开了一种奥美拉唑有关物质的分析方法,采用高效液相色谱法,用有机溶剂和盐的混合相溶解样品,以乙腈和pH=7.6的磷酸盐为流动相,在辛烷基键和硅胶色谱柱上进行梯度洗脱。本方法简单有效地解决了有关物质和溶剂峰重叠、杂质检出数量少等缺点。
现有的国家药典标准中并未收载复方奥美拉唑干混悬剂的检测方法;因此,建立一种分离度高、专属性强、灵敏度高、操作简单的新的复方奥美拉唑干混悬剂的杂质分离检测方法,实现复方奥美拉唑干混悬剂中各杂质的检出和分离,有利于提高该品种的安全性、有效性及质量可控性,在临床上有着重要的现实意义。
发明内容
本发明针对现有技术存在的问题,提供了一种复方奥美拉唑干混悬剂中杂质的分离检测方法,复方奥美拉唑干混悬剂中的杂质包括杂质A、杂质B、杂质C、杂质D、杂质E、杂质H、杂质I,该检测方法为特定的HPLC法,专属性强、灵敏度高、操作方便,检测结果准确可靠,可有效控制药品品质。
为实现以上目的,本发明提供的技术方案如下:
一方面,本发明提供了一种复方奥美拉唑干混悬剂中杂质的分离检测方法,该分离检测方法为采用HPLC法;
所述HPLC法的色谱条件为:
色谱柱:十六烷基键合硅胶填料色谱柱;
流动相:流动相A为戊烷磺酸钠溶液;流动相B为含三氟乙酸的四氢呋喃;
洗脱:梯度洗脱,洗脱程序如下所示:
流速:0.5-1.5mL/min;
柱温:10-30℃;
检测波长:260-268nm。
优选地,所述色谱柱为十六烷基键合硅胶填料色谱柱(4.6mm×150mm,5μm)。
优选地,所述流动相A,戊烷磺酸钠溶液的浓度为10-40mmol/L,pH为5.0-6.5。
进一步优选地,所述戊烷磺酸钠溶液的浓度为20-30mmol/L,pH为5.5-6.0。
更进一步优选地,所述戊烷磺酸钠溶液的浓度为25mmol/L,pH为6.0。
进一步优选地,所述流动相A,使用磷酸、硫酸或盐酸进行pH的调节;更进一步优选为磷酸。
优选地,所述流动相B为含0.1-0.4%三氟乙酸的四氢呋喃。
进一步优选地,所述流动相B为含0.2-0.3%三氟乙酸的四氢呋喃。
最优选地,所述流动相B为含0.2%三氟乙酸的四氢呋喃。
优选地,所述梯度洗脱,洗脱程序如下所示:
优选地,所述流速为0.6-1.0mL/min;所述柱温为15-25℃;所述检测波长为262-265nm。
进一步优选地,所述流速为0.8mL/min;所述柱温为20℃;所述检测波长为264nm。
优选地,所述HPLC法,分离检测的进样体积为15-25μL;进样盘温度为3-5℃。
优选地,分离检测方法,在进样前需对样品进行稀释,稀释剂为流动相A和流动相B的混合物。
进一步优选地,所述稀释剂中,流动相A和流动相B的体积比为2-4:1。
更进一步优选地,所述稀释剂中,流动相A和流动相B的体积比为3:1。
优选地,所述分离检测方法,包括步骤:
(1)样品配制:取对照品、供试品用乙腈溶解,然后用稀释剂进行稀释,分别制得对照品溶液和供试品溶液;
(2)采用上述HPLC法的色谱条件,对对照品溶液和供试品溶液进行高效液相分析。
另一方面,本发明提供上述分离检测方法在复方奥美拉唑干混悬剂质量检测中的应用。
相对于现有技术,本发明具有以下有益效果:
本发明提供的分离检测方法,采用特定条件的HPLC法能高效分离测定复方奥美拉唑干混悬剂中杂质A、杂质B、杂质C、杂质D、杂质E、杂质H、杂质I的含量,且灵敏度高、专属性强,检查结果可靠准确,有利于对复方奥美拉唑干混悬剂中杂质的检出提供技术手段,从而对其进行质量控制,为复方奥美拉唑干混悬剂的质量检测提供新的分离检测方法。
附图说明
图1为专属性-空白溶液色谱图。
图2为专属性-空白辅料溶液色谱图。
图3为专属性-对照品溶液色谱图。
