一种癌症检测模型及其构建方法和试剂盒
文献发布时间:2023-06-19 13:49:36
技术领域
本发明涉及癌症检测技术领域,具体而言,涉及一种癌症检测模型及其构建方法和试剂盒。
背景技术
恶性肿瘤已经成为严重威胁世界人群健康的主要公共卫生问题之一,防控形势严峻。2020年全球最新癌症负担数据报告显示,全球新发癌症病例约1929万例,死亡病例996万例,癌症在全球112个国家是人类死亡原因第一或第二位主因。若保持2020年的增长水平,预测到2040年全球新发病例将达到2840万例,比2020年的1929万例增加47%,2040年新发病例将净增加410万例,这一预测由于人口的增长和老龄化因素,可能会进一步增加。随着经济的快速发展,许多已知的癌症风险因素流行率也随之增加,包括吸烟、不健康饮食、肥胖流行,以及缺乏运动等。因此,为了减轻癌症负担,医疗领域急需对癌症预防和治疗两个层面的干预措施。实际上,癌症发现得早和晚,在癌症治疗的方式选择、生活质量、经济花费和预后效果等方面差别巨大。所以,及早发现与治疗是目前治疗癌症最有效的手段。
现有临床常用的肿瘤筛查和检测手段主要包括影像学检查、血清学检查和病理学诊断等,而这几种筛查和检测手段均存在一定的技术局限性,例如:(1)常用的血清学标志物诊断灵敏度和特异性都不够理想,容易受到炎症影响,出现短暂的异常的信息,导致假阳性或假阴性;(2)影像学检查只能发现直径1cm以上的肿瘤病灶,发现时基本到了中晚期;(3)病理学诊断是癌症诊断的金标准,但需要进行穿刺活检,一般用于疑似癌症患者的确诊。而液体活检因不具备侵袭性,且可以在疾病治疗的不同阶段重复进行,使得目前肿瘤精准诊疗的焦点转向液体活检。而液态活检的分析物中,以cfDNA(circulating cell-freeDNA,cfDNA)的应用最为广泛。cfDNA是游离于血液循环系统中的来自细胞的DNA片段,主要来自于细胞凋亡进程中片段化的DNA、坏死细胞的DNA碎片、细胞分泌的外泌体。cfDNA中最重要的一类是循环肿瘤DNA(Circulating Tumor DNA,ctDNA),它是进入血液循环系统中的来自肿瘤基因组的DNA片段,因具有以下优点而能够成为肿瘤早筛的潜力标志物:(1)携带肿瘤信息,提供有关肿瘤大小的信息,还可提供对肿瘤基因组的综合描述,从而反映出疾病的发展状态;(2)深度测序可以发现肿瘤内异质性和只在部分细胞中出现的基因突变。目前ctDNA应用于肿瘤筛查和检测的主要研发方向包括ctDNA突变检测、表观遗传学方向(甲基化、羟甲基化)、及多组学检测等。
ctDNA突变检测。从目前的研究进展来看,ctDNA突变检测遇到的主要问题是ctDNA本身在血液中含量就比较低,并且还会被实时清理大约只有游离血浆DNA的0.1%到1%,其次如果使用超深度测序,其检测费用也很昂贵,因此ctDNA点突变多应用于中晚期癌症的伴随诊断和用药指导。目前关于ctDNA突变检测在领域内的一项典型研究为TEC-Seq,平均测序深度30000X,是一个超灵敏度的检测方法。作者首先使用该方法检测正常人的血浆样本,没有发现肿瘤相关突变。随后作者使用该方法检测了4种肿瘤(乳腺癌,结肠癌,肺癌,卵巢癌)的194个病人的血浆样本。ctDNA的含量在在晚期肿瘤中的浓度更高。晚期肿瘤(stageIII and IV)检出率为>75%,早期肿瘤检出率为62%。
ctDNA有别于cfDNA的其他物理化学特性。发表于Science TranslationalMedicine的“Enhanced detection of circulating tumor DNA by fragment sizeanalysis”的文章指出,ctDNA的片段长度要小于cfDNA的长度,并以此为依据通过片段特异富集进行液体活检,在多癌种中验证了可以比CT更早检出肿瘤。在本文章中,作者并未纳入早期肿瘤患者的队列,因为这篇文章主要讨论的应用方向并不是早期患者的筛查和检测,因此这个方法具体的应用情况也没有具体的定论。
综合上述情况来看,针对于肿瘤早筛早诊,目前的各个研究方向的灵敏度和特异性也是各有优劣,整体看来并没有一个特别理想的结果,能够达到人们的预期。
鉴于此,特提出本发明。
发明内容
