有机电致器件用化合物、利用该化合物的有机电致器件及其电子装置
文献发布时间:2023-06-19 11:32:36
技术领域
本发明涉及一种有机电致器件用化合物、利用该化合物的有机电致器件及其电子装置。
背景技术
一般而言,有机发光现象是指利用有机物质将电能转化为光能的现象。利用有机发光现象的有机电致器件通常具备阳极、阴极以及二者间包含有机物层的结构。这里的有机物层以提高有机电子原件的效率和稳定性为目的,由各类不同物质构成的多层结构所组成,比如可由空穴注入层、空穴传输层、发光层、电子传输层以及电子注入层等组成。
按照功能的不同,在有机电致器件中被用作有机物层的材料可分为:发光材料、电荷传输材料,例如空穴注入材料、空穴传输材料、电子传输材料、电子注入材料等。
就内含杂原子的多环化合物而言,由于物质结构所带来的特性差异巨大,作为有机电致器件的材料正在应用于各种层。尤其,随着环的个数与稠合(fused)位置、杂原子的种类和排列的不同,具有带隙(HOMO、LUMO)、电性能、化学性能、物理性能等不同的特征,因此正在开展针对各种有机电致器件的层的应用开发。
其典型的例子为,专利文献1到专利文献4中公开了多环化合物中的五环化合物因杂原子的种类及排列、取代基种类、稠和位置等而具有不同的性能。
专利文献1:美国注册专利5843607
专利文献2:日本公开专利1999-162650
专利文献3:韩国公开专利2008-0085000
专利文献4:美国公开专利2010-0187977
专利文献5:韩国公开专利2011-0018340
专利文献6:韩国公开专利2009-0057711
在专利文献1与专利文献2采用了五环化合物中杂原子仅由氮(N)构成的吲哚咔唑核,同时公开了使用被吲哚咔唑的N取代或未被替代的芳基的实施例。然而,所述专利文献1中只存在烷基、氨基、烷氧基等被取代基所取代或未被取代的纯芳基,不足以证明多环化合物的取代基效果,同时只说明了空穴传输材料的用途,而未说明作为磷光主体材料的用途。
专利文献3与专利文献4虽然说明了在与所述专利文献1和专利文献2相同的五环化合物中杂原子为N的吲哚咔唑4核取代分别含有芳基和N的吡啶、嘧啶、三嗪等的化合物,但只说明了磷光绿色主体材料的使用例,并未对被吲哚咔唑核取代的其他杂环化合物的性能进行说明。
专利文献5虽然将五环化合物中的杂原子记为氮(N)、氧(O)、硫(S)、碳等,但性能检测数据中仅说明了全部采用相同的同构杂原子的实施例,无法能确认内含异构杂原子的五环化合物的性能特点。
因而,所述专利文献未对内含同构杂原子的五环化合物所具备的低电荷迁移度和低氧化稳定性的解决方案进行说明。
五环化合物分子一般在堆叠时随着相邻π-电子的增多而获得强烈的电气性相互作用,这与电荷迁移度存在紧密联系。尤其,N-N形式的同构五环化合物的分子间排列顺序在分子堆叠时分子之间的排列顺序变成边对面形态;而杂原子各不相同的异构五环化合物则与之相反,分子的堆积结构变成逆向相对的反平行π-堆叠结构(antiparallel cofacialπ-stacking structure),因此分子间的排列顺序变为面对面的形态。该堆叠结构的原因为非对称分布的杂原子N而被杂原子N所取代的取代基的立体效果带来了相对较高的迁移度和高水平的氧化稳定性。(Org.Lett.2008,10,1199)
专利文献6对7环以上的各类多环化合物被用作荧光主体材料的案例进行了说明。
正如上述内容,针对多环化合物的稠合(fused)位置、环数量、杂原子的排列和种类所引发的特性变化,尚未得到充分的开发。
其原因在于,尤其对采用磷光发光掺杂剂材料的磷光型有机电致器件而言,主体材料的LUMO以及HOMO能级是严重影响有机电致器件的效率和寿命的主要因素,而它能够依据是否可高效调节发光层内的电子和空穴注入,来防止由调节发光层内电子平衡、掺杂剂淬火(quenching)以及空穴传输层表面发光所导致的效率低下和寿命缩短问题。
对于荧光和磷光发光用的主体材料,最近正在研究利用TADF(Thermalactivateddelayed fluorescent、热活化延迟荧光)、exciplex(激光复合物)等提高有机电致器件的效率以及寿命等,尤其是正在对明确能量从主体材料传递到掺杂剂材料的方法进行大量的研究。
TADF(Thermal activated delayed fluorescent、热活化延迟荧光)、exciplex(激光复合物)的发光层内传递能量有各种明确的方法,可通过PL lifetime(TRTP,发光寿命)测试方法轻易确认到。