图4为专属性-供试品溶液色谱图。
图5为专属性-专属性溶液色谱图。
图6为奥美拉唑的线性标准曲线图。
图7为杂质A线性标准曲线图。
图8为杂质B线性标准曲线图。
图9为杂质C线性标准曲线图。
图10为杂质D线性标准曲线图。
图11为杂质E线性标准曲线图。
图12为杂质H线性标准曲线图。
图13为杂质I线性标准曲线图。
图14为实施例2专属性-专属性溶液色谱图。
图15为实施例3专属性-专属性溶液色谱图。
图16为对比例1专属性-专属性溶液色谱图。
图17为对比例2专属性-专属性溶液色谱图。
图18为对比例3专属性-专属性溶液色谱图。
具体实施方式
以下非限制性实施例可以使本领域的普通技术人员更全面的理解本发明,但不以任何方式限制本发明。下面以具体实施例的方式对本发明作进一步的说明。优选实例中未注明具体条件的试验方法,通常按照常规条件,所举实施例是为了更好地对本发明的内容进行说明,但并不是本发明的内容仅限于所举实施例。下述内容仅仅是对本申请要求保护的范围的示例性说明,本领域技术人员可以根据所公开的内容对本申请的发明做出多种改变和修饰,而其也应当属于本申请要求保护的范围之中。
本发明实施例中所使用的各种化学试剂如无特殊说明均通过常规商业途径获得。本发明中的具体实施过程中使用的试剂信息如下:
复方奥美拉唑干混悬剂的来源为浙江和沐康医药科技有限公司,批号:Z-OML2207039;规格为40mg/1.68g;
对照品来源信息如下:
奥美拉唑对照品:浙江和沐康医药科技有限公司;批号:WRSC309(T)-12107007C;含量:99.7%;
杂质A对照品:SINCO PHARMACHEM;批号:18-06-1725;含量:98.36%;
杂质B对照品:SINCO PHARMACHEM;批号:22-08-0109;含量:98.82%;
杂质C对照品:SINCO PHARMACHEM;批号:18-06-1726;含量:98.74%;
杂质D对照品:SINCO PHARMACHEM;批号:19-04-0912;含量:96.20%;
杂质E对照品:SINCO PHARMACHEM;批号:20-04-1419;含量:98.57%;
杂质H对照品:SINCO PHARMACHEM;批号:19-06-1007;含量:99.24%;
杂质I对照品:SINCO PHARMACHEM;批号:21-05-1018;含量:96.90%。
实施例1
(1)色谱条件
仪器:Thermo Vanquish;
色谱柱:用十六烷基硅烷键合硅胶为填充剂(Supelco SUPELCOSIL ABZ+Plus,4.6mm×150mm,5μm);鬼峰捕集柱Welch Ghost-Buster,4.6×50mm;
流动相A:25mmol/L戊烷磺酸钠溶液,用磷酸调节pH值至6.0(精密称取戊烷磺酸钠适量,用水溶解并配置成25mmol/L的溶液,用磷酸调pH值至6.0,过滤,超声脱气,即得);
流动相B:0.2%三氟乙酸的四氢呋喃;
稀释剂:流动相A-流动相B(体积比为3:1)。
洗脱梯度如下所述:
检测器:UV;检测波长:264nm;流速:0.8mL/min;柱温:20℃;进样体积:20μL;进样盘控温:4℃。
对该分离检测方法进行方法学验证,验证项目包括专属性、定量限、检测限、线性、耐用性,具体如下:
(2)专属性
空白溶液:流动相A:流动相B=3:1(体积比)。
空白辅料溶液:取空白辅料(包含碳酸氢钠1.68g、蔗糖1.02g、木糖醇1.3g、黄原胶0.06、三氯蔗糖0.06、香精0.035)4.1550g,精密称定,置50mL容量瓶中,加入乙腈适量,超声使溶解,期间不时振摇,取出放冷,用乙腈稀释至刻度,摇匀,滤过(针式过滤器使用为领航、
