一种烯丙基硅烷类化合物的制备方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于有机合成技术领域,具体涉及一种烯丙基硅烷类化合物的制备方法。
背景技术
通过过渡金属催化C–Si键的形成来构建复杂有机硅化合物在有机合成中占据着非常重要的地位中。硅基杂环芳烃结构单元常见于一些具有生物活性有机硅小分子化合物中,例如P38 MAP Kinase inhibitor,DB-67,Ctopoisomerase inhibitor,GnRH agonist等。
硅烷的的烯丙基化一直以来都是很有挑战性的研究。
烯丙基的引入能使原化合物理化性质发生改变,因此烯丙基化反应广泛应用于有机中间体和药物分子合成。但传统的过渡金属催过的烯丙基化需要严格控制反应条件,以及使用化学计量比的还原剂,这往往会降低官能团的耐受性。随着光和金属催化的结合,很好的解决了这些问题,文献报道了(Cartwright,K.C.;Tunge,Organophotoredox/palladium dual catalytic decarboxylative Csp
通过过渡金属催化C–Si键的形成来构建复杂有机硅化合物在有机合成中占据着非常重要的地位。而有机硅兼备了无机材料与有机材料的性能,具有耐高低温、电气绝缘、耐氧化、耐腐蚀、无毒无味以及生理惰性等优异特性。文献只报道过硅烷的烯基化反应(Dong,J.;Yuan,X.-A.;Yan,Z.;Mu,L.;Ma,J.;Zhu,C.;Xie,J.Manganese-catalyseddivergent silylation ofalkenes.Nat.Chem.2021,13(2),182–190.)。通过配体调节硅自由基活性,实现了高选择性的自由基C-Si偶联。但是通过硅的烯丙基化反应很少被报道。
发明内容
本发明的目的是提供一种烯丙基硅烷类化合物的制备方法,无需适用昂贵的贵金属催化剂和光催化剂,反应条件温和,底物普适性好,反应产率较高。
为实现上述目的,本发明采用如下技术方案:
一种烯丙基硅烷类化合物的制备方法,以乙酸烯丙酯和叔丁基二苯基硅烷甲酸为起始原料,在光催化剂、金属催化剂、配体、碱存在的条件下进行反应,得到烯丙基硅烷类化合物。
优选的,所述光催化剂为2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈,所述金属催化剂为乙二醇二甲醚溴化镍,所述配体为联吡啶,所述碱为磷酸氢二钾,反应式为:
优选的,所述乙酸烯丙酯和叔丁基二苯基硅烷甲酸的投料摩尔比为1:1.5-1:3。
优选的,所述乙酸烯丙酯和磷酸氢二钾的投料摩尔比为1:0.75-1:1。
优选的,所述2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈的投料摩尔比为乙酸烯丙酯的1.5%。
优选的,所述乙二醇二甲醚溴化镍的投料摩尔比为乙酸烯丙酯的10-20%。
优选的,所述联吡啶的投料摩尔比为乙酸烯丙酯的12-24%。
优选的,所述反应的条件为:反应溶剂为乙醇,反应光源为蓝光,反应气体氛围为氩气,反应时间为18~24个小时。
有益效果:本发明的一种烯丙基硅烷类化合物的制备方法,无需适用昂贵的贵金属催化剂和光催化剂,反应条件温和,底物普适性好,反应产率较高。
附图说明
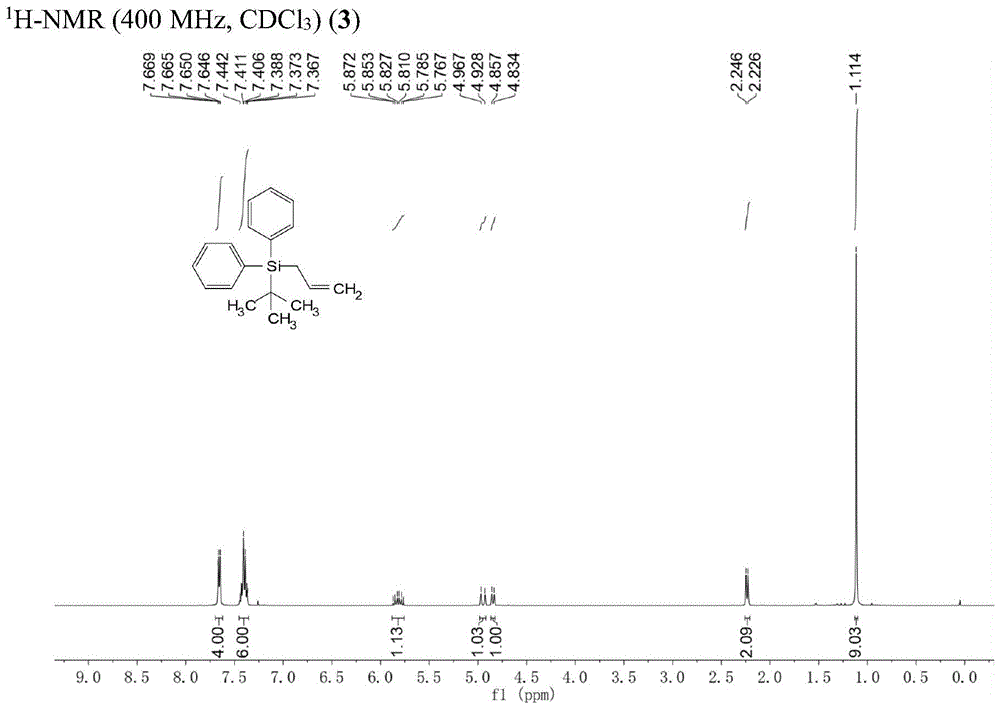
