5′-(E)-乙烯基磷酸酯的合成方法
文献发布时间:2024-04-18 20:01:23
技术领域
本发明涉及化学合成技术领域,具体而言,涉及一种5′-(E)-乙烯基磷酸酯的合成方法。
背景技术
RNA干扰是一种对双链RNA的生物学反应,介导对内源性寄生和外源性致病核酸的抗性,并调节蛋白质编码基因的表达。近年来,RNA干扰取得了一系列进展,基于RNAi的治疗方法已经进入临床研究。然而,RNAi疗法最大的影响因素是如何将siRNA传送到目标细胞以及siRNA在人体内的分解。
对siRNA的修饰来改善其传递和稳定性成为目前研究的热点。报道使用5’-乙烯基磷酸盐修饰siRNA能有效提高其稳定性。但是将乙烯基磷酸盐引入5’位羟基的合成方法却不多。
目前常见的5’-乙烯基亚磷酰胺单体合成路线如下(文献:J. Med. Chem. 2018,61, 3, 734–744. Facile Synthesis, Geometry, and 2’-Substituent-Dependent inVivo Activity of 5’-(E)- and 5’-(Z)-Vinylphosphonate-Modified siRNAConjugates):
目前该路线主要问题为化合物3的稳定性,该醛类化合物不稳定,后处理过程中遇水容易形成水合物,对后续反应有影响。且浓缩破坏反应发现该化合物浓缩过程中不稳定,容易变质导致放大收率明显降低。
发明内容
本发明旨在提供一种5′-(E)-乙烯基磷酸酯的合成方法,以解决现有技术中5′-(E)-乙烯基磷酸酯制备收率较低的技术问题。
为了实现上述目的,根据本发明的一个方面,提供了一种5′-(E)-乙烯基磷酸酯的合成方法。该合成方法包括以下步骤:使用moffatt氧化
进一步地,使用moffatt氧化
。
进一步地,使用moffatt氧化
第一反应釜中加入DMSO和
第二反应釜中加入四氢呋喃和
将第一反应釜中物料滴加到第二反应釜中,保温反应;
向第二反应釜中加入氯化铵水溶液淬灭反应;
采用MTBE对第二反应釜中的物料进行萃取,得到
进一步地,合成方法还包括对有机相进行纯化的步骤;
优选的,采用柱层析纯化
进一步地,第一反应釜中的DMSO加入量为
优选的,吡啶的加入量为0.8~1.5当量,优选1当量,三氟乙酸的加入量为吡啶当量的一半;
优选的,加入吡啶和三氟乙酸,搅拌0.5~1h后,加入二环己基碳二亚胺;更优选的,二环己基碳二亚胺的加入量为1.5~2.0当量,进一步优选为1.6当量;
优选的,加入二环己基碳二亚胺后,第一反应釜的反应温度保持在0~25℃;更优选的,HPLC跟踪至原料≤5%。
进一步地,第二反应釜中四氢呋喃的加入量为8~15V,优选10V,
优选的,第二反应釜中的反应温度为-20~0℃℃;
优选的,第二反应釜中加入的碱为氢化钠、叔丁醇钾或叔丁醇钠中的一种或多种,优选为氢化钠;
更优选的,氢化钠的加入量为1.3 eq. 60%氢化钠;更优选的,加入氢化钠后保温反应的时间为0.5~1h;
优选的,将第一反应釜中物料滴加到第二反应釜中保温反应后取样跟踪HPLC,每1~4h取样一次直至
进一步地,第二反应釜中氯化铵水溶液的加入量为8~25V V,优选20 V,淬灭后搅拌0.5~1h;
优选的,采用MTBE对第二反应釜中的物料进行萃取包括:控温0~25℃,向第二反应釜中加入5~20 V,优选10V MTBE, 搅拌1~2h后静置分层后分液,水相再次使用5~20 V,优选10 V MTBE萃取,将得到的有机相合并后,使用5~10 V,优选5V饱和氯化钠水溶液反洗
优选的,有机相合并后,浓缩至1~2V,柱层析纯化
进一步地,脱保护基反应的反应式为:
。
进一步地,脱保护基反应包括:
向第三反应釜中加入甲酸、纯化水和
采用DCM对第三反应釜中的物料进行萃取,得到
保温45~55℃跟踪反应,HPLC跟踪至
将第三反应釜中体系温度降温至0~10℃,优选降温速度10~30℃/h;