主成分贮备液:精密称取奥美拉唑对照品7.998mg,置200mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,即得。
对照品溶液:精密移取主成分贮备液1mL,置100mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
供试品溶液:取本品1袋(40mg/1.68g,复方奥美拉唑干混悬剂),倒出内容物,置于50mL量瓶中,用少量乙腈润洗包装袋内壁,润洗液转移至上述容量瓶中,加入乙腈适量,超声使奥美拉唑溶解,期间不时振摇(超声过程放冰袋防止温度变高),取出放冷,用乙腈稀释至刻度,摇匀,滤过,再精密移取续滤液5mL,置20mL量瓶中,用稀释剂稀释至刻度,摇匀,即得(按奥美拉唑计约含0.2mg/mL)。
杂质A贮备液:精密称取杂质A对照品1.490mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质B贮备液:精密称取杂质B对照品1.496mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质C贮备液:精密称取杂质C对照品1.562mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质D贮备液:精密称取杂质D对照品1.531mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质E贮备液:精密称取杂质E对照品1.526mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质H贮备液:精密称取杂质H对照品1.500mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质I贮备液:精密称取杂质I对照品1.571mg,置10mL量瓶中,用乙腈溶解并稀释至刻度,摇匀即得。
杂质A定位溶液:精密移取杂质A贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质A:1.5μg/mL)。
杂质B定位溶液:精密移取杂质B贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质B:1.5μg/mL)。
杂质C定位溶液:精密移取杂质C贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质C:1.5μg/mL)。
杂质D定位溶液:精密移取杂质D贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质D:1.5μg/mL)。
杂质E定位溶液:精密移取杂质E贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质E:1.5μg/mL)。
杂质H定位溶液:精密移取杂质H贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质H:1.5μg/mL)。
杂质I定位溶液:精密移取杂质I贮备液1mL,置100mL量瓶,用稀释剂稀释至刻度,摇匀,即得(杂质I:1.5μg/mL)。
杂质贮备液-1:精密称取杂质A、E、I、B、H、C对照品分别为1.052mg、1.061mg、1.045mg、1.031mg、1.049mg、1.015mg,置同一25mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,即得。杂质D贮备液-1:精密称取杂质D对照品1.990mg,置20mL量瓶中,用乙腈溶解并稀释至刻度,摇匀,即得。
