一种电催化胺氧化脱氢制备腈类化合物的催化剂、制备方法及应用
文献发布时间:2024-04-18 20:01:30
技术领域
本发明属于有机电化学领域,涉及一种电催化胺氧化脱氢制备腈类化合物的催化剂、制备方法及应用。
背景技术
腈类化合物不仅本身是一种天然活性物质,而且是一种非常重要的化学原料,其衍生物作为多功能中间体,是合成许多药物、农用化学品、染料和精细化学品的基本组成部分,在国民经济的各个领域都有着广泛的应用。
目前有几种合成腈类化合物的方法,首先,传统严格氰化物的使用,主要是通过卤代烃氰基化或胺化合物经过Sandmeyer重氮盐法制备,但由于安全和环境问题,HCN或金属及有机氰化物的实际应用被严重限制;其次,可以通过牺牲性强氧化剂,如t-BuOOH、臭氧、O
针对上述不足,开发一种廉价,低毒,无污染,简便快速,且高选择性和高产率的制备腈类化合物的催化剂及合成方法显得尤为重要,且对工业化生产具有一定的实际意义。
发明内容
本发明的目的旨在克服现有技术的不足,制备一种高效的单一金属镍基催化剂,采用电催化方式在三电极体系中将胺类化合物稳定、高效的转化为腈类化合物。本发明体系具有催化剂催化范围广,催化剂制备成本低,产物产率和选择性高,反应条件温和,操作简单,绿色安全,反应可控性高等优点,具有工业应用前景。
本发明的技术目的通过如下技术方案予以实现:
一种电催化胺氧化脱氢制备腈类化合物的催化剂制备方法,包括步骤如下:
1)对泡沫镍进行预处理:去除表面氧化层后冲洗干净干燥备用;
生长溶液的配备:将Ni
2)Ni(OH)
3)Ni(II)Ni
进一步地,步骤3)所述次氯酸钠水溶液质量浓度为25%。
进一步地,步骤2)所述水热反应温度为80℃。
本发明还提供了,上述方法制备得到的电催化胺氧化脱氢制备腈类化合物的Ni(II)Ni
本发明还提供了,一种电催化胺氧化脱氢制备腈类化合物的方法,以上述的Ni(II)Ni
进一步地,所述胺类化合物为苄胺、对甲基苄胺、对甲氧基苄胺、4-甲基哌啶、正丁胺或正庚胺中的一种。
进一步地,所述碱性电解液为0.1M KOH溶液。
进一步地,所述胺类化合物在电解液中的浓度为10mM。
相对于现有技术,本发明技术方案取得的有益效果是:
1)本发明提出通过电化学方式催化胺氧化脱氢制腈,通过改进催化剂制备方式,制备高效单一金属镍基催化剂,降低催化剂制备成本,增加催化剂的活性位点密度的同时,提高催化剂本征催化活性,进而提高催化效率,为镍基催化剂电催化胺脱氢制腈的实际应用提出一个有意义的研究思路。
2)在三电极体系中,Ni(II)Ni
3)本发明催化体系工艺过程更加简单,反应条件宽松,避免了其他合成法方法中操作复杂,反应耗时长,产率低,选择性差,试剂毒性强,以及对反应温度和压力的设备要求较高等问题。
附图说明
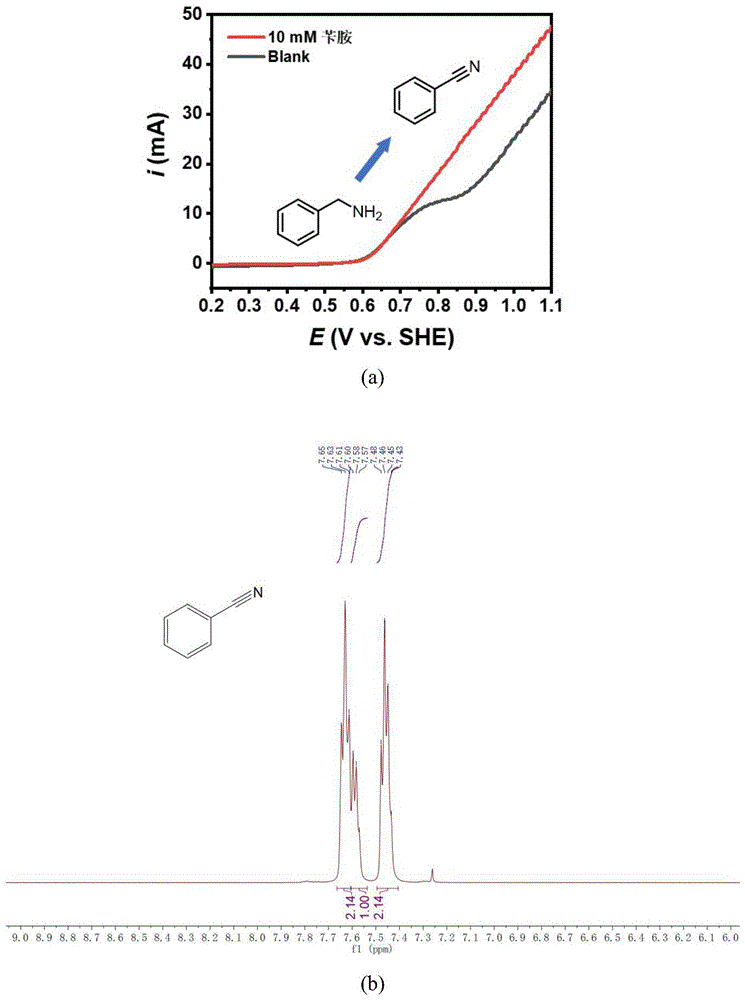
图1(a)为空白条件和苄胺电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
图2(a)为空白条件和对甲基苄胺电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
图3(a)为空白条件和对甲氧基苄胺电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
图4(a)为空白条件和4-甲基哌啶电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
图5(a)为空白条件和正丁胺电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
图6(a)为空白条件和正庚胺电催化脱氢过程LSV曲线。(b)为产物的核磁共振氢谱图。
具体实施方式:
为了使本发明所要解决的技术问题、技术方案及有益效果更加清楚、明白,下面结合附图和实施例对本发明做进一步详细说明。
实施例1:Ni(II)Ni
采取如下条件制备Ni(II)Ni
1)泡沫镍(NF)预处理:将市售泡沫镍按需裁剪为1×2.5cm
2)生长溶液的配备:将8mmol NiSO
3)Ni(OH)
4)Ni(II)Ni
实施例2:Ni(II)Ni
采取如下条件制备Ni(II)Ni
1)泡沫镍(NF)预处理:将市售泡沫镍按需裁剪为1×2.5cm
2)生长溶液的配备:将8mmol NiSO
3)Ni(OH)
4)Ni(II)Ni
实施例3:Ni(II)Ni
采取如下条件制备Ni(II)Ni
1)泡沫镍(NF)预处理:将市售泡沫镍按需裁剪为1×2.5cm
2)生长溶液的配备:将8mmol NiSO
3)Ni(OH)
4)Ni(II)Ni