图1为实施例1中化合物3的
图2为实施例1中化合物3的产物的
图3为实施例2中化合物6的产物的
图4为实施例2中化合物6的
图5为实施例2中化合物7的产物的
图6为实施例2中化合物7的
图7为实施例2中化合物7的HRMS谱图;
图8为实施例3中化合物10的产物的
图9为实施例3中化合物10的
图10为实施例3中化合物11的产物的
图11为实施例3中化合物11的
图12为实施例3中化合物11的HRMS谱图;
图13为实施例4中化合物13的产物的
图14为实施例4中化合物13的
图15为实施例4中化合物14的产物的
图16为实施例4中化合物14的
图17为实施例4中化合物14的HRMS谱图。
具体实施方式
下面结合实施例对本发明作进一步描述。以下实施例只是用于更加清楚地说明本发明的性能,而不能仅局限于下面的实施例。
实施例1
化合物3的制备
选取两个10mL装有磁力搅拌子的带支管的史莱克管,分别加入化合物1(10.1mg,0.1mmol),化合物2(42.7mg,0.15mmol),2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈(1.2mg,0.0015mmol,1.5mol%),乙二醇二甲醚溴化镍(3.1mg,0.01mmol,10mol%),联吡啶(1.9mg,0.012mmol,12mol%),磷酸氢二钾(13.4mg,0.075mmol,0.75eq.)。以及1.5mL的无水乙醇。抽换三次氩气后,在蓝光照射下常温反应18个小时。反应结束后合并两个反应,加入水5mL,再用5mL乙酸乙酯萃取3次,合并有机相加入无水硫酸镁干燥,旋转蒸发仪上旋蒸除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚),即可得到化合物3(46.6mg,82%)。反应式如下:
产物结构表征数据如下:
1
13
实施例2
1)化合物5的制备
选取一个50mL装有磁力搅拌子的带支管的史莱克管,管口接入恒压滴液漏斗,密封反应管后,在支管口处抽换三次氮气后,在氮气氛围下往史莱克管中加入正癸醛4(0.7814g,5mmol),再用注射器注入无水的四氢呋喃10mL,开启磁力搅拌,溶解后放置于冰浴中,冷至0℃后,用带针管的注射器往恒压滴液漏斗滴加入1.0mol/L的乙烯溴化镁四氢呋喃溶液7.5mL(浓度为1.0mol/L,1.5当量),在冰浴条件下缓慢滴乙烯溴化镁四氢呋喃溶液于史莱克管中,加完之后即可撤除冰浴,在室温(25℃)下反应,8小时以后,加入20mL饱和的氯化铵溶液,再加入10mL乙酸乙酯萃取3次,合并有机相后,通过旋转蒸发仪除去溶剂后,得到粗产物5,无需纯化,直接进行下一步。反应式如下:
2)化合物6的制备
选取一个50mL装有磁力搅拌子的带支管的史莱克管,管口接入恒压滴液漏斗,密封反应管后,在支管口处抽换三次氮气后,在氮气氛围下往史莱克管中加入4-二甲氨基吡啶(244.4mg,2mmol,40mol%),粗产物5,三乙胺3.0357g(30mmol,6当量),再加入二氯甲烷10mL,开启磁力搅拌,溶解后,用带针管的注射器缓慢加入乙酸酐(2.0458g,20mmol,4当量),在室温(25℃)下滴加结束后,将反应在40℃下反应8小时以后,加入20mL饱和的碳酸钠溶液,再加入10mL乙酸乙酯萃取3次,合并有机相后,通过旋转蒸发仪除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚:乙酸乙酯体积比=20:1),即可得到化合物6(827.7mg,73%)。反应式如下:
产物结构表征数据如下:
1
10.5Hz,1H),2.03(s,3H),1.55(tt,J=14.4,7.0Hz,2H),1.25(d,J=7.6Hz,14H),0.85(t,J=6.8Hz,3H)ppm.如图3。
13
3)化合物7的制备
选取两个10mL装有磁力搅拌子的带支管的史莱克管,分别加入化合物6(22.7mg,0.1mmol),化合物2(42.7mg,0.15mmol),2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈(1.2mg,0.0015mmol,1.5mol%),乙二醇二甲醚溴化镍(3.1mg,0.01mmol,10mol%),联吡啶(1.9mg,0.012mmol,12mol%),磷酸氢二钾(13.4mg,0.075mmol,0.75eq.)。以及1.5mL的无水乙醇。抽换三次氩气后,在蓝光照射下常温反应18个小时。反应结束后合并两个反应,加入水5mL,再用5mL乙酸乙酯萃取3次,合并有机相加入无水硫酸镁干燥,旋转蒸发仪上旋蒸除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚),即可得到化合物7(69.9mg,86%)。反应式如下:
产物结构表征数据如下:
1
13
HRMS(ESI):[M+Na]
实施例3
1)化合物9的制备
选取一个50mL装有磁力搅拌子的带支管的史莱克管,管口接入恒压滴液漏斗,密封反应管后,在支管口处抽换三次氮气后,在氮气氛围下往史莱克管中加入环己基甲醛8(0.4908g,5mmol),再用注射器注入无水的四氢呋喃10mL,开启磁力搅拌,溶解后放置于冰浴中,冷至0℃后,用带针管的注射器往恒压滴液漏斗滴加入1.0mol/L的乙烯溴化镁四氢呋喃溶液7.5mL(浓度为1.0mol/L,1.5当量),在冰浴条件下缓慢滴乙烯溴化镁四氢呋喃溶液于史莱克管中,加完之后即可撤除冰浴,在室温(25℃)下反应,8小时以后,加入20mL饱和的氯化铵溶液,再加入10mL乙酸乙酯萃取3次,合并有机相后,通过旋转蒸发仪除去溶剂后,得到粗产物9,无需纯化,直接进行下一步。反应式如下:
2)化合物10的制备
选取一个50mL装有磁力搅拌子的带支管的史莱克管,管口接入恒压滴液漏斗,密封反应管后,在支管口处抽换三次氮气后,在氮气氛围下往史莱克管中加入4-二甲氨基吡啶(244.4mg,2mmol,40mol%),粗产物9,三乙胺(3.0357g,30mmol,6当量),再加入二氯甲烷10mL,开启磁力搅拌,溶解后,用带针管的注射器缓慢加入乙酸酐(2.0458g,20mmol,4当量),在室温(25℃)下滴加结束后,将反应在40℃下反应8小时以后,加入20mL饱和的碳酸钠溶液,再加入10mL乙酸乙酯萃取3次,合并有机相后,通过旋转蒸发仪除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚:乙酸乙酯体积比=20:1),即可得到化合物10(402.4mg,48%)。反应式如下:
产物结构表征数据如下:
1
3)化合物11的制备