本发明的目的在于提供一种癌症检测模型及其构建方法和试剂盒。
本发明是这样实现的:
第一方面,本发明实施例提供了一种癌症检测模型的构建方法,其包括:获取每个分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;将每个分类指标的测试数据作为输入数据构建单指标分类模型,获得对样本的单指标预测得分;采用逻辑回归模型对所有分类指标的单指标预测得分进行整合,获得对样本的逻辑回归评分;将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分作为输入数据,构建癌症检测整合模型。
第二方面,本发明实施例提供了检测分类指标和拷贝数差异的试剂在制备用于癌症检测的试剂盒中的应用,所述分类指标为前述实施例所述的构建方法构建的癌症检测模型中的分类指标。
第三方面,本发明实施例提供了一种通过全基因组测序识别癌症特征的癌症检测试剂盒,其包括:检测分类指标和拷贝数差异的试剂,所述分类指标为前述实施例所述的构建方法构建的癌症检测模型中的分类指标。
第四方面,本发明实施例提供了一种癌症检测模型构建装置,其包括:数据获取模块、预测得分获取模块、逻辑回归评分获取模块以及癌症检测模型构建模块;
其中,数据获取模块用于获取待测样本的分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
预测得分获取模块用于将每个分类指标的测试数据输入单指标分类模型,获得对样本的单指标预测得分,所述单指标分类模型为如前述实施例所述的构建方法构建的单指标分类模型;
逻辑回归评分获取模块用于将所有分类指标的单指标预测得分输入逻辑回归模块,获得对样本的逻辑回归评分;
癌症检测模型构建模块用于将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分作为输入数据,构建癌症检测模型。
第五方面,本发明实施例提供了一种测试数据的处理方法,其包括:获取样本的每个分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;将每个分类指标的测试数据对应输入如前述实施例所述的构建方法构建的单指标分类模型,获取样本的单指标预测得分;将所有分类指标的单指标预测得分输入逻辑回归模型,获取对样本的逻辑回归评分;将逻辑回归评分、拷贝数变异数据以及所有分类指标的单指标预测得分输入如前述实施例所述的构建方法构建的癌症检测模型中。
第六方面,本发明实施例提供了一种测试数据的处理装置,其包括:数据获取模块、第一执行模块、第二执行模块以及预测模块;
数据获取模块用于获取待测样本的分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
第一执行模块用于将所述分类指标的测试数据输入如前述实施例所述的构建方法构建的单指标分类模型中,获得对样本的单指标预测得分;
第二执行模块用于将所有分类指标的单指标预测得分输入逻辑回归模型中,获得对样本的逻辑回归评分;
预测模块用于将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分输入如前述实施例所述的构建方法构建的癌症检测模型中,获得样本的预测结果。
第七方面,本发明实施例提供了一种电子设备,其包括:处理器和存储器,所述存储器用于存储一个或多个程序,当所述程序被所述处理器执行时,使得所述处理器实现如前述实施例所述的癌症检测模型的构建方法,或者,如前述实施例所述的测试数据的处理方法。
第八方面,本发明实施例提供了一种计算机可读介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现如前述实施例所述的癌症检测模型的构建方法,或者,如前述实施例所述的测试数据的处理方法。
本发明具有以下有益效果:
本发明通过对血浆游离DNA的全基因组测序,挖掘出了多个维度的可应用于癌症检测的基因组特征(核小体分布特征,末端序列特征,片段大小分布),通过对这3个特征的指标分别进行分类模型的构建,得到每个指标对于样本的预测得分,然后使用逻辑回归(logistic regression)模型,对这些得分进行整合并加入拷贝数变异特征信息,得到最终分类预测模型。