实施例4:苄胺的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的苄胺作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和苄胺电催化脱氢过程LSV曲线如图1a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图1b所示)进行定性分析,液相色谱进行定量分析。苄胺的转化率为99%、苯甲腈的选择性为98%。1H NMR(500MHz,Chloroform-d)δ7.63(t,J=8.1Hz,2H),7.58(t,J=6.7Hz,1H),7.46(q,J=7.2Hz,2H).
实施例5:对甲基苄胺的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的对甲基苄胺作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和对甲基苄胺电催化脱氢过程LSV曲线如图2a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图2b所示)进行定性分析,液相色谱进行定量分析。对甲基苄胺的转化率为98%、对甲基苯甲腈的选择性为95%。1H NMR(500MHz,Chloroform-d)δ7.51(d,J=7.8Hz,2H),7.24(s,2H),2.40(s,3H).
实施例6:对甲氧基苄胺的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的对甲氧基苄胺作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和对甲氧基苄胺电催化脱氢过程LSV曲线如图3a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图3b所示)进行定性分析,液相色谱进行定量分析。对甲氧基苄胺的转化率达98%、对甲氧基苯甲腈的选择性为98%。1HNMR(500MHz,Chloroform-d)δ7.67–7.49(m,2H),6.94(d,J=8.4Hz,2H),3.85(d,J=1.3Hz,3H).
实施例7:4-甲基哌啶的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的4-甲基哌啶作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和4-甲基哌啶电催化脱氢过程LSV曲线如图4a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图4b所示)进行定性分析,液相色谱进行定量分析。4-甲基哌啶的转化率为99%、产物的选择性为99%。
实施例8:正丁胺的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的正丁胺作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和正丁胺电催化脱氢过程LSV曲线如图5a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图5b所示)进行定性分析,液相色谱进行定量分析。正丁胺的转化率为99%、丁腈的选择性为98%。1H NMR(600MHz,Chloroform-d)δ2.29(t,J=7.1Hz,2H),1.67(p,J=7.3Hz,2H),1.04(t,J=7.5Hz,3H).
实施例9:正庚胺的催化脱氢
以实施例2的条件制备Ni(II)Ni
以30mL密封单池电解池作为容器,电解池内加入25mL 0.1M KOH溶液,然后加入10mM的正庚胺作为底物,并用磁子不断搅拌,转速为500rpm。以制备的Ni(II)Ni
空白条件和正庚胺电催化脱氢过程LSV曲线如图6a所示。反应结束后,收集产物,用乙酸乙酯进行萃取,减压蒸馏,再用液质联用、核磁(如图6b所示)进行定性分析,液相色谱进行定量分析。正庚胺的转化率为98%、庚腈的选择性为98%。1H NMR(600MHz,Chloroform-d)δ2.32(t,J=7.2Hz,2H),1.69–1.57(m,2H),1.43(ddd,J=15.0,8.8,6.1Hz,2H),1.35–1.23(m,4H),0.88(t,J=6.9Hz,3H)。
本发明的实施例是为了示例和描述起见而给出的,而并不是无遗漏的或者将本发明限于所公开的形式。很多修改和变化对于本领域的普通技术人员而言是显而易见的。选择和描述实施例是为了更好说明本发明的原理和实际应用,并且使本领域的普通技术人员能够理解本发明从而设计适于特定用途的带有各种修改的各种实施例。