一种检测人类PKD1基因变异的探针和引物组合物及其应用
文献发布时间:2024-04-18 19:52:40
技术领域
本发明涉及基因检测技术领域,尤其涉及一种检测人类PKD1基因变异的探针和引物组合物及其应用。
背景技术
常染色体显性遗传性多囊肾(Autosomal Dominant Polycystic KidneyDisease,ADPKD)是一种常见的迟发性系统疾病,其发病率为1/1000-1/400。随着ADPKD的发展,到60岁时,多达50%的患者会发展为终末期肾病(End-Stage Kidney Disease,ESKD)。因此,ADPKD是ESKD最常见的原因之一。在已报道的ADPKD的病例中,发现致病突变分别为PKD1(78%)、PKD2(15%)、GANAB(0.3%)和NAJB11(0.1%),此外,还有约7%为未知的遗传因子。ADPKD的诊断主要通过肾的影像学来确定,但是该病在新生儿中通常没有临床症状,因此得到诊断的患者估计不到1/2。
PKD1基因位于16号染色体(16p13.3),包含约52000bp。其中14148bp的46个外显子编码4304个氨基酸。由于16号染色体的重排现象,导致16p在16p13.1产生了与PKD1基因高度同源的区域。Bogdanova等人的研究表明,这些高度同源的区域,均为PKD1的假基因。最近的研究表明,虽然PKD1基因将近三分之四的区域(1-33号外显子)存在基因序列相似程度高达97.7%的6条假基因,但是在不同的外显子区域,他们的假基因的占比从14%~33%不等。PKD1基因结构的复杂性,除了具有高度同源的假基因以外,1号外显子的GC含量也高达85%。
由于假基因与高GC区域的存在,常规的检测方法对于PKD1基因的基因检测存在诸多的障碍。目前,主流的分析方法,是使用长距离聚合酶链式反应(Long-Range PCR,LR-PCR)结合下一代测序(Next Generation Sequencing,NGS)的方法,对PKD1基因进行检测。NGS技术具有高通量、快速、和成本低的优点,可以实现对多个样本、多个基因同时检测。但是,NGS技术在对高GC的区域不能很好的得到有效的数据。此外,由于NGS的读序较短,很难区分PKD1基因和其假基因。与此同时,Sanger测序是确定致病基因突变的金标准,仅使用NGS技术,无法提供准确的测序结果,无法完全满足临床的实际需求。
人口增速变缓已经成为现状,随着三胎政策的落实,优生优育的国策得到进一步加强,男女高龄等因素导致胎儿新发突变比例升高现象突显,辅助生殖、产前诊断技术成为重要的解决生育困难和优生优育的有力手段,占所有生育解决方案的50%以上,辅助生殖技术中涉及到对胚胎的筛选等环节,需要利用单个或者少量细胞及其扩增产物进行致病位点的鉴定,目前使用的单细胞全基因组扩增技术会造成等位基因脱扣现象,即,在基因组中呈现杂合的位点经过全基因扩增后,严重偏离50%:50%的比例关系,呈现出0.01%~99.99%:99.99%~0.01%的关系,具体表现在PKD1基因上,可以描述为,由于6个假基因的存在,原始基因组样本中,PKD1真基因杂合位点中致病基因型的比例1/7*50%=7%,又由于全基因扩增脱扣现象的存在,具体呈现出0.01%~99.99%:99.99%~0.01%的比例关系,传统的LR-PCR联合NGS的技术已经检测不到这样的比例,造成误诊;另一种胚胎移植前单基因病遗传连锁检测技术虽然能在一定程度上克服脱扣现象造成的困扰,但由于需要制定单倍型家系谱图,会导致检测成本高昂、最多浪费一半的胚胎等结果;在胚胎筛选环节之后,胚胎移入子宫发育,传统的无创检测也无法很好进一步确定胎儿是否含有PKD1有害突变,具体表现为母体外周血游离DNA中通常含有5~10%的胎源游离DNA,PKD1真基因致病基因型比例从7%下降至0.35%~0.7%,此时,传统的LR-PCR联合NGS的技术和遗传连锁检测技术无法使用,只能进行羊水穿刺在产前进一步确定胎儿是否含有PKD1真基因致病突变,羊水穿刺是一种手术,具有2%-5%的流产风险,给患者家庭带来较大的经济和精神压力。