本发明构建的癌症检测模型能显著提高癌症检测的效率和准确性,且分析所需数据量较少,仅需要满足平均测序深度为全基因组的0.25×,检测的成本和/或效果超过现有技术,能够对不同癌症进行检测,适用于分析和预测各个时期的肿瘤,尤其是针对癌症的早期检测。
本发明提供的试剂盒能完成检测模型所需指标的检测,使之在ctDNA检测中能够创新地应用于癌症领域。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
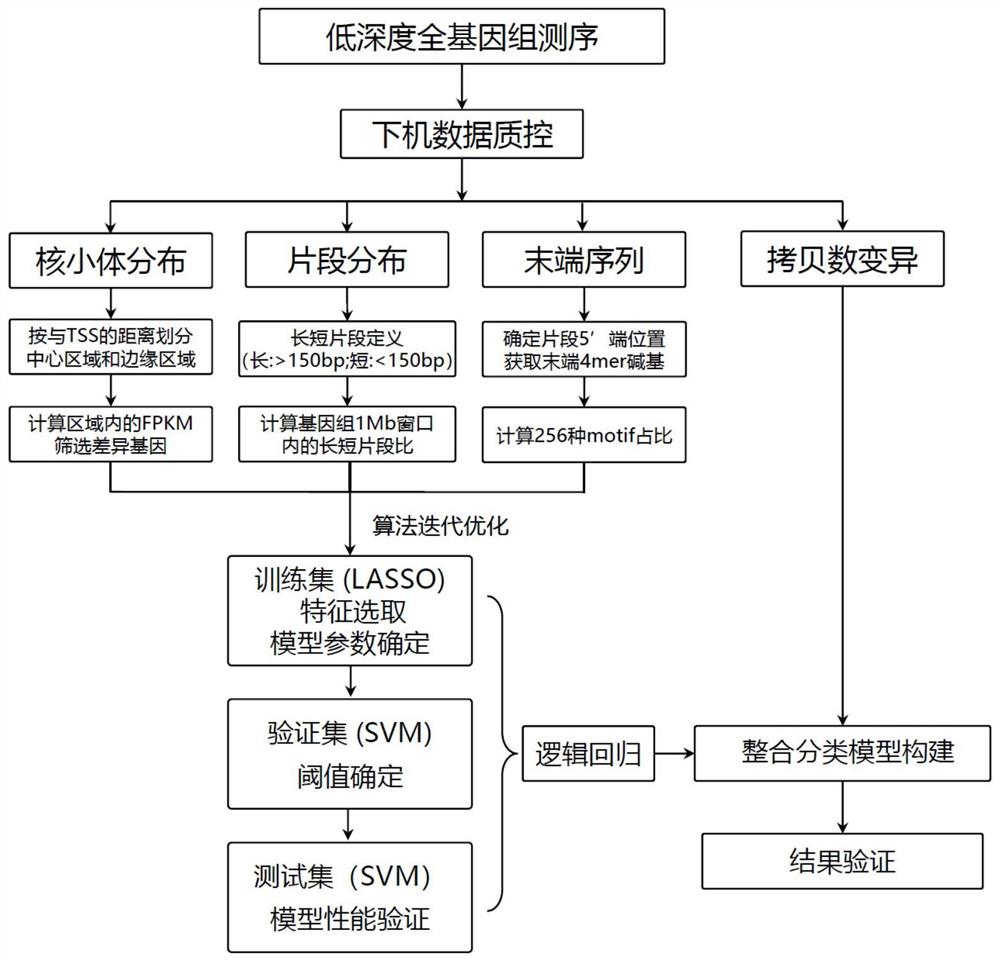
图1为实施例1中癌症检测模型的构建流程图;
图2为实施例1中不同组对应的核小体的分布图;
图3为实施例1中不同样本之间,不同种类motif的占比;
图4为实施例1中对肿瘤患者和健康人群的血浆低深度WGS分析结果;
图5为实施例1中不同样本之间片段大小分布特征的检测数据;
图6为实施例2中使用整合模型与肝癌相关的临床指标(AFP)的结果统计AUC绘制柱状图对比肝癌的分类结果;
图7为实施例2中采用整合模型与通过数据挖掘得到的单个分类指标构建的模型的结果计算AUC值;
图8为实施例3中整合分数对于处在不同的BCLC分期的肿瘤病人的预测得分;
图9为本发明实施例4中整合模型与直接使用挖掘到的4个分类指标组合到一起后使用SVM进行模型构建的结果进行比较;
图10为实施例5中整合模型对胰腺癌样本的预测结果。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
现有研究中大多只研究单个指标用于早期肿瘤检测,很少从多个维度进行联合研究,且在多种肿瘤检测指标已知的情况下,现有技术所能达到检测结果仍然存在灵敏度低、特异性不高和/或检测成本昂贵等问题。
本发明数据分析方面从多个维度对数据进行分析,对原始特征数据进行降维和特征提取,定义合理的模型对应参数,使用网格搜索的方法选取最优的参数组合,通过迭代训练得到优良的分类模型(整合模型),并定义出了整合的评分规则,最后使用验证集和测试集验证模型效果。
相对于现有技术而言,具有灵敏度高、特异性好的优点,且检测成本低,适于推广。
具体地,本发明实施例提供的一种癌症检测模型的构建方法包括以下步骤:
获取每个分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
将每个分类指标的测试数据作为输入数据构建单指标分类模型,获得对样本的单指标预测得分;
采用逻辑回归模型对所有分类指标的单指标预测得分进行整合,获得对样本的逻辑回归评分;
将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分作为输入数据,构建癌症检测整合模型。
核小体分布特征(NF):通过从血浆中分离的cfDNA的全基因组测序,可以发现cfDNA中核小体的分布与细胞核结构、基因结构和在细胞中观察到的基因表达密切相关。在转录活跃的基因(House Keeping)的启动子区域,其染色体结构较为松散,核小体分布较为稀疏,而在沉默基因(Silent)则不存在如此现象,染色体结构较为紧密,核小体分布较为均匀密集。因此,可根据核小体分布的差别推测基因表达,因为在组织细胞DNA降解进入血液的过程中,未受核小体保护的DNA更易被降解而更不易被检测。因此,在对cfDNA测序时,活跃基因的启动子中心区域较其周围区域检测到DNA分子会更少,进而导致该基因启动子中心区域测序深度会降低,因此,可依此原理对核小体分布差异进行量化。同样的肿瘤细胞也应具有特有的核小体分布特征,从而可以作为肿瘤检测的指标。
根据启动子的区域,定义中心区域和边缘区域,中心区域为转录起始位点(TSS)前120~170bp至转录起始位点(TSS)后30~70bp;边缘区域为中心区域两侧边缘各向外延伸1800~2200bp。
对每个区域统计检测到的DNA片段数,用于计算所述核小体分布特征的测试数据。优选地,所述核小体分布特征的测试数据为核小体分布差异值,所述核小体分布差异值=边缘区域片段数/片段总数(百万)×边缘区域长度(Kb)-中心区域片段数/片段总数×中心区域长度(Kb)。以此分值表示核小体的分布以及基因的转录活跃程度,且用于模型构建的输入特征。