TRTP(Time resolved transient PL,时间分辨瞬态光致发光)测试法是将脉冲光源照射于薄膜之后随着时间推移观察光谱的衰变(Decay time)的方法,通过观察能量传递以及发光延迟时间可明确能量传递方式。所述TRTP测试是可区分荧光与磷光的区分以及mixed主体材料中的能量传递方式、exciplex能量传递方式、TADF能源传递方式等的测试方法。
如此,有许多因素随着从主体材料将能量传递至掺杂剂物质的方式的不同而对效率和寿命产生影响,同时不同物质的能量传递方式也各不相同,目前用于稳定高效的电子器件的主体材料尚未得到充分开发。因此,不断产生开发新材料的要求,尤其迫切需要开发出发光层的主体材料。
发明内容
(要解决的问题)
本发明为解决所述磷光主体材料的问题而开发,其目的在于提供提高如下的有机电致器件用化合物、利用该化合物的有机电致器件及其电子装置:通过对包括磷光掺杂剂在内的磷光发光型有机电致器件的主体材料调节HOMO能级,据此调节charge balance(电荷平衡),以及提高效率、寿命。
(解决问题的手段)
本发明为了有效地调节磷光发光型有机电致器件发光层内空穴注入,在特定的第一主体材料组合特定的第二主体材料来作为主要成分并且包含该主要成分,进而能够缩小能量屏障,并且最大限度地提高发光层内的电荷平衡(charge balance),提高有机电致器件的效率、寿命。
本发明在提供一种有机电致器件,其特征在于,包含第一电极、第二电极以及形成在所述第一电极与所述第二电极之间的有机物层的有机电致器件中,所述有机物层包含发光层,所述发光层包含由以下化学式1表示的第一主体化合物以及由以下化学式2表示的第二主体化合物。
另外,本发明提供利用由所述化学式表示的化合物的有机电致器件及其电子装置。
(发明的效果)
利用本发明所涉及的混合物为磷光主体材料,可实现有机电致器件的高发光效率、低驱动电压,同时还可大幅提高器件的寿命。
附图说明
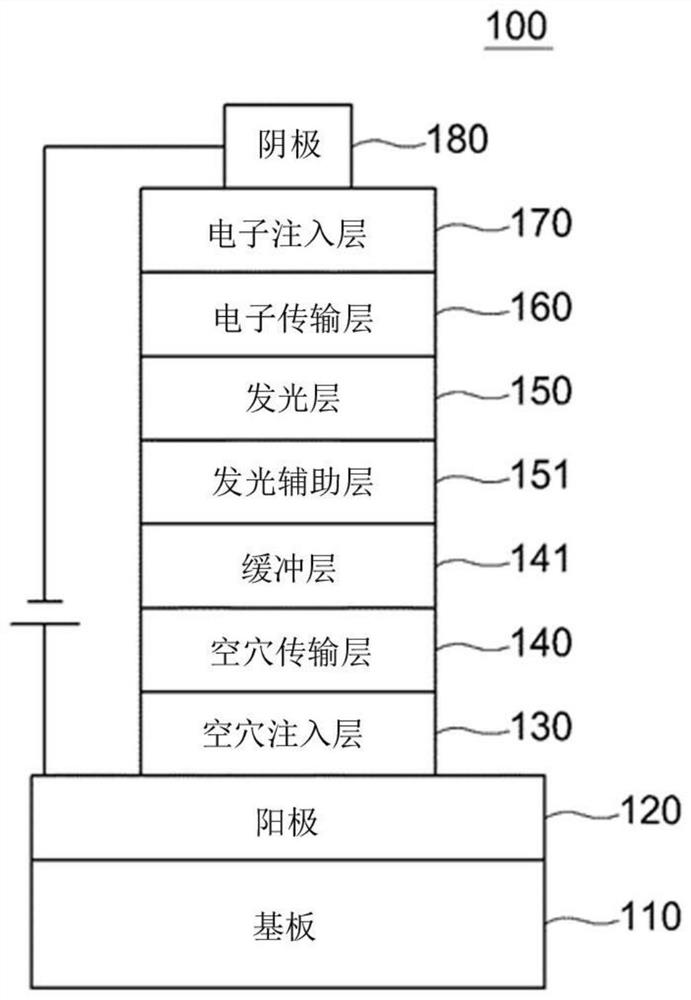
图1是本发明的一种有机电致发光器件的示例图。
图2是示出本发明的化合物1-54的PL lifetime(发光寿命)结果。
图3是示出本发明的化合物3-6的PL lifetime结果。
图4是示出混合本发明的化合物1-54与化合物3-6情况下的PL lifetime结果。
(附图标记说明)
100:有机电致器件 110:基板
120:第一电极(阳极) 130:空穴注入层
140:空穴传输层 141:缓冲层
150:发光层 151:发光辅助层
160:电子传输层 170:电子注入层
180:第二电极(阴极)
具体实施方式
下面结合本发明的实施例对本发明作出详细说明。在对本发明进行说明时,如果认为针对相关的已知结构和功能的具体说明可能混淆本发明的要点,则省略该详细说明。
此外,在介绍本发明的组成部分时可能会采用第一、第二、A、B、(a)、(b)等术语。此类术语只是为了区分该组成部分和其他组成部分,此类术语并不限于相应组成部分的本质或次序、顺序等。当某个组成部分与其他组成部分“连接”、“结合”或“接触”时,应理解为该组成部分直接与其他组成部分相连,或者虽然能够连接,但各组成部分之间还可能“连接”、“结合”或“接触”其他组成部分。
如本说明书及其附带的权利要求范围中的用法,除非另有说明,否则下列术语具有如下含义:
除非另有说明,否则本说明书所用术语“卤代”或“卤素”是指氟(F)、溴(Br)、氯(Cl)或碘(I)。
除非另有说明,否则本发明所用术语“烷基”具有碳数为1至60的单键,意味着直链烷基、支链烷基、环烷基(脂环族)、烷基-取代环烷基、环烷基-取代烷基等饱和脂肪族官能团的自由基。
除非另有说明,否则本发明所用术语“卤代烷基”或“卤素烷基”意味着被卤素取代的烷基。
本发明所用术语“杂烷基”是指构成烷基的碳原子中超过一个碳原子被杂原子取代后的产物。
除非另有说明,否则本发明所用术语“烯基”、“链烯基”或“炔基”分别具有碳数在2至60之间的双键或三键,并内含直链型或侧链型链条,但并不限于此。
除非另有说明,否则本发明所用术语“环烷基”意味着形成碳数在3至60之间的环状的烷基,但并不限于此。
本发明所用术语“烷氧基”意味着附有氧自由基的烷基,且在无另外说明的情况下碳数在1至60之间,但并不限于此。
本发明所用术语“烯氧基”意味着附有氧自由基的烯基,且在无另外说明的情况下碳数在2至60之间,但并不限于此。
本发明所用术语“芳氧基”意味着附有氧自由基的芳基,且在无另外说明的情况下碳数在6至60之间,但并不限于此。
除非另有说明,否则本发明所用术语“芳基”与“亚芳基”的碳数分别在6至60之间,但并不限于此。本发明中的芳基或亚芳基意味着单环或侧链的芳香族,且内含相邻的取代基参与结合或反应后形成的芳香族环。举例来说,芳基可以是苯基、非苯基、芴基、螺双芴基。