杂质贮备液-2:精密移取杂质贮备液-1、杂质D贮备液-1各1mL,置同一10mL量瓶,用乙腈稀释至刻度,摇匀,即得。(含杂质A、E、I、B、H、C各4μg/mL,杂质D10μg/mL)。
专属性溶液:取本品(复方奥美拉唑干混悬剂)1袋(40mg/1.68g),倒出内容物,置于50mL量瓶中,用少量乙腈润洗包装袋内壁,润洗液转移至上述容量瓶中,加入乙腈适量,超声使奥美拉唑溶解,期间不时振摇(超声过程放冰袋防止温度变高),取出放冷,用乙腈稀释至刻度,摇匀,滤过(针式过滤器使用为领航、
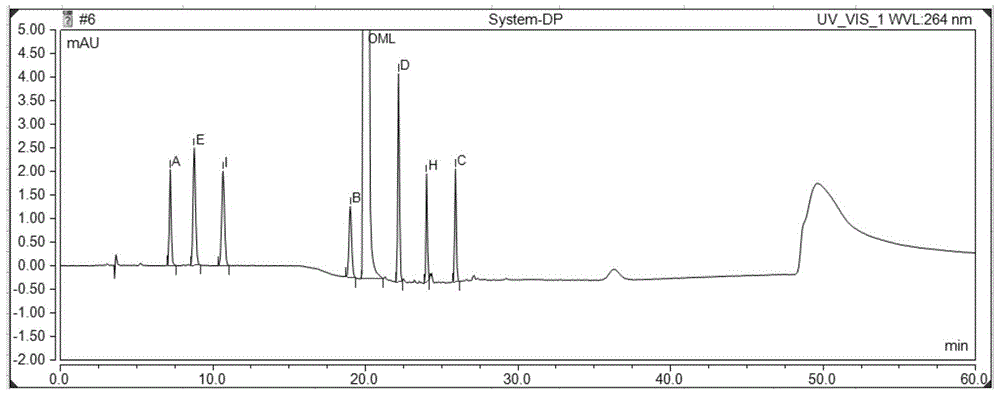
按上述色谱条件,精密量取空白溶液、空白辅料溶液、对照品溶液、供试品溶液、各杂质定位溶液及专属性溶液10μL,注入液相色谱仪,记录色谱图,专属性结果见表1、专属性溶液色谱图见图1-5。图1是专属性-空白溶液色谱图;图2是专属性-空白辅料溶液色谱图;图3是专属性-对照品溶液色谱图;图4是专属性-供试品溶液色谱图;图5是专属性-专属性溶液色谱图。
表1专属性
结果表明:专属性溶液中各杂质与前后杂质分离度均大于1.5,分离度良好,本发明提供的分离检测方法专属性好。
(3)定量限
杂质贮备液-1、杂质D贮备液-1:配制同“专属性”项下。
对照品贮备液:配制同“专属性”项下“主成分贮备液”。
定量限溶液:分别精密移取对照品贮备液0.1mL、杂质贮备液-1 0.1mL、杂质D贮备液-10.1mL,置100mL量瓶中,用稀释剂稀释至刻度,摇匀,即得,平行配制6份。
按上述色谱条件(1),精密量取20μL,注入液相色谱仪,记录色谱图,计算6份奥美拉唑和杂质峰面积的RSD%。定量限结果见表2-表9。
表2定量限试验-杂质A
表3定量限试验-杂质B
表4定量限试验-奥美拉唑
表5定量限试验-杂质C
表6定量限试验-杂质D
表7定量限试验-杂质E
表8定量限试验-杂质H
表9定量限试验-杂质I
结果表明:6份定量限溶液各峰信噪比均大于10,各杂质峰峰面积相对标准偏差(RSD%)均小于10%。当样品浓度为0.2mg/mL时,杂质A、杂质E、杂质I、杂质B、杂质H、杂质C、其他杂质(以奥美拉唑计)定量限均为0.02%,杂质D定量限为0.05%。
(4)检测限
检测限溶液:精密移取5mL定量限溶液,置10mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。取上述溶液,按上述色谱条件(1),精密量取20μL,注入液相色谱仪,记录色谱图,检测限结果见表10。
表10检测限试验
结果表明:当样品浓度为0.2mg/mL时,杂质A、杂质E、杂质I、杂质B、杂质H、杂质C、其他杂质(以奥美拉唑计)的检测限为0.010%,杂质D检测限为0.024%,信噪比均大于3。综上,本法检测灵敏度较高。
(5)线性与范围
杂质贮备液-1、杂质D贮备液-1:配制同“专属性”项下。
对照品贮备液:配制同“专属性”项下“主成分贮备液”。
线性溶液1(定量限):同“定量限”项下定量限溶液;
线性溶液2(50%):分别精密移取对照品贮备液0.5mL、杂质贮备液-1 0.5mL、杂质D贮备液-1 0.5mL,置100mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
线性溶液3(100%):分别精密移取对照品贮备液1mL、杂质贮备液-1 1mL、杂质D贮备液-1 1mL,置100mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