末端序列特征(Motif):cfDNA的断裂并非是随机事件,尤其对于来源于肿瘤细胞释放的ctDNA,具有偏好性的断裂位点,即会形成特异性的断裂末端。深度测序可以找到这些肿瘤特异的断裂末端,但对于低深度测序数据来说,一般很难看到。本发明发现,统计片段末端的碱基组合类型占比能更有效的富集这一差异。找到与健康人群存在显著差异的末端序列种类,使用这些占比存在差异的motif作为模型构建的输入特征。
优选地,所述末端序列特征的测试数据为差异末端序列占比,所述差异末端序列占比=(与健康样本的cfDNA片段末端的碱基排列相比具有显著差异的碱基排列的类型/末端碱基排列类型的总和)。
“具有显著差异的碱基排列的类型”具体可以通过以下方式确认:基于健康样本和待测样本中末端碱基排列的分布情况,统计每一种末端碱基排列的数量占比,当一种末端碱基排列的数量占比在健康样本和待测样本之间具有显著差异时,则将该末端碱基排列标记为具有显著差异的末端碱基排列。
本文中“具有显著差异”可以指P<0.05,或P<0.01的情况,即差异具有统计学意义。
优选地,所述末端碱基排列是指cfDNA片段末端最后3~6个碱基的排列,具体可以为3、4、5或6个碱基的排列。
更优选为4个,根据末端4个碱基的分布差异更能有效地体现肿瘤样本和健康样本之间的差异,从而更准确有效地实现肿瘤样本的检测。当末端碱基排列为末端4个碱基的排列(末端4mer碱基)时,末端碱基排列的总和为4×4×4×4=256。其他碱基数以此类推,不再赘述。
片段大小分布特征(Fragmentation):肿瘤患者cfDNA的片段长度比健康人的cfDNA的片段长度要短,与非肿瘤个体相比,肿瘤患者cfDNA短片段与长片段比例不稳定,在基因组不同区域也存在差异。
优选地,所述片段大小分布特征的测试数据为片段差异分布占比,片段差异分布占比=(片段大小分布差异区域的数量/划分区域的总数),其中,片段大小分布差异区域是指与健康样本相比,短片段和长片段的比例具有显著差异的划分区域,所述划分区域是指将样本基因组按特定长度划分所获得的区域,长片段的长度>150bp,短片段的长度小于150bp。
优选地,所述特定长度为0.5~3M,更优选为1M。
本文中的“预测得分”可以选自分类模型输出的样本患有癌症的概率、癌症的分类结果。
本发明提供的癌症检测模型对于癌症的种类没有特殊限定,在构建方法不变的情况下,不同的癌症可以采用对应癌症类型的样本对模型进行构建,参数可能会对应发生微小变化,以更特异性地针对不同癌症的样本进行预测和分析。
癌症的类型可以选自已知的所有癌症,根据肿瘤的发病部位,癌症可分为喉癌、脑癌、食管癌、胃癌、肺癌、乳腺癌、肝癌和直肠癌等,根据癌症的癌细胞的组织来源分为以下几类:腺癌、鳞状细胞癌,大细胞癌,小细胞癌,类癌等。根据癌症的癌细胞的分化程度分为:高分化癌,中分化癌,低分化癌和未分化癌等。
优选地,所述构建方法采用支持向量机(SVM)、随机森林,多层感知机,弹性网络,决策树和深度神经网络中的任意一种分类算法对每个分类指标进行分类模型的构建,优选为SVM。
优选地,本申请还包括使用特征降维的方法对每个分类指标的原始数据进行降维和特征提取,定义合理的分类模型对应参数,使用网络搜索的方法选取最优的参数组合,通过迭代训练得到优良的分类模型。
可选地,所述特征降维的方法选自:LASSO、PCA(主成分分析)、弹性网络、随机森林和递归消除中的任意一种,优选为LASSO。
本发明实施例提供的构建方法中,分类指标的测试数据是基于样本的测序数据获得,测序可以是高深度测序、中深度测序或低深度测序,优选为低深度测序,优选为0.25~5×测序,优选为0.25×。
逻辑回归模型把单指标整合后的模型的ROC曲线绘制在同一坐标中,以直观地鉴别优劣,靠近左上角的ROC曲线所代表的模型的预测最准确。并且使用AUC对模型的性能进行量化,通过直接比较AUC的大小也可以对模型性能进行比较。
优选地,所述逻辑回归模型的公式见表1。
优选地,所述构建方法包括使用验证集和测试集的数据对分类模型进行性能的评估和优化。
优化时,将单个分类指标对应的预测得分与各个输入特征对应的权重相乘,计算特征项乘以权重并与截距项加和,训练时会根据误差的大小来进一步调整权重的值,直到迭代次数到达指定次数或者误差小于一定的阈值就会停止迭代。