前缀“芳”是指被芳基取代的自由基。例如,芳烷基是被芳基取代的烷基,芳烯基是被芳基取代的烯基,被芳基取代的自由基具有本说明书所介绍的碳数。
此外,连续以前缀命名的情况意味着按照书写顺序罗列取代基。例如,芳烷氧基意味着被芳基取代的烷氧基,烷氧羰基意味着被烷氧基取代的羰基,芳羰基烯基意味着被芳羰基取代的烯基,而这里的芳羰基则是被芳基所取代的羰基。
除非另有说明,否则本说明书所用术语“杂烃基”意味着至少内含1个杂原子的烷基。除非另有说明,否则本发明所用术语“杂芳基”或“杂亚芳基”分别意味着内含1个以上杂原子且碳数在2至60之间的芳基或亚芳基,但不仅限于此,而且至少包含单环和多环中的一种,相邻的官能团结合后也能形成。
除非另有说明,否则本发明所用术语“杂环基”至少内含1个杂原子,碳数在2至60之间,且至少包含单环和多环中的一种,还内含杂脂肪族环和杂芳香族环。相邻的官能团结合后也能形成。
除非另有说明,否则本说明书所用术语“杂原子”表示N、O、S、P或Si。
此外,“杂环基”也可能含有内含SO
除非另有说明,否则本发明所用术语“脂肪族”表示碳数为1至60的脂肪族碳化氢,而“脂环”则意味着碳数为3至60的脂肪族碳化氢环。
除非另有说明,否则本发明所用术语“环”是指碳数为3至60的脂环、碳数为6至60的芳香环、碳数为2至60的杂环或者由这些环组合而成的稠环,其中包括饱和或不饱和环。
上述杂化合物之外的其他杂化合物或杂自由基至少包含1个杂原子,但并不限于此。
除非另有说明,否则本发明所用术语“羰基”是指-COR'所表示的物质,这里的R'是指氢、碳数在1至20的烷基、碳数在6至30的芳基、碳数在3至30的环烷基、碳数在2至20的烯基、碳数在2至20的炔基以及它们形成的组合。
除非另有说明,否则本发明所用术语“醚”是指-R-O-R'所表示的物质,这里的R或R'分别独立地指氢、碳数在1至20的烷基、碳数在6至30的芳基、碳数在3至30的环烷基、碳数在2至20的烯基、碳数在2至20的炔基以及它们的组合。
此外,除非明确说明,否则本发明所用的术语“取代或未取代”中的“取代”表示被选自重氢、卤素、氨基、氰基、硝基、(C
此外,除非明确说明,否则本发明所用的化学式同样适用基于下列化学式的指数定义的取代基定义。
当这里的a为0的整数时取代基R
另外,除非另有说明,否则本发明所用术语“邻位(ortho)”、“间位(meta)”、“对位(para)”是指所有取代基的取代位置,邻位(ortho)是指取代基的位置紧邻的化合物,举例说明苯的情况,是指1、2位置;间位(meta)是指紧邻取代位置的下一个取代位置,在举例说明苯时,是指1、3位置;对位(para)作为间位(meta)的下一个取代位置,在举例说明苯时,是指1、4位置。更详细如下举例说明取代位置:可以确认到邻位(ortho)、间位(meta)是非线性(non-linear)的类型(type);对位(para)是线性(linear)的类型(type)。
(对位(ortho)的示例)
(间位(meta)的示例)
(对位(para)的示例)
下面,说明根据本发明一个侧面的化合物及包含所述化合物的有机电致器件。
本发明在提供一种有机电致器件,其特征在于,包含第一电极、第二电极以及形成在所述第一电极与所述第二电极之间的有机物层的有机电致器件中,所述有机物层包含发光层,所述发光层包含由以下化学式1表示的第一主体化合物以及由以下化学式2表示的第二主体化合物。
{在上述化学式1以及2中,
Ar
a为0至4的整数;b为0至3的整数;c以及e为0至10的整数;d为0至2的整数;
3)R
4)L
5)A以及B是相互独立地为C
但是,在A以及B全部为取代或者未取代的C
6)i以及j为0或者1并且相互独立;
但是,i+j为1以上,在此,若i或者j是0,则意味着直接结合;
7)X
R’以及R”为氢、C
8)n为1或者2的整数;在n为2的情况下,2个Ar
(在此,芳基、芴基、亚芳基、杂环基、稠环基、烷基、烯基、烷氧基和芳氧基还能够分别被选自由下列基团组成的基团群的一个以上的取代基取代:重氢、卤素、被C
另外,本发明提供由所述化学式2表示的化合物。
在本发明的具体的另一方面,由所述化学式1表示的第一主体化合物包含由以下化学式3或者化学式4表示的化合物。
{在所述化学式3以及4中,R
并且,提供如下的有机电致器件:在所述化学式1中,所述L
{在所述化学式(A-1)至(A-12)中,
1)a’、c’、d’以及e’为0至4的整数;b’是0至5的整数;f’以及g’是0至3的整数;h’是0至1的整数;
2)R
