一种吲哚衍生物的合成方法
文献发布时间:2023-06-19 13:30:50
技术领域
本发明属于吲哚合成技术领域,具体涉及一种吲哚衍生物的合成方法。
背景技术
吲哚又叫苯并吡咯,由苯环和吡咯环构成。吲哚及其衍生物广泛存在于各种生物体内,最初由靛蓝降解发现。煤焦油、香料、氨基酸、激素中均含有吲哚及吲哚衍生物,粪便的臭味与其中含有的3-甲基吲哚有很大关系。此外,吲哚及其衍生物作为非常重要的中间体或特征结构,在医药领域应用广泛。
已有合成吲哚及其衍生物方法中的催化体系均为多组分催化,主催化剂均为贵金属,且结构复杂,价格昂贵。由苯胺和乙二醇体系一步催化合成吲哚及其衍生物因具有反应原料价廉、反应步骤少、副产物无害且易分离(副产物为水和氢气)等特点而倍受关注。苯胺和乙二醇合成吲哚的反应存在诸多优势,如反应步骤少、原料易得、副产物只有水和氢气、对环境没有污染等,从而被广泛研究应用于吲哚的工业化生产。
鉴于国内煤制乙二醇产量逐年大幅增加,且质量指标尚未达到生产聚酯的要求,针对乙二醇的高值精细转化开展研究具有重要的市场需求和经济意义。吲哚及其衍生物作为乙二醇下游产物之一,在生物、药物、食品、染料、香料、农药等领域应用广泛,吲哚及其衍生物的合成研究将为乙二醇的高值转化提供有效途径。
发明内容
本发明为了解决目前以乙二醇为原料合成吲哚及其衍生物的反应,条件相对苛刻,主催化剂的结构复杂、价格昂贵等问题,提供了一种吲哚衍生物的合成方法。
本发明由如下技术方案实现的:一种吲哚衍生物的合成方法,以酸性Al
所述Pt/Al
所述苯胺类化合物为:烷基、甲氧基、甲酸甲酯基、硝基或氟取代的苯胺,以及1-萘胺或2-萘胺。
所述苯胺类化合物为:
所述苯胺类化合物为:供电子基团取代的苯胺或2-萘胺。
所述苯胺类化合物为2-萘胺。
所述苯胺类化合物为1mmol,乙二醇为54mmol,催化剂用量为0.015mmol;柱层析按照石油醚与乙酸乙酯的体积比为90:10进行洗脱。
本发明对乙二醇苯胺反应中催化效果较好的Pt/Al
为了增大催化剂中的贵金属铂的利用效率,对催化剂进行了几步回收重复利用。结果显示,在前4次反应中,催化剂的催化效果减弱较小,当反应次数达到5次时,反应的收率降低较大,说明催化剂的稳定性相对较好,可以重复利用4次,累计参与反应时间96 h而催化效果降低幅度不大,可大幅降低催化剂成本,进而使本发明的经济效益更加突出,且大大增加了本发明技术的核心竞争力及工业化生产的可能。对原催化剂与分别经历1、3、5次反应的回收催化剂进行了电镜微观形貌表征,结果显示原催化剂中载体上的铂颗粒分布较均匀,随着反应次数的增加,分布与粒径都变得混乱。由此得出结论:铂颗粒参与反应过程会发生流失,在高温条件下,铂颗粒会熔融结合成更大的颗粒使催化剂有效接触面积减少,这是催化效率降低的主要原因。
附图说明
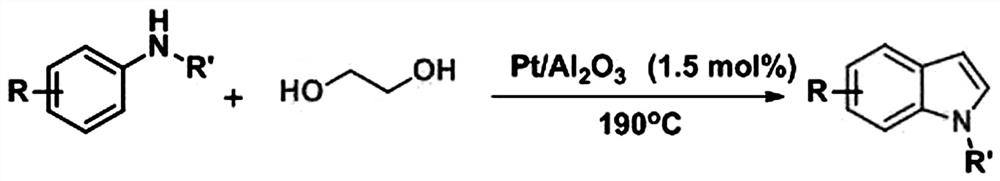
图1为苯胺衍生物合成吲哚衍生物反应方程式;
图2为
图3为2-萘胺与乙二醇反应生成副产物质谱图与结构式;
图4为2-萘胺与乙二醇反应中间体以及副产物的变化趋势;
图5为推测2-萘胺与乙二醇反应过程图;
图6为2-萘胺与乙二醇反应生成4,5-苯并吲哚的反应方程式;
图7为催化剂经过四次循环使用后的质量及收率变化;
图8为新制备负载型催化剂Pt/Al
图9为一次反应后Pt/Al
图10为三次反应后Pt/Al
图11为五次反应后Pt/Al
图12为催化剂中铂的粒径、粒径方差与反应次数的关系;
图13为7-甲基吲哚的核磁谱图;
图14为7-甲氧基吲哚的核磁谱图;