优选地,所述构建方法还包括将加和的结果使用sigmoid函数映射到[0,1],即将结果转化为百分比。
拷贝数变异(CNV):健康人基因组为二倍体,肿瘤组织基因组在复制过程会产生非正常的拷贝,因此肿瘤组织基因组在某些染色体或某些区域会产生拷贝数的扩增或缺失即发生拷贝数变异,肿瘤组织中的CNV信号在血浆中可检测,信号在低至平均1×基因组覆盖深度下仍可稳定检出。
优选地,拷贝数变异的数据可由以下方式获得:将样本基因组按一定长度(1~5K,优选为2K)划分为若干区域,统计每个小区域的平均测序深度,并进行GC校正;以健康样本数据构建阴性池,以计算获得的健康样本在每个小区域的经过GC矫正的平均测序深度作为基线;将肿瘤样本与阴性池比较,找到存在拷贝数差别超过特定阈值的小区域,并进行连接形成最终的存在拷贝数变异的区域;根据拷贝数变异类型(扩增和缺失)以及所在区域涉及到的原癌基因与抑癌基因,计算CNV score,具体公式见表1。
本发明将拷贝数变异的测试数据(CNV score)作为定性指标,通过上述设定的规则将三个分类指标以及使用逻辑回归整合过后的模型结果,进行进一步整合,构成的癌症检测整合模型,能够更加精准地对癌症进行检测,对提高轻症癌症患者的治愈率以及重症患者的存活时间有非常积极的作用。
优选地,所述构建方法包括将所有分类指标对应的单指标预测得分整合后,获得单指标评分,然后与逻辑回归评分和拷贝数变异数据一同作为输入数据用于癌症检测模型的构建。即,癌症检测模型评分包括3个部分:单指标评分、逻辑回归评分和拷贝数变异评分。
只要是通过机器学习方法,基于单指标评分、逻辑回归评分和拷贝数变异评分,实现对样本预测结果的输出的技术方案均属于本申请的保护范围内。
优选地,癌症检测模型的整合的评分规则如下:拷贝数变异特征评分为检测到拷贝数变化超过特定阈值(0.03~0.15)的区域计为1分;逻辑回归评分为使用逻辑回归算法整合核小体分布特征、末端序列特征和片段大小分布特征,得到的分值大于其阈值计为1分;单指标评分为各单项指标分值的加和,其中各单项指标分值为各指标分类模型获得的单指标预测得分大于其各自阈值计0.5分。在本申请设定的评分规则能更有效地提高检测的灵敏度和特异性。
表1计算公式
其中,logistic Score为逻辑回归的评分,B为截距项,x1为NF权重,x2为Motif权重,x3为Fragment权重,Z为各特征项乘以权重的加和后与截距项的总和;Single Score为单指标评分,cutoff为对应的阈值;整合评分为对样本最终的预测得分;TSG为抑癌基因;OG为原癌基因;ESS为常规功能基因,i,j为人类基因组染色体臂和基因。
需要说明的是,分类模型、逻辑回归模型以及整合分类模型的计算公式中的对应参数并非仅限制于上述参数的设定,上述对应参数属于优选方案。在其他实施例中,也可以基于验证集和测试集的数据对参数进行调整。
只要是基于上述技术构思实现对癌症检测模型的构建或对测序数据的处理,即属于本发明的保护范围内。
本发明实施例还提供了检测分类指标和拷贝数差异的试剂在制备用于癌症检测的试剂盒中的应用,所述分类指标为前述任意实施例所述的构建方法构建的癌症检测模型中的分类指标。
本发明对试剂的具体类型不作具体限定,能够用于获取样本分类指标和拷贝数差异的测试数据即可。
本发明实施例还提供了一种癌症检测的试剂盒,其包括:检测分类指标和拷贝数差异的试剂,所述分类指标为前述任意实施例所述的构建方法构建的癌症检测模型中的分类指标。
本发明实施例还提供了一种癌症检测模型构建装置,其包括:数据获取模块、预测得分获取模块、逻辑回归评分获取模块以及癌症检测模型构建模块。
其中,数据获取模块用于获取待测样本的分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
预测得分获取模块用于将每个分类指标的测试数据输入分类模型,获得对样本的单指标预测得分,所述分类模块为如前述任意实施例所述的构建方法构建的分类模型;
逻辑回归评分获取模块用于将所有分类指标的单指标预测得分输入逻辑回归模块,获得对样本的逻辑回归评分;