或者,若所述f’以及g’为2以上,则分别作为复数相互相同或者各异,并且多个R
邻接的R
3)Y为NR’、O、S或者CR’R”;
4)R’以及R”与所述权利要求2定义的相同;
5)Z
另外,本发明的特征在于,由所述化学式1表示的第一主体化合物为以下化学式5至8中的一个。
{在所化学式5至8中,
1)R
R
3)a’、c’以及d’为0至4的整数,f’以及g’为0至3的整数;
4)Y为NR’、O、S或者CR’R”。}
另外,本发明提供如下的有机电致器件:由所述化学式1表示的第一主体化合物为由以下化学式9至20中的一种表示的化合物。
{在所述化学式9至20中,
R
另外,本发明所述化学式1表示的第一主体化合物包含由以下化学式21至化学式22表示的化合物。
{在所述化学式21以及化学式22中,R
另外,本发明提供如下的有机电致器件:在所述化学式1中n为1的情况下的化合物;在所述化学式2中n为2的情况下的化合物。
本发明提供如下的有机电致器件:由所述化学式1表示的第一主体化合物包含由以下化学式23表示。
{在所述化学式23中,
1)R
2)f为0至3的整数,g为0至4的整数;
R
4)Y为NR’、O、S或者CR’R”;
5)R’以及R”与所述权利去要求2定义的相同。}
本发明提供如下的有机电致器件:所述化学式2表示的第二主体化合物包含由以下化学式24或者化学式25表示的化合物。
{在所述化学式24以及25中,R
另外,本发明提供如下的有机电致器件:所述化学式2的A以及B选自由化学式(B-1)至(B-7)组成的群中的化合物。
{所述化学式(B-1)至(B-7)中,
1)Z
2)R’与所述权利要求2定义的相同;
3)*表示缩合的位置。}
作为本发明的具体示例,提供如下的有机电致器件:包含所述化学式2的R
举另一示例,本发明提供如下的有机电致器件:由所述化学式2表示的第二主体化合物由以下化学式26至化学式45中一种表示的化合物。
{在所述化学式26至45中,
Ar
本发明提供如下的有机电致器件:由所述化学式2表示的第二主体化合物包含由以下化学式46至化学式53表示的化合物中的一个。
{在所述化学式46至53中,
R
举本发明的具体示例,由所述化学式1表示的第一主体化合物为以下化合物1-1至1-68以及化合物2-1至2-68中的一种。
另外,在本发明中提供如下的有机电致器件,由所述化学式2表示的第二主体化合物包含以下化合物3-1至3-86中的一种。
下面参照附图1进行说明,本发明的有机电致器件100具有:形成于基板110上的第一电极120、第二电极180,以及置于所述第一电极120和所述第二电极180之间有机物层,其中所述有机物层包含由化学式1表示的化合物。此时,第一电极120可以是阳极,第二电极180可以是阴极。若为变换器型,则第一电极为阴极,第二电极为阳极。
有机物层在第一电极120上可依次包括:空穴注入层130、空穴传输层140、发光层150、电子传输层160及电子注入层170。此时,除发光层150外,可不形成其他层,也可额外包括空穴阻挡层、电子阻挡层、发光辅助层151、电子传输辅助层、缓冲层141等,电子传输层160等也可起到空穴阻挡层的作用。
虽然附图中没有示出,本发明的有机电致器件还可包括在第一电极和第二电极中至少在一侧中所述有机物层和相反侧形成的保护层。
另一方面,即使是同一核,根据在哪个位置结合何种取代基,将体现出不同的带隙、电子特性、表面特性等,因此核的选择以及与此结合的副取代体组合也非常重要,尤其是当各个有机物层间的能级及T1值、物质的固有特性(迁移率、表面特性等)等达到最佳组合时,可同时达到寿命长、效率高的目标。
根据本发明一实施例的有机电致发光器件可利用物理气相沉积(PVD,physicalvapor deposition)方法进行制造。例如,可在基板蒸镀金属或具有传导性的金属氧化物、其合金来形成阳极后,在其上形成包括空穴注入层130、空穴传输层140、发光层150、电子传输层160及电子注入层170的有机物层,之后再沉积可使用为阴极的物质而制造。
另外,在空穴传输层140和发光层150之间还可形成发光辅助层151,而在发光层150和电子传输层160之间还可形成电子传输辅助层。
由此,本发明提供一种还包括光效率改善层的有机电致器件,所述光效率改善层在所述第一电极的一侧中与所述有机物层相反的一侧或在所述第二电极的一侧中与所述有机物层相反的一侧中的至少一侧形成。
另外,本发明中所述有机物层可通过旋涂、喷嘴式涂布、喷墨涂布、狭缝涂布、浸渍涂布或双滚涂布方式中的任何一种方法形成。本发明的有机物层可通过各种方式形成,但所述形成方法不限制本发明的权利要求范围。