图15为4-甲氧基吲哚的核磁谱图;
图16为6-甲氧基吲哚的核磁谱图;
图17为6-甲酸甲酯基吲哚的核磁谱图;
图18为5-甲酸甲酯基吲哚的核磁谱图;
图19为4,5-苯并吲哚的核磁谱图;
图20为6-氨基-6,7-苯并吲哚的核磁谱图;
图21为N-甲基-6,7-苯并吲哚的核磁谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例;基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
一、实验试剂及仪器:实验仪器见表1,试剂见表2。
表1:实验仪器
表2:实验试剂
二、吲哚衍生物的合成
1、实验方法:
负载型催化剂的制备:先将酸载体性Al
分离条件:反应结束后,先加水搅拌均匀,然后再用二氯甲烷进行萃取,有机相经旋蒸浓缩除去溶剂后以石油醚、乙酸乙酯为洗脱剂,经柱层析分离得到目标产物。反应总方程式如图1所示。
2、实验结果:
选取烷基、甲氧基、甲酸甲酯基、硝基、氟取代的苯胺以及萘胺,进行底物拓展性研究,反应条件为:0.015mmol催化剂,3ml乙二醇、1mmol苯胺下反应,控制温度为190℃,反应24h,催化剂负载量为3wt%。所得吲哚衍生物的结构和收率如表3所示。
表3:吲哚衍生物的合成实验结果
所得产物的核磁分析结果如下:
1、7-甲基吲哚
2、7-甲氧基吲哚
3、4-甲氧基吲哚
4、6-甲氧基吲哚
5、吲哚-6-甲酸甲酯
6、吲哚-5-甲酸甲酯
7、4,5-苯并吲哚
8、6-氨基-6,7-苯并吲哚
9、
由上述结果可以看出此反应体系的底物普适性较好,多种苯胺衍生物可以在此催化体系下与乙二醇反应生成对应的吲哚衍生物。但反应的收率相差较大,氨基邻位存在甲酸甲酯基团的苯胺衍生物可能由于空间位阻影响,反应不能顺利进行或者反应收率不佳。苯胺上有强吸电子基团硝基与氰基取代时,反应几乎不能发生,但供电子基团取代的苯胺,反应收率较高,2-萘胺的反应收率可达到95%,其转化率接近100%。推测此现象是由于苯胺的氨基氢与乙二醇羟基脱水过程中,当苯环上电子云密度更高时时,此过程进行得更快。而1-萘胺与2-萘胺相比,2-萘胺空间位阻相对更小,1-位的反应活性相对更高,因此反应产率更高。
2-萘胺与乙二醇的反应收率优秀,生成的4,5-苯并吲哚化合物具有多种光热特性,可以应用于有机电致发光器件、有机太阳能电池、钙钛矿电池、有机薄膜晶体管或有机光感受器领域。本反应路线为有光热应用的4,5-苯并吲哚分子以后的化学转化提供了可能性。
3、对反应机理的推测:
选择反应收率最高的2-萘胺作为研究对象:采用气相色谱-质谱联用(GC-MS)对2-萘胺与乙二醇的反应液进行了分析,结合文献分析所获得的原始数据,解析出了几种可能的中间体以及副产物的结构。中间体的质谱峰面积占比随时间变化如表4所示。
反应以及取样方法:2-萘胺用量为1 mmol,乙二醇为54 mmol,催化剂用量为0.015mmol,反应温度为190℃,反应经过一定时间(1 h、2 h、3 h、4 h、8 h、12 h、16 h、20 h、24h)后冷却至室温,分别从上清液取0.2 mL留样备用检测,取样后继续加热反应。
采用配有AOS-5000 plus自动进样器的日本岛津GC MS-QP 2010 ultra 气相色谱-质谱联用仪(GC-MS)联用仪对反应体系进行了跟踪分析,所用分析条件为:RestekRxi-5ms色谱柱(30 m×0.25 mm×0.25 μm),初始温度60℃,以6℃/min升至250℃(后保持10min,进样温度为300℃,载气线速度为36.9 cm/s,柱前为压55.5 kPa,分流比为70:1,载气为高纯氮气,流量为1.00 mL/min;MS离子源为EI源,温度为230℃,质谱与色谱接口温度280℃,质量扫描范围(m/z)29~250 amu。