癌症检测模型构建模块用于将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分作为输入数据,构建癌症检测模型。
本发明实施例还提供了一种测试数据的处理方法,其包括:
获取样本的每个分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
将每个分类指标的测试数据对应输入如前述任意实施例所述的构建方法构建的分类模型,获取样本的预测得分;
将所有分类指标的预测得分输入逻辑回归模型,获取对样本的逻辑回归评分;
将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分输入如前述任意实施例所述的构建方法构建的癌症检测模型中。
所述数据的处理方法不以疾病的诊断或治疗为直接目的,而是机械性地根据测试的数据进行处理。
本发明实施例还提供了一种测试数据的处理装置,其包括:数据获取模块、第一执行模块、第二执行模块以及预测模块。
其中,数据获取模块,用于获取待测样本的分类指标和拷贝数变异的测试数据,所述分类指标包括核小体分布特征、末端序列特征和片段大小分布特征;
第一执行模块,用于将所述分类指标的测试数据输入如前述任意实施例所述的构建方法构建的分类模型中,获得对样本的单指标预测得分;
第二执行模块,用于将所有分类指标的单指标预测得分输入逻辑回归模型中,获得对样本的逻辑回归评分;
预测模块,用于将逻辑回归评分、拷贝数变异的数据以及所有分类指标的单指标预测得分输入如前述任意实施例所述的构建方法构建的癌症检测模型中,获得样本的预测结果。
本发明实施例还提供了一种电子设备,其包括:处理器和存储器,所述存储器用于存储一个或多个程序,当所述程序被所述处理器执行时,使得所述处理器实现如前述任意实施例所述的癌症检测模型的构建方法,或者,如前述任意实施例所述的测试数据的处理方法。
该电子设备可以包括存储器、处理器、总线和通信接口,该存储器、处理器和通信接口相互之间直接或间接地电性连接,以实现数据的传输或交互。例如,这些元件相互之间可通过一条或多条总线或信号线实现电性连接。处理器可以处理与目标识别有关的信息和/或数据,以执行本申请中描述的一个或多个功能。例如,处理器可以获取待识别图像,并根据上述数据进行目标识别,进而实现本申请提供的目标识别方法。
存储器可以是但不限于,随机存取存储器(Random Access Memory,RAM),只读存储器(Read Only Memory,ROM),可编程只读存储器(Programmable Read-Only Memory,PROM),可擦除只读存储器(Erasable Programmable Read-Only Memory,EPROM),电可擦除只读存储器(Electric Erasable Programmable Read-Only Memory,EEPROM)等。
处理器可以是一种集成电路芯片,具有信号处理能力。该处理器可以是通用处理器,包括中央处理器(Central Processing Unit,CPU)、网络处理器(Network Processor,NP)等;还可以是数字信号处理器(Digital Signal Processing,DSP)、专用集成电路(Application Specific Integrated Circuit,ASIC)、现场可编程门阵列(Field-Programmable Gate Array,FPGA)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件。
在实际应用中,该电子设备可以是服务器、云平台、手机、平板电脑、笔记本电脑、超级移动个人计算机(ultra-mobile personal computer,UMPC)、手持计算机、上网本、个人数字助理(personal digital assistant,PDA)、可穿戴电子设备、虚拟现实设备等设备,因此本申请实施例对电子设备的种类不做限制。
本发明实施例还提供了一种计算机可读介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现如前述任意实施例所述的癌症检测模型的构建方法,或者,如前述任意实施例所述的测试数据的处理方法。