作为另一个具体示例,本发明提供在所述有机物层中所述发光层为磷光发光层的有机电致器件。
另外,本发明提供如下的有机电致器件:在所述有机物层的发光层中由所述化学式1以及所述化学式2表示的化合物以1:9至9:1中任意一种比例混合而成来包含在发光层。
另外,本发明提供如下的有机电致器件:在所述有机物层的发光层中由所述化学式1以及所述化学式2表示的化合物以1:9至5:5中任意一种比例混合而成来包含在发光层。更加优选为,由所述化学式1以及所述化学式2表示的化合物以2:8或者3:7的混合比例包含在发光层。
根据本发明一个实施例的有机电致器件,可根据使用材料,可为前侧发光型、后侧发光型或两面发光型。
白光有机发光二极管(WOLED,White Organic Light Emitting Device)可轻松实现高清晰度,且可加工性优秀,同时可利用现有LCD的彩色滤光技术进行制造。现有的专利主要公开用于背光源装置的白色有机发光器件。具代表性的是,从平面上并行排列R(Red)、G(Green)、B(Blue)发光部位的方式(side-by-side),上下堆积R、G、B发光层的叠层(stacking)方式,还有利用使用蓝色(B)有机发光层的电致发光和由此的光的无机荧光体的自发光(photo-luminescence)的颜色转换材料(color conversion material、CCM)方式等。而本发明亦可适用于此类WOLED上。
同时,本发明提供一种电子装置,包括:包含所述有机电致器件的显示装置;以及驱动所述显示装置的控制部。
另一方面,本发明还提供一种电子装置,其特征在于,所述有机电致器件至少是有机电子发光器件、有机太阳能电池、有机光电导体(OPC)、有机晶体管及,单色或白色照明器件中至少一种。此时,电子装置可以是当前或未来的通讯终端,包括手机等移动终端、PDA、电子词典、PMP、遥控器、导航、游戏机、各种电视机、各种电脑等所有电子装置。
以下,以实施例具体说明本发明中所述化学式1表示的化合物的合成例以及本发明的有机电致器件之制造例,但本发明不限于下列实施例。
(合成例1)
本发明的所述化学式1表示的化合物(final products 1)是通过如下反应式1反应Sub1与Sub2制造而成。
<反应式1>
Sub1合成例
在反应式1的Sub1中,若L
<反应式2>
将3-溴-9-苯基-9H-咔唑(45.1g、140mmol)溶解于DMF980mL,之后依次加入二硼酸频哪醇(39.1g、154mmol)、氯化钯(dppf)催化剂(3.43g、4.2mmol)、乙酸钾(41.3g、420mmol)之后搅拌24小时合成硼酸化合物,之后将得到的化合物经过硅胶柱层析(silicagelcolumn)及再结晶进行分离,之后得到硼酸化合物35.2g(反应收率:68%)。
将2-溴-9-苯基-9H-咔唑(76.78g、238.3mmol)、联硼酸频那醇脂(66.57g、262.1mmol)、氯化钯(dppf)(5.84g、7.1mmol)、乙酸钾(70.16、g、714.9mmol)通过与Sub1-3(1)相同的试验方法进行试验得到生成物Sub1-3(2)73.92g(反应收率:84%)。
Sub1(10)合成例
将9-苯基-3-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-9H-咔唑(29.5g、80mmol)溶解于THF 360mL,之后添加3-溴-3'-碘-1,1'-联苯(30.16g、84mmol)、四(三苯基膦)钯(2.8g、2.4mmol)、氢氧化钠(9.6g、240mmol)、水180mL之后进行搅拌回流。等反应结束,之后用乙醚(ether)与水提取,之后用硫酸镁干燥浓缩有机物层,之后对生成的有机物经过硅胶柱层析(silicagel column)及再结晶得到生成物26.56g(反应收率:70%)。
9-苯基-2-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-9H-咔唑(29.5g、80mmol)、THF 360mL、1-溴-4-碘苯(23.8g、84mmol)、四(三苯基膦)钯(2.8g、2.4mmol)、氢氧化钠(9.6g、240mmol)、水180mL通过与所述Sub1(10)相同的试验方法进行试验的得到生成物Sub1(3)22.9g(反应收率:72%)。