综上所述可以看到,在胚胎移植前检测领域和产前诊断领域中,由于技术局限性以及发育生物学特点的客观存在,现实要求我们能够高效方便低成本的检测到0.01%含量的PKD1致病突变,同时排除其假基因以及高GC含量的干扰。
发明内容
为解决上述技术问题,本发明提供了一种能够稳定检测到0.01%、灵敏度达到0.001%的PKD1真基因变异信息的试剂盒,不仅适合于常规样本的检测,而且更适合于单细胞级别样本、单细胞扩增样本、游离DNA样本的检测。
本发明的第一个目的是提供一种检测人类PKD1基因变异的探针和引物组合物:
所述引物为扩增人类PKD1基因1-33号任意一个或多个外显子的上游引物和下游引物;
所述探针覆盖一个或多个PKD1真假基因碱基差异位点;
在0.25mol/L盐浓度、55℃下:
所述探针的吉布斯自由能ΔG符合-20.7073kcal/mol≤ΔG≤-44.6782kcal/mol;
所述上游引物的吉布斯自由能ΔG符合-15.1985kcal/mol≤ΔG≤-19.5029kcal/mol;
所述探针和上游引物的吉布斯自由能之差ΔΔG在-3.6107×N/Tr范围内,其中,N为54~457的整数,Tr为PCR循环的退火温度。
进一步地,所述探针的序列如下:
/>
其中X1为CCC、CCCG、CGGG、CGCG、GCGG、GGCG、GGGG、CCGC、CCCC、GCGG、GGG、CGCC、GGCC、GGTT、GGC、GGT、CCG、CGC、GGGC、CCTT、GCG中的一种。
进一步地,所述上游引物的序列如下:
/>
/>
进一步地,所述下游引物的序列如下:
进一步地,所述探针的3′端修饰有C3 spacer、C6 spacer、Biotin、PO
进一步地,PCR循环的退火温度为摄氏度,优选为60℃。
进一步地,所述上游引物和探针的分子数比例为1:1~1:100。
进一步地,体系中终浓度为10μM~300μM,优先为100μM。
进一步地,所述上游引物的3′端和探针的5′端有3~30个碱基相同。
进一步地,真假基因特征位点(即真假基因碱基差异位点)在人类参考基因组的位置如下表所示,探针需要覆盖这些位点:
/>
进一步地,扩增产物长度为30-1500bp,扩增产物优选使用一代测序、二代测序、三代测序、PCR、qPCR、ddPCR等平台进行检测。
本发明的第二个目的是提供一种检测人类PKD1基因超低比例变异的试剂盒,所述试剂盒包括上述探针和引物组合物。
本发明的第三个目的是提供上述探针和引物组合物在制备常染色体显性遗传性多囊肾病检测试剂中的应用。
借由上述方案,本发明至少具有以下优点:
本发明用于扩增样本中的PKD1真基因的一个或多个不同外显子,包括不可延伸的寡合苷酸链BL(即探针)中的一条或多条、可延伸的寡合苷酸链FP(即上游引物)中的一条或多条和可延伸的寡核苷酸链RP(即下游引物)中的一条或者多条,该组合能够检测超低比例PKD1基因变异,实现灵敏度0.01%~0.001%的检测效果。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合详细附图说明如后。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明。
图1为单细胞级别样本中检测PKD1真基因的荧光信号结果;