表4:2-萘胺与乙二醇反应生成4,5-苯并吲哚的GC-MS分析结果
根据质谱结果,并对照标准谱图,推测
上述中间体、副产物和产物的峰面积随时间的变化趋势表示如图4所示,可以看出反应产物4,5-苯并吲哚在4 h以后呈持续减少趋势,说明此反应的反应时间仍需优化,在4h以前已经反应到达峰值,4 h之后反应的副产物也逐渐增多,且副产物均以反应产物4,5-苯并吲哚为原料发生的,几种副产物多是与吡咯环上的N-H发生了取代反应。
由此推测在反应初期,氨基更容易与乙二醇进行脱水缩合:乙二醇在酸性氧化铝的诱导下自身消除一分子水,转化为乙烯醇,经过质子转移转化为乙醛,乙醛与萘上的氨基脱除一分子水,完成醛胺缩合,缩合后的亚胺容易转化为烯胺,随后在铂的作用下同萘的1号位C-H作用,脱除一分子氢气,最终生成产物4,5-苯并吲哚。
反应机理推测如图5所示。根据上述可能的反应过程,图2中的化合物
4、催化剂Pt/Al
Pt/Al
负载型催化剂的活性降低可能存在多种原因,例如反应混合物中的配位性物质会使催化剂中毒失去活性;或者高温反应之后催化剂的晶粒变大,比表面积减小,活性降低;或是反应之后活性物质表面出现碳沉积,导致活性组分不能充分接触反应物,其他还包括流失和破碎等原因。在此反应中,催化剂经历较高温度,可能发生积碳、烧结、流失等状况。将反应过后回收的催化剂在高温下重新焙烧,可基本排除由于中毒与碳沉积等因素导致催化剂失活。
A、实验方法:基于底物拓展中收率最高的2-萘胺和乙二醇在Pt/Al
反应结束后反应液静置降温至室温,反应液离心后取上层清液,以石油醚(PE):乙酸乙酯(EtOAc)= 90:10(体积比) 进行柱层析分离,计算收率。固体催化剂用水洗涤、抽滤后于500℃焙烧5 h,冷却后称量得到回收率,并进行循环使用。
B、实验结果
图7为催化剂经过四次循环使用后的质量及收率变化。反应收率由最初的95.1%逐步降低,第二次产率为89.5 %,第三次产率为83.1 %,最后一次反应收率为72.8%;催化剂回收效率也由91.7 %降低至70.0 %。由此可知,经过反应的Pt/Al
为了验证猜想,用美国FEI公司Tiecnai G2 F20 S-Twin透射电子显微镜对原始Pt/Al
根据未经反应催化和经反应1次、3次、5次的Pt/Al
图12是Pt/Al
对乙二醇苯胺反应中催化效果较好的Pt/Al
为了增大催化剂中的贵金属铂的利用效率,对催化剂进行了几步回收重复利用。结果显示,在前4次反应中,催化剂的催化效果减弱较小,当反应次数达到5次时,反应的收率降低较大,说明催化剂的稳定性相对较好,可以重复利用4次,累计参与反应时间96 h而催化效果降低不大。对原催化剂与分别经历1、3、5次反应的回收催化剂进行了电镜微观形貌表征,结果显示原催化剂中载体上的铂颗粒分布较均匀,随着反应次数的增加,分布与粒径都变得混乱。由此得出结论:铂颗粒参与反应过程会发生流失,在高温条件下,铂颗粒会熔融结合成更大的颗粒使催化剂有效接触面积减少,这是催化效率降低的主要原因。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 吡啶并异吲哚衍生物或苯并吡啶并异吲哚衍生物及其合成方法
- 一种能高效降解全氟化合物的吲哚衍生物的合成方法及其应用