一种吡啶并1,2-a吲哚类化合物的制备方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于光催化合成技术领域,具体涉及吡啶并[1,2,a]吲哚类化合物的合成方法。
背景技术
含氮杂环骨架广泛存在于具有生物活性的天然产物、药物分子中,其中,吡啶并[1,2-a]吲哚结构合成方法的探索具有重要的研究价值。目前关于吡啶并[1,2-a]吲哚衍生物的合成方法大多需要以吲哚、吲哚嗪等稠环化合物作为起始原料。吡啶并[1,2-a]吲哚类化合物因其表现出来优异的药理活性而受到了广泛关注,如何构筑吡啶并[1,2-a]吲哚类化合物是有机合成化学领域的热点研究内容之一。目前,通过三氟甲基可以成功合成吡啶并[1,2-a]吲哚类化合物。但是引入三氟甲基对于分子性质有较大改变,如增加代谢稳定性、极性、溶解性等。另外,光催化三氟甲基化反应往往需要额外添加光催化剂或者过渡金属催化剂,导致生产成本的上升、金属离子残留以及分离困难等不利结果。
发明内容
本发明是要解决现有合成吡啶[1,2,a]并吲哚类化合物的方法,需要引入光催化剂或金属催化剂,导致底物复杂,分离困难的技术问题,而提供一种吡啶并[1,2-a]吲哚类化合物的制备方法。
一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将N-取代吲哚类化合物和Umemoto试剂加入到盛有溶剂的石英反应管中;
二、在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;
三、将步骤二反应获得的混合液进行旋蒸处理,得到粗产品;
四、将步骤三获得的粗产品进行柱层析分离处理,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
进一步的,步骤一中所述N-取代吲哚类化合物的结构式如下:
式中,R
或者R
或者R
或者R
进一步的,步骤一所述Umemoto试剂为三氟甲基试剂,结构式如下:
进一步的,步骤一所述溶剂为二氯甲烷。
其中N-取代吲哚类化合物的制备反应式如下:
其中R包括R
R
或者R
或者R
或者R
本发明反应式如下:
本发明无需光催化剂和金属催化剂,直接光照下便可发生反应。操作简单,底物适用性好。
本发明以N-取代吲哚1a(R
N-取代吲哚1a在光照下发生聚合,成为低聚物poly-1a,可以吸光引发反应。三氟甲基试剂2a中S-CF
本发明有益效果:
本发明首次合成了此类化合物,使用自由基串联环化策略一步反应成两个键构建了一个新的六元环。本发明采用简单、高效、绿色的光催化合成策略,在无外加光催化剂和氧化剂的条件下实现了吡啶并[1,2-a]吲哚类化合物合成。反应操作简单、条件温和,官能团兼容性较好。与现有技术相比具有以下优点:
(1)、无光催化剂、金属催化剂发生自由基串联环化反应构建带三氟甲基的吲哚并环产物,降低成本、避免可能的金属残留、简化方法
(2)、反应底物均是廉价易于合成的,底物范围广,所得产物易于分离,反应高效。
本发明方法用于合成吡啶并[1,2-a]吲哚类化合物。
附图说明
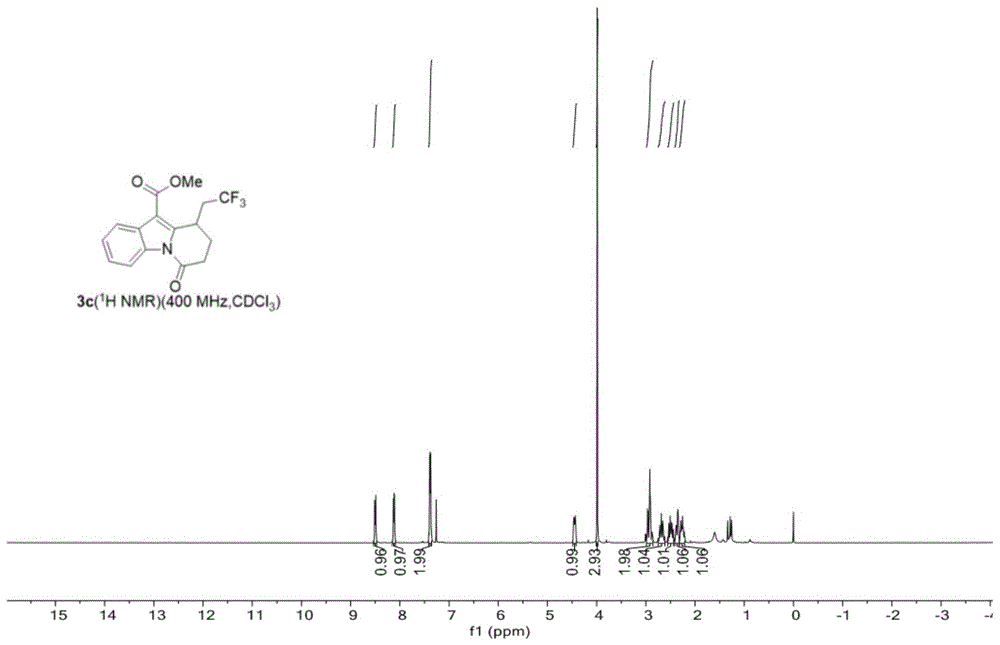
图1为实施例三制备的吡啶并[1,2-a]吲哚类化合物3c谱图;
图2为实施例三制备的吡啶并[1,2-a]吲哚类化合物3c碳谱图;
图3为实施例三制备的吡啶并[1,2-a]吲哚类化合物3c氟谱图;
图4为实施例六制备的吡啶并[1,2-a]吲哚类化合物3f氢谱图;
图5为实施例六制备的吡啶并[1,2-a]吲哚类化合物3f碳谱图;
图6为实施例六制备的吡啶并[1,2-a]吲哚类化合物3f氟谱图。
具体实施方式
具体实施方式一:本实施方式一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将N-取代吲哚类化合物和Umemoto试剂加入到盛有溶剂的石英反应管中;
二、在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;
三、将步骤二反应获得的混合液进行旋蒸处理,得到粗产品;
四、将步骤三获得的粗产品进行柱层析分离处理,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
具体实施方式二:本实施方式与具体实施方式一不同的是:步骤一中所述N-取代吲哚类化合物的结构式如下:
式中,R
或者R
或者R
或者R
具体实施方式三:本实施方式与具体实施方式一或二不同的是:步骤一所述Umemoto试剂为三氟甲基试剂,结构式如下:
其它与具体实施方式一或二相同。
具体实施方式四:本实施方式与具体实施方式一至三之一不同的是:步骤一所述溶剂为二氯甲烷。其它与具体实施方式一至三之一相同。