图2为单细胞级别样本中检测PKD1真基因的qPCR产物一代测序结果;
图3为囊胚1添加Blocker的反应结果;
图4为囊胚2添加Blocker的反应结果;
图5为囊胚3添加Blocker的反应结果;
图6为检测Phi29单细胞扩增产物中PKD1真基因的一代测序结果;
图7为单盲验证结果;
图8为含有PKD1 c.348_352delTTTAA变异的囊胚单细胞扩增产物验证结果;
图9为对PKD1 Exon5区域0.001%的PKD1真基因检测的qPCR结果;
图10-11为对PKD1 Exon5区域0.001%的PKD1真基因检测的一代测序结果;
图12为对PKD1 Exon24区域0.001%的PKD1真基因检测的qPCR结果;
图13-14为对PKD1 Exon24区域0.001%的PKD1真基因检测的一代测序结果;
图15为cfDNA的PKD1真基因检测结果;
图16为检测PKD1真基因中GC含量高的外显子的结果;
图17-18为优化引物与Blocker浓度比的结果;
图19-22为优化引物与Blocker的能量设计的结果;
图23为实施例1中的qPCR程序;
图24为实施例4中的热循环程序;
图25为实施例5中的热循环程序;
图26为实施例6中的热循环程序。
具体实施方式
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
下述实施例涉及的材料如下:
每一条寡核苷酸链BL具有一个或数个X1核心,寡核苷酸链BL在盐浓度为0.25mol/L、温度为55℃时,吉布斯自由能ΔG在-20.7073kcal/mol≤ΔG≤-44.6782kcal/mol范围内;优选的寡核苷酸链BL的序列如下:
寡核苷酸链FP需要与对应的寡核苷酸链BL配合使用时才能取得相应的技术效果:寡核苷酸链FP在盐浓度为0.25mol/L、温度为55℃时,吉布斯自由能ΔG在-15.1985kcal/mol≤ΔG≤-19.5029kcal/mol范围内;其中,BL与FP的吉布斯自由能之差ΔΔG在-3.6107×N/Tr范围内,其中Tr为PCR循环的退火温度(摄氏度)的值,优选为60,N为54~457范围内的整数,ΔΔG可通过X1进行微调;优选的寡核苷酸链FP的序列如下:
/>
检测超低比例PKD1基因变异时需要寡核苷酸链RP中的一条或者多条配合实施完整的PCR反应,优选的寡核苷酸链RP序列如下表:
/>
实施例1在单细胞级别样本中检测PKD1真基因
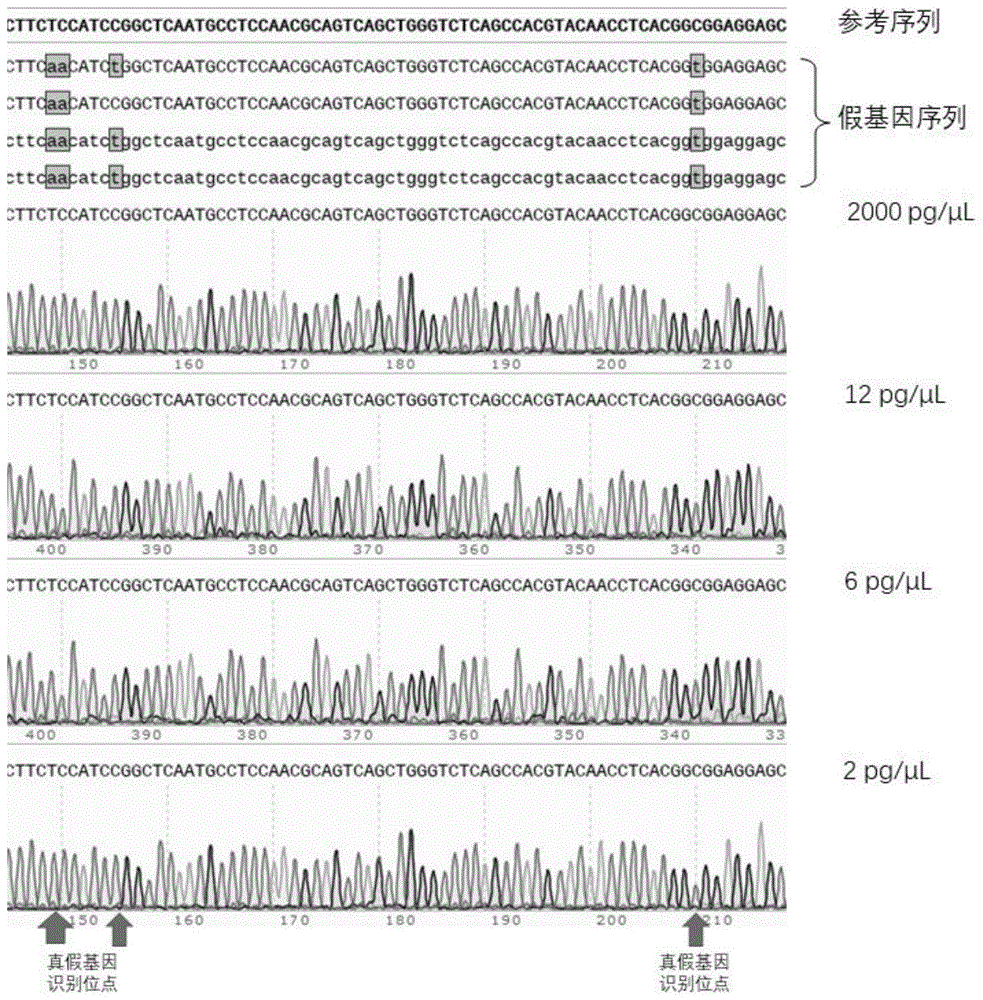
目的:一般来说,普通PCR反应中的DNA模板量在十几纳克到几百纳克级别,DNA模板量越少,PCR越不容易进行。一个人类细胞的DNA大约是6.67pg,10ng大约等1500个细胞所含的DNA的量。在胚胎移植前检测领域和产前诊断领域的具体场景中,经常需要检测1~10个细胞的样本,本实施例将外周血基因组DNA稀释到低浓度,取约等于1.5个细胞、4.5个细胞和9个细胞的DNA来做检测,目的是为了证明本发明公开的方案和体系可以直接针对模拟单细胞级别的DNA模板进行PKD1真基因的检测。
1、需要准备的样本和试剂有:
将提取好的外周血基因组DNA用ddPCR定量,并合理稀释,获得2pg/μL、6pg/μL、12pg/μL以及2000pg/μL的标准品,以及PKD1 15号外显子的引物组合,其中,FP:RP:BL=1:1:10、ZoomBDA Master Mix和nuclease free water。
2、30μL qPCR反应体系如下:
3、将PCR管混匀离心后放置在7500实时荧光定量PCR系统中,选取“