将9-苯基-2-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-9H-咔唑(73.92g、200.2mmol)放入圆底烧瓶内用THF 880mL溶解,之后将1-溴-2-碘代苯(85.0g、300.3mmol)、四(三苯基膦)钯(11.6g、10mmol)、碳酸钾(83g、600.6mmol)、水440mL通过与所述Sub1(10)相同的试验方法进行试验得到生成物Sub1(5)55.8g(反应收率:70%)。
将9-苯基-2-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-9H-咔唑(73.92g、200.2mmol)放入圆底烧瓶内用THF880mL溶解,之后将2-溴-7-碘二苯并[b,d]呋喃(112.0g、300.3mmol)、四(三苯基膦)钯(11.6g、10mmol)、碳酸钾(83g、600.6mmol)、水440mL通过与所述Sub1(10)相同的试验方法进行试验得到生成物Sub1(15)72.4g(反应收率:74%)。
将9-苯基-2-(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)-9H-咔唑)放入圆底烧瓶内用THF 880mL溶解,之后将1,3-二溴-5-碘苯(108.65g、300.3mmol)、四(三苯基膦)钯(11.6g、10mmol)、碳酸钾(83g、600.6mmol)、水440mL通过与所述Sub1(10)相同的试验方法进行试验得到生成物Sub1(22)69.7g(反应收率:73%)。
Sub1的示例如下,但是并不限定于此:
(表1)
Sub2合成例
反应式1的Sub2可通过如下的反应式3的反应途径合成,而且并不限定于此。
(反应式3)
在圆底烧瓶放入溴苯(37.1g、236.2mmol)用甲苯(2200
mL)溶解,之后依次添加苯胺(20g、214.8mmol)、三(二亚苄基丙酮)二钯(9.83g、10.7mmol)、三叔丁基膦(4.34g、21.5mmol)、叔丁醇钠(62g、644.3mmol)在100℃下进行搅拌。反应结束之后用乙醚与水提取,之后用硫酸镁干燥浓缩有机物层,对生成的化合物经过硅胶柱层析及再结晶,得到生成物28g(反应收率:77%)。
对3-溴二苯并[b,d]噻吩(42.8g、162.5mmol)、甲苯(1550mL)、[1,1'-联苯]-4-胺(25g、147.7mmol)、三(二亚苄基丙酮)二钯(6.76g、7.4mmol)、三叔丁基膦(3g、14.8mmol)、叔丁醇钠(42.6g、443.2mmol)使用所述Sub2-1合成法得到生成物37.9g(反应收率:73%)。
Sub2的示例如下,但并不限定于此:
(表2)
最终成品1的合成例
将Sub2-1(8.0g、47.3mmol)放入圆底烧瓶用甲苯(500mL)溶解,之后添加Sub1(6)(20.7g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(13.6g、141.8mmol)在100℃下进行搅拌。反应结束之后用二氯甲烷和水提取,再用硫酸镁干燥并浓缩有机物层,之后对生成的化合物进行硅胶柱层析及再结晶,得到生成物16.1g(反应收率:70%)。
对Sub2-35(19.4g、47.3mmol)、甲苯(500mL)、Sub1(5)(20.7g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(13.6g、141.8mmol)通过与所述1-37相同的试验方法进行试验得到生成物1-10 24.1g(反应收率:70%)。
对Sub2-2(11.6g、47.3mmol)、甲苯(500mL)、Sub1(22)(24.8g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(13.6g、141.8mmol)通过与所述1-37相同的试验方法进行试验得到生成物Inter_A-1 22.8g(反应收率:75%)。
对Sub2-13(8g、29.05mmol)、所述Inter_A-1(20.5g、32mmol)、甲苯(305mL)、三(二亚苄基丙酮)二钯(1.5g、1.6mmol)、三叔丁基膦(0.65g、3.2mmol)、叔丁醇钠(8.4g、87.2mmol)通过与所述1-37相同的试验方法进行试验得到生成物1-54 18g(反应收率:74%)。
将Sub2-46(7.2g、20mmol)、Sub1(33)(8.73g、22mmol)、三(二亚苄基丙酮)二钯(1g、1.1mmol)、三叔丁基膦(0.4g、2.2mmol)、叔丁醇钠(5.74g、60mmol)、甲苯(210mL)与所述1-37相同的试验方法进行试验得到生成物2-5 11.5g(反应收率:85%)。