选取两个10mL装有磁力搅拌子的带支管的史莱克管,分别加入化合物10(16.9mg,0.1mmol),化合物2(42.7mg,0.15mmol),2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈(1.2mg,0.0015mmol,1.5mol%),乙二醇二甲醚溴化镍(3.1mg,0.01mmol,10mol%),联吡啶(1.9mg,0.012mmol,12mol%),磷酸氢二钾(13.4mg,0.075mmol,0.75eq.)。以及1.5mL的无水乙醇。抽换三次氩气后,在蓝光照射下常温反应18个小时。反应结束后合并两个反应,加入水5mL,再用5mL乙酸乙酯萃取3次,合并有机相加入无水硫酸镁干燥,旋转蒸发仪上旋蒸除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚),即可得到化合物11(69.9mg,72%)。反应式如下:
产物结构表征数据如下:
1
13
HRMS(ESI):[M+Na]
实施例4
1)化合物13的制备
选取一个50mL装有磁力搅拌子的带支管的史莱克管,管口接入恒压滴液漏斗,密封反应管后,在支管口处抽换三次氮气后,在氮气氛围下往史莱克管中加入4-二甲氨基吡啶(146.7mg,1.2mmol,40mol%),异植物醇12(889.6mg,3mmol),三乙胺(1.8215g,18mmol,6当量),再加入二氯甲烷10mL,开启磁力搅拌,溶解后,用带针管的注射器缓慢加入乙酸酐(1.2275g,12mmol,4当量),在室温(25℃)下滴加结束后,将反应在40℃下反应8小时以后,加入20mL饱和的碳酸钠溶液,再加入10mL乙酸乙酯萃取3次,合并有机相后,通过旋转蒸发仪除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚:乙酸乙酯体积比=20:1),即可得到化合物13(577.0mg,57%)。反应式如下:
产物结构表征数据如下:
1
13
2)化合物14的制备
选取两个10mL装有磁力搅拌子的带支管的史莱克管,分别加入化合物13(33.9mg,0.1mmol),化合物2(85.4mg,0.3mmol),2,4,5,6-四(9H-咔唑-9-基)间苯二甲腈(2.4mg,0.003mmol,3.0mol%),乙二醇二甲醚溴化镍(6.2mg,0.02mmol,20mol%),联吡啶(3.8mg,0.024mmol,24mol%),磷酸氢二钾(17.5mg 0.01mmol,1.0eq.)。以及1.5mL的无水乙醇。抽换三次氩气后,在蓝光照射下常温反应18个小时。反应结束后合并两个反应,加入水5mL,再用5mL乙酸乙酯萃取3次,合并有机相加入无水硫酸镁干燥,旋转蒸发仪上旋蒸除去溶剂后,通过快速硅胶柱层析(洗脱剂石油醚),即可得到化合物14(75.3mg,73%)。反应式如下:
产物结构表征数据如下:
1
13
HRMS(ESI):[M+Na]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种含(2-烯丙基苯氧基)三甲硅烷添加剂的高压功能电解液及其制备方法与应用
- 一种4-甲基-N-苯基-N-(2-苯基烯丙基)苯磺酰胺类化合物的合成方法
- 一种氯丙基三乙氧基硅烷的制备方法
- 一种γ异氰酸酯丙基硅烷及其制备方法
- 一种烯丙基阴-非离子乳化剂及其制备方法
- 一种(Z)-2-烷基-3-烯丙基硫基-3-芳基烯丙醇类化合物的制备方法
- 一种(Z)‑2‑烷基‑3‑烯丙基硫基‑3‑芳基烯丙醇类化合物的制备方法