4、荧光信号结果如图1显示,结果显示,当DNAinput低至10pg(2pg/μL组的标准品)时,仍然可以获得稳定的荧光信号。
5、将qPCR产物进行一代测序,结果返回如图2。
真假基因识别位点信息从左到右为TC/AA、C/T、C/T,由一代测序结果可以看出来,我们公开的体系在对单细胞级别DNA样本成功进行PCR扩增的同时,也将PKD1的假基因进行很好的抑制,真假基因识别位点在一代测序结果中显示出TC、C和C碱基纯峰,说明在一代测序结果中只出现了PKD1真基因的特征信息。
实施例2检测MALBAC单细胞扩增产物中的PKD1真基因信息
目的:在胚胎移植前检测领域和产前诊断领域等的具体临床应用场景中,1~10个细胞的DNA量通常还是太少了,因此在进行下游检测前都会先需要进行全基因扩增,单细胞的全基因扩增试剂盒较多,但都普遍存在脱扣的现象。本实施例展示了用MALBAC试剂盒进行单细胞全基因之后,先进行了常规的PGT-M检测获得每个胚胎样本的位点信息,然后从本发明公开的引物中选择合适的引物组,利用该引物组进行单盲验证,最后再与PGT-M结果进行核对。
1、需要准备的样本和试剂有:
某一个家系中的三个囊胚单细胞样本,经过MALBAC试剂盒扩增后,先进行相应的临床PGT-M检测,剩余产物稀释成2ng/μL,待用。本实施例需要用到的引物组合为PKD1_Exon15_FP:-18:-1、PKD1_Exon15_B:-6:+14和PKD1_Exon15_RP(1),其中FP:RP:BL=1:1:20,其他试剂包括ZoomBDA MasterMix和nuclease free water。
2、30μL qPCR反应体系如下:
3、将PCR管混匀离心后放置PCR热循环仪上,PCR程序与实施例1相同:
4、将PCR产物进行Sanger测序。经过Sanger测序之后,1号和3号囊胚发现突变,而2号囊胚未发现突变,我们的结果与PGT-M的结果是一致的。其测序结果如下:
囊胚1添加Blocker的反应结果(图3),即本发明的方案中的管核苷酸BL(下同),其中的真假基因识别位点信息说明Sanger所测到的序列是PKD1真基因的序列,突变位点信息为C/A的杂合,整个测序结果显示该突变和PKD1真基因特征位点在同一条链上,即C>A的突变发生在PKD1真基因上。囊胚1未加Blocker序列结果,即普通PCR的结果中,真假基因识别位点是T/C双峰,说明真假基因混合在一起无法分开,同时可能发生突变的突变位点是没有A的信号的,也就是说本发明的方案获得了压制PKD1假基因、识别出真基因上突变的技术效果。
囊胚2的结果解读同囊胚1(图4),但囊胚2在突变位点没有发现A的信号,因此,囊胚2的PKD1真基因上没有发生C>A的突变。
囊胚3的结果解读同囊胚1(图5),即在囊胚3的单细胞全基因组扩增产物中检测到了PKD1真基因上发生C>A的突变。揭盲后,PGT-M的结果与我们的检测结果一致,也就是说本发明的方案获得了压制PKD1假基因、识别出真基因上突变并且与PGT-M技术结果一致的技术效果。
实施例3检测Phi29单细胞扩增产物中PKD1真基因的信息
目的:单细胞全基因组扩增方法除了基于多重PCR技术的MALBAC之外,还有基于Phi29酶的全基因组等温扩增方法,等温扩增比MALBAC的保真性更好,扩增产物长度更长,适合用本发明公开的产物长度较长的引物组进行PKD1真基因检测。
1、单盲验证含有PKD1 c.5014_5015del变异的囊胚单细胞扩增产物
某一个家系4个囊胚,与实施例2一样,Phi29进行单细胞全基因扩增之后,先进行PGT-M检测。使用的引物组是PKD1_Exon15.5_FP:-17:-1,PKD1_Exon15.5_RP,和PKD1_Exon15.5_B:-6:+19。浓度比为1:1:10。反应条件与其他的实施例相同。一代测序结果如图6所示。