将Sub2-12(9.7g、20mmol)、Sub1(34)(12.2g、22mmol)、三(二亚苄基丙酮)二钯(1.0g、1.1mmol)、三叔丁基膦(0.4g、2.2mmol)、叔丁醇钠(5.8g、60mmol)、甲苯(210mL)通过与所述1-37相同的试验方法进行试验得到生成物2-18 15.5g(反应收率:81%)。
对Sub1(35)(13.9g、24.1mmol)、Sub2-16(6.3g,28.9mmol)、三(二亚苄基丙酮)二钯(2.2g、2.4mmol)、三叔丁基膦(1g、4.8mmol)、叔丁醇钠(8.3g、86.7mmol)、甲苯(260mL)与所述1-37相同的试验方法进行试验得到生成物2-60 16.5g(反应收率:80%)。
(表3)
(合成例2)
如下反应式4所示,由本发明的化学式2表示的化合物(final product 2)是与Sub3与Sub4反应制造而成。
(反应式4)
Sub3合成例
反应式4的Sub3可通过以下反应式5的反应途径合成,而且并不限定于此。
(反应式5)
Sub3-2-1合成法
将5-溴苯并[b]萘并[1,2-d]噻吩(50g、155mmol)、联硼酸频那醇脂(43.4g、171mmol)、醋酸钾(46g、466mmol)、氯化钯(dppf)(3.8g、4.7mmol)溶解于DMF(980mL)之后在120℃下环流12小时。反应结束后,冷却反应物至常温,用二氯甲烷提取,再用水清洗。用硫酸镁干燥并浓缩有机物层,之后用二氯甲烷和甲醇溶剂对生成的有机物进行再结晶化,得到所需Sub3-2-1(45g、80%)。
将通过上述过程得到的Sub3-2-1(40g、111mmol)、溴-2-硝基苯(26.91g、133mmol)、碳酸钾(46.03g、333mmol)、四(三苯基膦)钯(7.7g、6.66mmol)放入圆底烧瓶,之后加入THF(490mL)和水(245mL)进行溶解,之后在80℃下环流12个小时。反应结束后,冷却反应物至常温,利用二氯甲烷提取,再用水清洗。用硫酸镁干燥并浓缩有机物层后利用硅胶柱层析法进行分离,得到所需Sub3-4-1(27.6g、70%)。
将在上述过程中得到的Sub3-4-1(20g、56.3mmol)和三苯基膦(37g、141mmol)溶解于邻二氯苯(235mL)后环流24个小时。反应结束后利用减压蒸馏方法除去溶剂,通过硅胶柱层析及再结晶浓缩生成物,得所需Sub3(1)(13.6g、75%)。
对5-溴苯并[b]萘并[2,1-d]噻吩(50g、160mmol)、联硼酸频那醇脂(44.6g、176mmol)、乙酸钾(47g、479mmol)、氯化钯(dppf)(3.91g、4.8mmol)、DMF(1L)通过与所述Sub3-2-1的实验方法相同的方法进行实验得到生成物Sub3-2-2(45g、78%)。
对通过上述方法得到的Sub3-2-2(40g、111mmol)、溴-2-硝基苯(26.91g、133mmol)、碳酸钾(46.03g、333mmol)、四(三苯基膦)钯(7.7g、6.7mmol)、THF(490mL)、水(245mL)通过与所述Sub3-4-1相同的试验方法进行实验得到生成物Sub3-4-2(25.6g、65%)。
对在上述过程中得到的Sub3-4-2(20g、56.3mmol)、三苯基膦(44.28g、0.17mol)、邻二氯苯(235mL)通过与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(2)(12g、66%)。
对9-溴-11-苯基-11H-苯并[a]咔唑(50g、134mmol)、联硼酸频那醇脂(37.5g、148mmol)、乙酸钾(40g、403mmol)、氯化钯(dppf)(3.3g、4.0mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-3(45.1g、80%)。
对在上述中得到的Sub3-2-3(46.1g、111mmol)、Sub3-3(26.7g、131mmol)、碳酸钾(45.6g、330mmol)、四(三苯基膦)钯(7.63g、6.6mmol)通过与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-3(29.6g、65%)。
对在上述中得到的Sub3-4-3(20.7g、50mmol)、三苯基膦(32.7g、125mmol)、邻二氯苯(205mL)通过与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(7)(13.0g、68%)。