具体实施方式五:本实施方式与具体实施方式一至四之一不同的是:步骤一中N-取代吲哚类化合物与溶剂的用量比为0.28~0.32mmol:1~1.5mL;
Umemoto试剂与溶剂的用量比为0.08~0.12mmol:1~1.5mL。其它与具体实施方式一至四之一相同。
具体实施方式六:本实施方式与具体实施方式一至五之一不同的是:步骤一中N-取代吲哚类化合物与溶剂的用量比为0.3mmol:1.5mL;
Umemoto试剂与溶剂的用量比为0.1mmol:1.5mL。其它与具体实施方式一至五之一相同。
具体实施方式七:本实施方式与具体实施方式一至六之一不同的是:步骤二中光照反应时,蓝光灯的波长为440~460nm,照射时间为12~14h。其它与具体实施方式一至六之一相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同的是:步骤三采用旋转蒸发仪进行旋蒸处理。其它与具体实施方式一至七之一相同。
具体实施方式九:本实施方式与具体实施方式一至八之一不同的是:步骤四所述柱层析分离处理,采用硅胶和流动相,其中流动相为石油醚和乙酸乙酯的混合。其它与具体实施方式一至八之一相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同的是:石油醚和乙酸乙酯按体积比为20:1的混合。其它与具体实施方式一至九之一相同。
本发明内容不仅限于上述各实施方式的内容,其中一个或几个具体实施方式的组合同样也可以实现发明的目的。
实施例一:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率72%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3a核磁数据:
实施例二:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率70%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3b核磁数据:
实施例三:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率51%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3c核磁数据:
实施例四:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率42%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3d核磁数据:
实施例五:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率79%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3e核磁数据:
实施例六:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率66%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3f核磁数据:
实施例七:
本实施例一种吡啶并[1,2-a]吲哚类化合物的制备方法,具体按以下步骤进行制备:
一、将0.3mmol N-取代吲哚类化合物和0.1mmol Umemoto试剂加入到盛有1.5mL溶剂无水DCM(二氯甲烷)的石英反应管中;
所述N-取代吲哚类化合物的结构式如下:
式中,R
二、除气后,在氮气氛围、室温条件下,将步骤一中的反应管放在蓝光灯下,进行光照反应;蓝光灯的波长为450nm,照射时间为12h;
三、将步骤二反应获得的混合液进行采用旋转蒸发仪旋蒸处理,得到粗产品;
四、点板确定过柱展开剂极性,将步骤三获得的粗产品进行柱层析分离处理,其中流动相为石油醚和乙酸乙酯按体积比为20:1的混合,得到所述一种吡啶并[1,2-a]吲哚类化合物,完成制备。
本反应中产生副产物二苯硫醚极性最小,剩余底物次之、三氟甲基化产物极性相比于原料增大。反应产率30%,产物为白色固体。
本实施例获得的吡啶并[1,2-a]吲哚类化合物3g核磁数据:
- 一种稠环1,2-a吲哚类化合物和2,3-二取代吲哚类化合物的合成方法
- 含有二氮杂螺环片段的咪唑并1,2-a吡啶-3-酰胺类化合物及其制备方法和应用
- 一种吲哚类化合物的制备方法
- 一种手性异吲哚啉酮类化合物及其制备方法
- 一种锰催化合成吡啶并1,2-a吲哚-6(1H)-酮类化合物的方法
- 一种锰催化合成吡啶并1,2-a吲哚-6(1H)-酮类化合物的方法