结果显示,除了2号囊胚的单细胞全基因扩增产物有质量问题之外,其他囊胚都没有检测到真基因上的c.5014_5015del突变,说明这些囊胚都是好囊胚。假基因序列中标红的方框是真假基因的特征位点,可以看到1/3/4号囊胚对应的位置都是真基因的碱基信息,说明我们扩增出来并进行Sanger检测的PKD1真基因序列,整个结果说明,1/3/4号囊胚中并没有突变发生在PKD1真基因上。揭盲的结果显示,本案例的结果与PGT-M结果一致。
2、单盲验证含有PKD1 c.2079_2080delinsA变异的囊胚单细胞扩增产物
某一个家系4个囊胚,Phi29进行单细胞全基因扩增之后,先进行PGT-M检测。使用的引物组是PKD1_Exon10_FP:-21:-3,PKD1_Exon10_RP,和PKD1_Exon10_B:-9:+10。浓度比为1:1:10。反应条件与其他的实施例同。
结果显示(图7),2号囊胚检测到PKD1真基因上含有c.2079_2080delinsA突变,其他囊胚都没有检测到真基因上的c.2079_2080delinsA突变,说明这些囊胚都是好囊胚。假基因序列中标红的方框是真假基因的特征位点,可以看到1/3/4号囊胚对应的位置都是真基因的碱基信息,说明我们扩增出来并进行Sanger检测的PKD1真基因序列,整个结果说明,1/3/4号囊胚中并没有突变发生在PKD1真基因上。揭盲的结果显示,本案例的结果与PGT-M检测结果一致。
3、验证含有PKD1 c.348_352delTTTAA变异的囊胚单细胞扩增产物
某一个家系4个囊胚,Phi29进行单细胞全基因扩增之后,先进行PGT-M检测。使用的引物组是PKD1_Exon2&3_FP:-26:-1,PKD1_Exon2&3_RP,和PKD1_Exon2&3_B:-10:+15。浓度比为1:15。反应条件与其他的实施例同。
结果显示(图8),只有4号囊胚的PKD1真基因上不含c.348_352delTTTAA突变,其他囊胚都检测到了真基因上发生的c.348_352delTTTAA突变。揭盲的结果显示,本案例的结果与PGT-M检测结果一致。
实施例4实现对0.001%的PKD1真基因信息的检测
目的:如前所述,单细胞全基因扩增容易发生脱扣的现象,也就是说,如果PKD1真基因发生了脱扣的现象,那么PKD1真基因及其携带的信息在全基因组扩增产物中将低于14%,甚至低于0.1%或0.01%,为了应对这种情况,本发明也公开了一种能检测到0.001%PKD1真基因的体系和实施方法,能够进一步减少临床应用场景中的假阴性。
1、实现对PKD1 Exon5区域0.001%的检测
标准品的制备方法:首先,按照PKD1 exon5假基因所在的基因序列,定制合成与引物组合所对应区域的质粒。在质粒到货后,制备成3×10
引物组合:PKD1_Exon5_FP:-25:-9、PKD1_Exon5_RP、PKD1_Exon5_B:-13:8和PKD1_Exon5_MGB(Cy5-CCTCCTTTGCCTGC-MGB),其中,FP:RP:BL=1:1:5。
热循环程序如图24所示。:
qPCR结果见图9,我们可以看到0.000%的样本是一条直线,意味着没有非特异性的信号,0.001%明显起峰,具有很好的特异性。同样,我们将PCR产物进行一代测序,从测序结果中我们可以看到对99.999%本底的抑制效果。