线性溶液4(150%):分别精密移取对照品贮备液1.5mL、杂质贮备液-1 1.5mL、杂质D贮备液-1 1.5mL,置100mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
线性溶液5(200%):分别精密移取对照品贮备液1mL、杂质贮备液-1 1mL、杂质D贮备液-1 1mL,置50mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
线性溶液6(600%):分别精密移取对照品贮备液3mL、杂质贮备液-1 3mL、杂质D贮备液-13mL,置50mL量瓶中,用稀释剂稀释至刻度,摇匀,即得。
精密量取上述线性溶液各20μL,注入液相色谱仪(Thermo Vanquish),记录色谱图,以浓度为横坐标,峰面积为纵坐标,作线性回归方程。计算杂质的校正因子。结果见表11-表18、图6-图13。其中,图6为奥美拉唑(对照品)的线性标准曲线图;图7为杂质A线性标准曲线图;图8为杂质B线性标准曲线图;图9为杂质C线性标准曲线图;图10杂质D线性标准曲线图;图11杂质E线性标准曲线图;图12杂质H线性标准曲线图;图13杂质I线性标准曲线图。
杂质的校正因子=主成分线性回归方程斜率/杂质线性回归方程斜率
表11线性-奥美拉唑-对照品
表12线性-杂质A
/>
表13线性-杂质B
表14线性-杂质C
表15线性-杂质D
表16线性-杂质E
表17线性-杂质H
表18线性-杂质I
结果表明:杂质A、杂质B、杂质C、杂质D、杂质E、杂质H、杂质I、奥美拉唑在各自的浓度范围内,线性回归方程的相关系数(r)均大于0.995,Y轴截距均在100%响应值的10%以内,响应因子的RSD%均在10%以内,均符合要求,本方法线性关系良好。
(6)耐用性
取专属性溶液,在4℃条件下放置0、2、4、7、9、28小时后进样,比较各时间点测定的峰面积与0h的峰面积的比值,考察溶液稳定性。
耐用性考察结果如表19所示。
表19耐用性
结果表明:专属性溶液在4℃条件下放置44小时,各时间点测定的峰面积与0h的峰面积的比值均在95%-105%范围内,溶液稳定性良好,本方法耐用性良好。
实施例2
色谱条件:
仪器:Thermo Vanquish;
色谱柱:用十六烷基硅烷键合硅胶为填充剂(Supelco SUPELCOSIL ABZ+Plus,4.6mm×150mm,5μm);鬼峰捕集柱Welch Ghost-Buster,4.6×50mm;
流动相A:20mmol/L戊烷磺酸钠溶液,用磷酸调节pH值至5.0(精密称取戊烷磺酸钠适量,用水溶解并配置成20mmol/L的溶液,用磷酸调pH值至5.0,过滤,超声脱气,即得);
流动相B:0.2%三氟乙酸的四氢呋喃;
稀释剂:流动相A-流动相B(体积比为3:1)。
洗脱梯度如下所述:
检测器:UV;检测波长:264nm;流速:0.5mL/min;柱温:15℃;进样体积:20μL;进样盘控温:4℃。
按实施例1所述的专属性检测方法,进行分离检测,专属性检测结果见表20。该实施例的专属性溶液色谱图见图14。
表20实施例2专属性
结果表明,使用本发明的分离检测方法和HPLC的色谱条件,可以高效的将专属性溶液中的各杂质与药品成分分离开,各杂质与前后杂质分离度均大于1.5,分离度高,分离效果好,无重叠。
实施例3
色谱条件:
仪器:Thermo Vanquish;
色谱柱:用十六烷基硅烷键合硅胶为填充剂(Supelco SUPELCOSIL ABZ+Plus,4.6mm×150mm,5μm);鬼峰捕集柱Welch Ghost-Buster,4.6×50mm;