计算机可读介质包括:U盘、移动硬盘、只读存储器、随机存取存储器、磁碟或者光盘等各种可以存储程序代码的介质。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
参照附图1,本实施例提供了一种癌症检测模型的构建方法,其包括以下步骤。
1、cfDNA的提取及低深度WGS测序。
血浆分离:外周血采集于无创抗凝管中,记录并核对样本信息、样本状态。采血管放于离心机离心,吸取上清液再次离心,再次吸取上清液获得血浆样本。
血液中cell-free DNA(cfDNA)的提取:用血浆游离DNA提取试剂盒提取血浆中的cell-free DNA。待血浆完全融化后高速离心,转移离心后的血浆,分别加入裂解液、磁珠等试剂,震荡混匀后反应一段时间,然后使用清洗液进行清洗,使用洗脱液回收DNA。
DNA质量检测:DNA浓度用Qubit核酸/蛋白质定量荧光计或酶标仪检测。
WGS文库构建和上机测序:使用全基因组测序文库构建试剂进行文库构建,主要包含末端修复(末端修复缓冲液、末端修复酶)和接头连接(接头连接缓冲液、接头连接酶、Index接头)两个步骤。
末端修复缓冲液、末端修复酶、连接缓冲液、连接酶以及Index接头于-20±5℃保存,使用前取出至2-8℃解冻,并充分混匀。磁珠需提前从4℃冰箱取出,室温平衡30min。
末端修复缓冲液及末端修复酶预混液与cfDNA混合,37℃下发生反应;接头连接缓冲液及连接酶预混液与上一步末端修复产物及接头Index接头混合,20℃下发生反应。接头连接后使用纯化磁珠进行纯化。
使用qPCR的方法对文库进行定量分析,查看溶解曲线等各项参数是否达到要求,并计算文库浓度。文库定量后上机测序。
2、整合的分类指标来自于上述实验方法得到的低深度全基因测序数据。
获取样本数据,本实施例使用数据为PreCar队列项目所招募的481名诊断为肝癌(HCC)的患者,患有肝硬化(LC)的患者2247例,健康对照(NC)476例的低深度全基因组二代测序(WGS);之后将测序数据进行质控,比对,进行分类指标的数据挖掘。
3、测序数据进行质控,比对。
使用Fastp软件对测序下机数据进行数据过滤,包括减去测序接头序列,去除测序读长小于20的DNA片段,去除测序质量较低的DNA片段;使用Bowtie将过滤后的数据与Hg19参考基因组进行比对,得到每个DNA片段基因组上对应的具体位置信息。
4、根据比对后的结果文件,获取核小体分布特征NF、末端序列特征Motif和片段大小分布特征Fragment的数据。
核小体分布特征的测试数据为核小体分布差异值,所述核小体分布差异值=边缘区域片段数/片段总数(百万)×边缘区域长度(Kb)-中心区域片段数/片段总数×中心区域长度(Kb);其中,中心区域为转录起始位点(TSS)前150bp~转录起始位点(TSS)后50bp;边缘区域为中心区域两侧边缘各向外延伸2000bp;
所述末端序列特征的测试数据为差异末端序列占比,所述差异末端序列占比=(与健康样本的cfDNA片段末端最后4位碱基排列相比具有显著差异的碱基排列的类型/256);
所述片段大小分布特征的测试数据为片段差异分布占比,片段差异分布占比=(片段大小分布差异区域的数量/划分区域的总数),其中,片段大小分布差异区域是指与健康样本相比,短片段和长片段的比例具有显著差异的划分区域,所述划分区域是指将样本基因组按特定长度划分所获得的区域。
如图2-5所示,核小体的分布在不同组之间存在差异(图2);不同种类motif的占比在不同组样本间也会有明显区别(图3);对肿瘤患者血浆低深度WGS分析发现,与健康人(cfDNA WGS)相比基因组的某些区段拷贝数存在明显变化(图4);血浆中ctDNA的片段长度会比正常的cfDNA片段长度要短,定义出肿瘤特征性片段分布区域(图5)。上述这些特征用作分类模型构建的指标。
5、把招募到的样本随机分成训练集,验证集和测试集,在训练的过程中对上述提到的3个分类指标的原始数据分别使用LASSO(least absolute shrinkage and selectionoperator)回归算法,减少数据的维度并且提取出相对重要的特征。对于提取以后的特征,分别使用SVM通过交叉验证的方法来构建分类模型,以获得单指标的预测得分。