从一代测序结果可以看出(图10-11),从左到右,真假基因特征识别位点分别是A/C,T/C和A/G,在0.000%样本的一代测序结果中,这三个识别位点分别是C、C、G,说明是假基因的序列,即对检测来说,检测到的是真阴性。0.001%~0.011%样本的一代测序结果中,真假基因特征识别位点分别是A/C,T/C和A/G的杂合峰,说明检测到了阳性信号,同时也能看出趋势,即随着PKD1真基因比例的提高,真基因上碱基信息的信号就越强,0.11%以上的样本看到的全部都是真基因的信号,意味着绝大部分的假基因都被抑制了,显示出优秀的背景信号抑制能力以及优秀的特异性。
2、实现对PKD1 Exon24区域0.001%的检测
因为Exon24的区域也存在着7条高通同源基因,因此标准品制备流程和方案以及qPCR条件同实施例4的1。PKD1_Exon24_FP:-26:-10、PKD1_Exon24_RP、PKD1_Exon24_B-14:+7和PKD1_Exon24_MGB(Cy5-CAGAGTCACTCCAGG-MGB),其中,FP:RP:BL=1:1:1。
qPCR结果如图12。我们可以看到0.000%的样本是一条直线,意味着没有非特异性的信号,0.001%明显起峰,具有很好的特异性。同样,我们将PCR产物进行一代测序,从测序结果中我们可以看到对99.89%本底的抑制效果。
一代测序结果见图13-14。从一代测序结果可以看出,从左到右,真假基因特征识别位点分别是T/C,A/G,T/C和T/C。在0.000%样本的PCR产物中,我们看到的都是假基因的特征信息,符合预期。从0.001%样本开始,真基因的特征信息逐渐增强,在0.110%样本的PCR产物测序结果中,我们在真假基因识别位点看到大部分是PKD1真基因的碱基信息,在0.532%以上的样本中,已经全部都是真基因的信号了,说明Exon24的引物组和反应条件在抑制假基因背景方面与Exon5相似,都取得了很好的效果,都能在qPCR和一代测序平台上获得很好的对假基因抑制、对真基因富集的效果。
实施例5检测外周血游离DNA(cfDNA)的PKD1真基因信息
目的:cfDNA片段大约在140+~160+bp左右,在产前诊断的应用场景中,会需要检测胎儿来源的PKD1真基因信息,cfDNA的应用场景要求我们能够对140+~160+bp的模板实行有效的扩增,同时还要求扩增子中包含有1个以上的真假基因识别位点,方便进行真基因序列的甄别。在本实施例中,我们实现了对cfDNA样本中PKD1真基因信息的检测。
cfDNA样本分别来自4名志愿者的外周血cfDNA,每个反应DNAinput20ng,引物组合:PKD1_Exon15_FP:-27:-10、PKD1_Exon15_RP(2)和PKD1_Exon15_B:-15:+7其中,FP:RP:BL=1:1:10。
热循环程序如图25所示。
PCR产物送测一代测序,序列比对结果如图15:在一代测序结果中,我们可以看到真假基因识别位点T/C,如果不加Blocker,即为普通的PCR,该位点的一代测序结果是T/C的双峰,说明产物是真假基因序列的混合物,在样本1~4中,我们公开的体系很好的抑制住了假基因,识别位点信息是真基因T的纯合峰。本实施例说明我们能够在cfDNA这样的短片段模板中抑制住假基因同时识别出PKD1真基因。
实施例6检测PKD1真基因中GC含量高的外显子
目的:PKD1基因的1号外显子的GC含量高达85%,目前检测的方法多为通过LongRange PCR结合NestedPCR获得PKD1基因的一号外显子的真基因信息,这种方法,虽然可以不针对高GC区域进行引物设计,但是需要耗费的时间较长,并且也无法完全排除PKD1基因假基因的干扰。本实施例展示了我们所公开的体系能够获得包括PKD1真基因Exon1在内的1048bp的目标序列,实现了高GC区域的短产物扩增。