流动相A:30mmol/L戊烷磺酸钠溶液,用磷酸调节pH值至6.5(精密称取戊烷磺酸钠适量,用水溶解并配置成30mmol/L的溶液,用磷酸调pH值至6.5,过滤,超声脱气,即得);
流动相B:0.2%三氟乙酸的四氢呋喃;
稀释剂:流动相A-流动相B(体积比为3:1)。
洗脱梯度如下所述:
检测器:UV;检测波长:264nm;流速:1.0mL/min;柱温:25℃;进样体积:20μL;进样盘控温:4℃。
按实施例1所述的专属性检测方法进行检测,专属性检测结果见表21。该实施例的专属性溶液色谱图见图15。
表21实施例3专属性
结果表明,使用本发明的分离检测方法和HPLC的色谱条件,可以高效的将专属性溶液中的各杂质与药品成分分离开,各杂质与前后杂质分离度均大于1.5,分离度高,分离效果好,无重叠。
对比例1
与实施例1不同的是,流动相A不同,具体为:
流动相A:25mmol/L甘氨酸溶液,用磷酸调节pH值至6.0(精密称取甘氨酸适量,用水溶解并配置成25mmol/L的溶液,用磷酸调pH值至6.0,过滤,超声脱气,即得)。
其余色谱条件和参数均与实施例1相同。
按实施例1所述的专属性检测方法进行检测,专属性检测结果见表22。该对比例的专属性溶液色谱图见图16。
表22对比例1专属性
结果表明,在该检测方法条件下,杂质B与杂质D峰重合,分离度小于1.5,不符合要求,分离度较差。
对比例2
与实施例1不同的是,洗脱梯度不同,具体为:
其余色谱条件和参数均与实施例1相同。
按实施例1所述的专属性检测方法进行检测,专属性检测结果见表23。该对比例的专属性溶液色谱图见图17。
表23对比例2专属性
结果表明,在该检测方法条件下,杂质I与杂质E峰重合,分离度小于1.5,不符合要求,分离度较差。
对比例3
与实施例1不同的是,色谱柱的填料不同,具体为:
色谱柱:Waters XBridge C8,4.6×150mm,5μm。
其余色谱条件和参数均与实施例1相同。
按实施例1所述的专属性检测方法进行检测,专属性检测结果见表24。该对比例的专属性溶液色谱图见图18。
表24对比例3专属性
结果表明,在该检测方法条件下,杂质I与杂质E峰重合,分离度小于1.5,不符合要求,分离度较差。
应用例
应用的检测方法:同实施例1。
结果:采用实施例1所述的检测方法检测济宁华能制药厂有限公司(国药准字H20233020)与南京海纳医药科技股份有限公司(国药准字H20213132)制剂,杂质检测结果如下:
表25不同厂家产品杂质检测结果
上述结果表明,实施例1中检测方法可检出不同水平的杂质E。杂质E为氮氧化物,会对人体的心血管系统、神经系统等造成危害:氮氧化物会影响心血管系统,导致心脏病、高血压、动脉粥样硬化等心血管疾病;可以影响神经系统的正常功能,导致头痛、眩晕、疲劳等症状,严重者可能产生神经衰弱、痴呆等疾病;除以上相对常见危害外,还可能导致其他,如口腔遗传、免疫系统异常、遗传能力丧失。故本方法对控制杂质,进而控制杂质产生的危害有重要作用。
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。
- 一种测定含色氨酸的复方氨基酸注射液中杂质B、C、D含量的检测方法
- 一种头孢克洛干混悬剂样品中的十二烷基硫酸钠的HPLC检测方法
- 一种高效液相色谱法分离分析阿伐斯汀原料药中间体Z3中Z3及有关杂质的方法
- 一种铝合金粉末中杂质元素含量的检测方法
- 一种奥美拉唑碳酸氢钠干混悬剂中杂质的制备方法及其检测方法
- 一种复方奥美拉唑干混悬剂及其制备方法