6、将三个分类模型对样本所有分类指标的单指标预测得分作为输入特征,使用逻辑回归模型对多个指标的预测得分进行整合。
使用验证集和测试集的数据对最终得到的模型进行性能的评估。单指标的分类模型的预测得分为输入项,各个输入特征对应相应权重,各特征项乘以权重与截距项的加和为最终输出,训练时会根据误差的大小来进一步调整权重的值,直到迭代次数到达指定次数或者误差小于一定的阈值就会停止迭代,最终把加和的结果使用sigmoid函数映射到[0,1],对应核小体分布权重x1为2.52,末端序列权重x2为1.96,片段大小分布权重x3为2.37,截距项B为-4.55。
逻辑回归模型的计算公式参照表1。
7、获取拷贝数变异的数据,计算公式参照表1。将所有分类指标的单指标预测得分整合后获得单指标评分;
将单指标评分、逻辑回归评分和拷贝数变异CNV数据作为输入数据,设定评分规则,构建癌症检测模型(整合模型)。
评分规则的计算公式参照表1,最终的整合评分包含三个部分:拷贝数变异特征评分,逻辑回归评分,单指标评分。其中拷贝数变异特征评分为检测到拷贝数变化超过0.03的区域计为1分;逻辑回归评分为使用逻辑回归算法整合核小体分布特征NF、末端序列特征Motif和片段大小分布特征Fragment,得到的分值大于其阈值计为1分;单指标评分为各单项指标分值的加和,其中各单项指标分值为各指标分类模型获得的单指标预测得分大于其各自阈值计0.5分。
实施例2
采用实施例1的构建方法进行癌症检测模型(整合模型)的构建,使用癌症检测模型(整合模型)与肝癌相关的临床指标(AFP)的结果计算AUC值对两者的分类结果进行比较(图6)。
如图所示,传统的AFP检测灵敏度和特异性为53.77%和90.81%,AUC低于0.85,而本发明验证集中灵敏度和特异性为95.79%和97.00%,AUC为0.996,测试集中灵敏度和特异性为95.52%和97.91%,AUC为0.996(最终以测试集效果为准),这大大提升检测准确性。
采用实施例1提供的癌症检测模型(整合模型)与通过数据挖掘得到的单个分类指标构建的模型的结果计算AUC值,对两种类型的结果进行比较,结果见图7和表2)。
表2检测结果
根据模型的AUC可以判断出整合分数的AUC明显大于其他分类指标的AUC。从结果中可以看出,整合后的模型的灵敏度和特异性也都优于单个指标的构建的模型的性能,大大提高检测的准确性。
实施例3
对于处在不同的BCLC分期的肿瘤病人,整合模型的预测分值基本没有差距,因此,可得出适用于肿瘤的各个时期(图8)。通过这个结果说明,本发明使用的整合模型评分方法明显优于目前的临床指标。
实施例4
本发明使用整合后的分类模型(整合模型)与直接使用挖掘到的4个分类指标(核小体分布特征、末端序列特征、片段大小分布特征以及拷贝数变异)组合到一起后使用SVM进行模型构建的结果进行比较(图9,灵敏度为0.841,特异性为0.858),从结果中可以看出,整合模型整体效果明显优于直接使用四个指标组合然后使用SVM进行模型构建的结果,进一步突出本发明的技术方法对分类结果明显的提升作用。
需要说明的是,实施例2-5使用样本信息均为:验证集包含肝癌95例,非肝癌200例(肝硬化100例,健康人100例);测试集包含肝癌131例,非肝癌1916例(肝硬化1800例,健康人116例)。
实施例5
本实施例提供给了一种癌症检测模型的构建方法,大致与实施例1相同,区别在于,采用胰腺癌样本对模型进行构建,获得整合模型与实施例1中模型的差异在于参数,模型中逻辑回归公式各单指标对应的权重发生变化,具体对应为核小体分布权重1.458,片段大小分布权重1.1052,末端序列权重为2.4305,逻辑回归公式中截距项数值变为-2.2985。
将获得的整合模型,对胰腺癌样本进行分类,样本包括胰腺癌样本124例,健康人200例,结果如图10和表3所示。随着样本数目的增加,后续提升效果会更加明显。
表3检测结果
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种癌症检测模型及其构建方法和试剂盒
- 一种用于检测热原的细胞模型的构建方法和细胞模型及热原检测试剂盒