控温0~10℃,向第三反应釜中加入氨水调pH~7;
控温0~20℃,加入5~15 V,优选10V DCM萃取,静置分液后再次使用5~15 V,优选10V DCM萃取后静置分液;
将有机相合并后控温T≤45℃,浓缩至1~2V;
柱层析过柱纯化得到
可选的,
应用本发明的技术方案,使用moffatt氧化(二环己基碳二亚胺和二甲亚砜氧化)
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1示出了实施例1中化合物4的色谱检测图;
图2示出了实施例2中化合物5的色谱检测图;以及
图3示出了实施例3中终产物的色谱检测图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
针对背景技术中提到的5’-乙烯基亚磷酰胺单体合成技术路线存在的问题,本申请对5’-乙烯基亚磷酰胺单体的合成方法进行了优化,为研究5’-乙烯基-siRNA的合成提供原料单体的优化合成方案。即在本发明中没有具体描述的步骤,可以参考背景技术中Facile Synthesis, Geometry, and 2’-Substituent-Dependent in Vivo Activity of5’-(E)- and 5’-(Z)-Vinylphosphonate-Modified siRNA Conjugates的技术方案完成。
根据本发明一种典型的实施方式,提供5′-(E)-乙烯基磷酸酯的合成方法。该方法包括以下步骤:使用moffatt氧化
应用本发明的技术方案,使用moffatt氧化(二环己基碳二亚胺和二甲亚砜氧化)
典型的,使用moffatt氧化
。
在本发明一实施例中,使用moffatt氧化
合成方法还包括对有机相进行纯化的步骤;
优选的,采用柱层析纯化
为了保证反应充分有效的进行,第一反应釜中的DMSO加入量为
为了充分的利用反应原料,保证反应效率,第二反应釜中四氢呋喃的加入量为8~15V,优选10V,
第二反应釜中氯化铵水溶液的加入量为8~25V,优选10V,淬灭后搅拌0.5~1h;优选的,采用IPAc或者MTBE(甲基叔丁基醚,优选甲基叔丁基醚)对第二反应釜中的物料进行萃取包括:控温0~25℃,向第二反应釜中加入5~20 V,优选10V IPAc或者MTBE(甲基叔丁基醚,优选甲基叔丁基醚), 搅拌1~2h后静置分层后分液,水相再次使用5~20 V,优选10 V IPAc或者MTBE(甲基叔丁基醚,优选甲基叔丁基醚)萃取,将得到的有机相合并后,使用5~10 V饱和氯化钠水溶液反洗
脱保护基反应的反应式为:
。
脱保护基反应包括:
向第三反应釜中加入甲酸、纯化水和
采用DCM对第三反应釜中的物料进行萃取,得到
第三反应釜中保持温度15~30℃,将5~15 V,优选12.5 V甲酸和5~15 V,优选12.5V纯化水混匀,加入包含1当量
保温45~55℃跟踪反应,HPLC跟踪至
将第三反应釜中体系温度降温至0~10℃,优选降温速度10~30℃/h;控温0~10℃,向第三反应釜中加入氨水调pH~7;控温0~20℃,加入5~15 V,优选DCM萃取,静置分液后再次使用5~15 V,优选DCM萃取后静置分液;将有机相合并后控温T≤45℃,浓缩至1~2V;柱层析过柱纯化得到
在本发明一种典型的实施方式中,
下面将结合实施例进一步说明本发明的有益效果。
本发明中化合物编号见背景技术中的描述。
实施例1
车间投料1Kg/80L反应化合物2到化合物4,反应IPC正常,2步连投收率~70%,产品(化合物4)过柱纯度~90%(产品:异构体~8:1)。
具体步骤:
1. 反应釜1中加入10 L DMSO(10 V)和原料2,15~25℃搅拌至全溶。
2. 降温至10~20℃。
3. 10~20℃加入158.44 g吡啶(1当量)和114.18 g三氟乙酸(0.5当量),搅拌0.5~1h后10~20℃缓慢加入661.27 g二环己基碳二亚胺(DCC)(1.5当量)。