需要准备的样本和试剂包括:三个不同志愿者的外周血基因组DNA样本、PKD1一号外显子的引物组合,其中FP(PKD1_Exon1_FP:-25:-6):RP(PKD1_Exon1_RP):BL(PKD1_Exon1_B:-13:+9)=1:1:10以及TaKaRa LA
PCR热循环仪程序如图26所示。
将1000多个碱基的PCR产物进行一代测序,结果返回如图16:结果表明,当BL未添加到反应预混液中时,由于假基因的影响,未呈现出预期的技术效果,且由于真假基因特征-/CGCGGGC的存在,一代测序信号呈现乱峰模样。相对地,含有BL的反应产物获得干净且只有真基因外显子1的测序结果。本实施例显示,本发明所公开的体系可以扩增富含GC的片段,同时抑制PKD1假基因的干扰,比Long PCR具有更好的便捷性和特异性。
实施例7通过优化引物与Blocker的浓度比来获得最佳技术效果的方法
目的:本实施例公开了引物和Blocker的优化策略与过程,目的在于说明并非设计出引物和Blocker之后马上就能获得最优的检测结果。特别是针对小于0.1%的检测目的,引物与Blocker的最佳效果仍需要精心的摸索才能获得。以实施例4为例,展现引物与Blocker的浓度比的优化方法。
为了探索最优的浓度比,分别准备以下引物与blocker的引物浓度组合:1:0、1:1、1:5、1:10、1:25、1:50和1:100。使用与实施例4相同的qPCR反应体系与热循环程序进行实验。
首先,根据SYBR的荧光信号结果进行第一步的筛选。
在qPCR的结果中(图17),我们可以看到当不添加blocker时,其Ct值为21,随着blocker浓度的增加,其Ct值逐渐增大,当引物与Blocker的浓度比增大到1:100时,已经无法有信号的产生。我们认为当Ct在25~28的范围时,blocker的效果最好。结果显示浓度比为1:5与1:10时,其Ct值均为26。因此,这两个浓度比将作为备选浓度。之后,根据MGB探针的信号进行第二步的筛选。
如图18的qPCR的结果,结果显示,当浓度比为1:1、1:5和1:10时,其Ct值均为29,但是1:5的荧光信号比1:10的荧光信号更强,因此,确定浓度比为1:5时为最优方案。
实施例8通过优化引物与Blocker的能量设计来获得最佳技术效果的方法
目的:实施例7展示了通过调节引物与Blocker之间的相对浓度来获得最优的技术效果,但有时候通过调节浓度,仍然无法获得最优的技术效果,需要根据实验情况,通过增减引物和Blocker的碱基数目来调整吉布斯自由能的大小,进而获得最佳的技术效果,本实施例展示了几组次优的引物和Blocker的技术效果来说明这一方法。
与实施例7相同,首先根据根SYBR的荧光信号结果进行第一步的筛选。
如qPCR结果所示(图19),当浓度比为1:50时,其Ct值为26.7,属于合格的浓度比范围,因此根据MGB探针的信号进行第二步的筛选。
但是,MGB探针的qPCR结果显示(图20),在浓度比1:50时,产生了荧光信号,但是其强度过于微弱。因此,说明原始的设计结果不佳,需要提升blocker的吉布斯自由能和/或降低引物的吉布斯自由能,既需要增加blocker3’端的碱基个数和/或减少引物5’端的碱基个数。
根据以上原则,在对引物和blocker进行重新设计后,再次进行相同步骤的筛选。
在对引物和blocker进行重新的设计后(图21),浓度比为1:1、1:5、1:10的Ct值分别为25.1、26.3、和27.5。因此,需要根据MGB探针的信息结果进行进一步的筛选。
MGB探针的结果显示(图22),当浓度比为1:1时,MGB探针的荧光信号最强。因此确定新设计的引物和blocker组合,在浓度比为1:1时,为最优组合方案。
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。