4. 保温15~25℃反应,HPLC跟踪至原料≤5%area。
5. 反应釜2中加入10 L四氢呋喃(10 V)和1393.82 g原料7(1当量),搅拌至全溶后反应釜2降温至-20~-10℃。
6. -20~-10℃反应釜2中加入120 g氢化钠(60%)(1.5当量),保温-20~-10℃反应0.5~1h。
7. 控温0~10℃,将反应釜1中物料通过管路缓慢滴加到反应釜2中,保温反应2h后取样跟踪HPLC,每1~4h取样一次直至原料3HPLC≤5%area。
8. 控温0~10℃,向反应釜2中加入20 V氯化铵水溶液淬灭反应。淬灭后搅拌0.5~1h。
9. 控温0~25℃,向反应釜2中加入10 V MTBE, 搅拌1~2h后静置分层后分液。水相再次使用10 V MTBE萃取。将有机相合并后,使用5 V盐水反洗有机相。
10. 有机相合并后,浓缩至1~2V。柱层析纯化(庚烷/IPAc=10/1~1/1)得到产品有机相。有机相浓缩得到1221.32 g淡黄色油状产品HPLC纯度为92.11%,内标含量为83%,直接用于下一步反应(Wittig反应反式产物:顺式异构体~15:1)。
化合物4的核磁检测结果:
化合物4的色谱检测图见图1。
实施例2
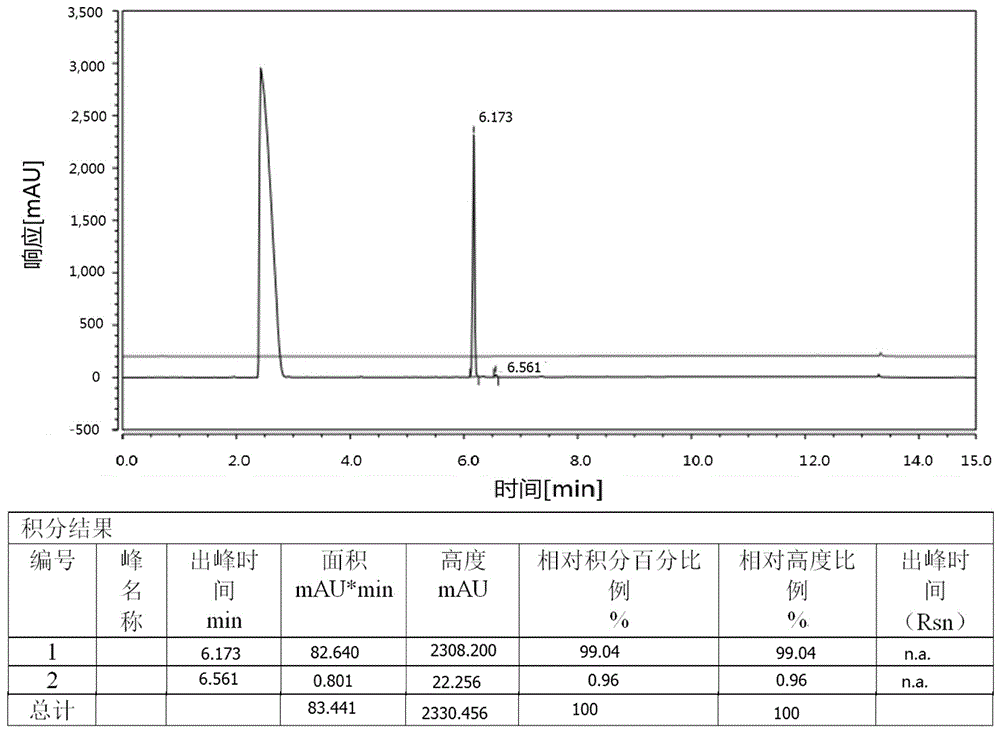
根据专利方法,车间投料1Kg/80L,将~90%HPLC纯度化合物4投水解反应至化合物5,反应IPC跟踪至原料反应HPLC≤5%area,产品过柱后收率~60%,产品(化合物5)95%HPLC纯度。
1.15~30℃,反应釜1中将12.5 L甲酸(12.5 V)和12.5 L纯化水(12.5 V)混匀,加入1 Kg原料。
2.将体系升温至45~55℃,参考升温速度5~15℃/h。
3.保温45~55℃跟踪反应,跟踪至原料4≤2%area且原料4异构体≤1%area。
4.将体系温度降温至0~10℃,参考降温速度10~30℃/h。
5.控温0~10℃,向反应釜1中加入氨水调pH~7。
6.控温0~20℃,加入10V MTBE萃取产品。静置分液后再次使用10V MTBE萃取后静置分液。
7.将有机相合并后控温T≤45℃,浓缩至1~2V。
8.柱层析过柱纯化(庚烷/IPAc=10/1~1/1)得到纯品溶液,浓缩后得到606.70 g产品,HPLC纯度为99.04%,内表含量为84.85%,用于下一步。
化合物5的核磁检测结果:
化合物5的色谱检测图见图2。
实施例3
车间投料0.5 Kg/80L反应化合物5到化合物6,反应IPC跟踪至原料反应HPLC≤1%area,经过反向制备后得到产品纯度为100%,纯化后收率为80%。
1.在反应釜中加入2.50 L二氯甲烷(5 V),再加入327.98 g磷试剂(1.5当量)和68.54 g 4,5-二氰基咪唑(0.8当量),15~25℃搅拌至溶解。
2.使用2.50 L二氯甲烷(5 V)溶解原料,加入反应釜中。
3.保温15~25℃反应1~3 h,直至原料检测≤1%area。
4.控温15~25℃,使用5 L饱和氯化钠水溶液(10 V)搅洗两次,分液。
5.使用分子筛进行干燥有机相,直至KF≤1%。
6.将有机相控温T≤45℃,浓缩至1~2V。
7.进行反向制备,产品体系冻干即得到516.13 g白色固体产品,HPLC纯度为100%,内标含量为97%。
终产物的核磁膦谱检测结果:
终产物的核磁检测结果:
终产物的色谱检测图见图3。
上述检测结果表明本发明成功的合成了各中间产物及终产物。
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
应用本发明的技术方案,使用moffatt氧化(二环己基碳二亚胺和二甲亚砜氧化)
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种治疗动脉粥样硬化的干细胞药物
- 一种治疗动脉粥样硬化的干细胞药物