对Sub3-1-4(55.6g、160mmol)、联硼酸频那醇脂(44.7g、176mmol)、乙酸钾(47.1g、480mmol)、氯化钯(dppf)(3.92g、4.8mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-4(47.9g、76%)。
对得到的Sub3-2-4(43.4g、110mmol)、Sub3-3(26.7g、132mmol)、碳酸钾(45.6g、330mmol)、四(三苯基膦)钯(7.63g、6.6mmol)通过与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-4(27.4g、64%)。
对在上述中得到的Sub3-4-4(19.5g、50.1mmol)、三苯基膦(32.8g、125.2mmol)、邻二氯苯(205mL)通过与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(13)(11.5g、64%)。
将Sub3-1-5(51.7g、160mmol)、联硼酸频那醇脂(44.7g、176mmol)、乙酸钾(47.1g、480mmol)、氯化钯(dppf)(3.92g、4.8mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-5(46.2g、78%)。
对在上述中得到的Sub3-2-5(40.7g、110mmol)、Sub3-3-1(33.2g、132mmol)、碳酸钾(45.6g、330mmol)、四(三苯基膦)钯(7.62g、6.6mmol)以与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-5(30.6g、67%)。
对得到的Sub3-4-5(20.8g、50.1mmol)、三苯基膦(32.8g、125mmol)、邻二氯苯(205mL)通过与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(26)(11.9g、62%)。
对Sub3-1-6(59.6g、160mmol)、联硼酸频那醇脂(44.7g、176mmol)、乙酸钾(47.1g、480mmol)、氯化钯(dppf)(3.92g、4.8mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-6(50.4g、75%)。
对在上述中得到的Sub3-2-6(46.1g、110mmol)、Sub3-3-2(33.3g、132mmol)、碳酸钾(45.6g、330mmol)、四(三苯基膦)钯(7.63g、6.6mmol)通过与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-6(32.2g、63%)。
对得到的Sub3-4-6(23.2g、50mmol)、三苯基膦(32.7g、125mmol)、邻二氯苯(205mL)通过与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(39)(14.1g、65%)。
对Sub3-1-7(47.5g、160mmol)、联硼酸频那醇脂(44.7g、176mmol)、乙酸钾(47.1g,480mmol)、氯化钯(dppf)(3.92g、4.8mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-7(43.0g、78%)。
对在上述过程中得到的Sub3-2-7(37.9g、110mmol)、Sub3-3-1(33.3g、132mmol)、碳酸钾(45.7g、330mmol)、四(三苯基膦)钯(7.64g、6.6mmol)通过与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-7(27.4g、64%)。
将在上述中得到的Sub3-4-7(19.5g、50.1mmol)、三苯基膦(32.8g、125mmol)、邻二氯苯(205mL)以与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(45)(12.0g、67%)。
对Sub3-1-8(43.7g、160mmol)、联硼酸频那醇脂(44.7g、176mmol)、乙酸钾(47.1g、480mmol)、氯化钯(dppf)(3.9g、4.8mmol)通过与所述Sub3-2-1的试验方法相同的方法进行试验得到生成物Sub3-2-8(38.4g、75%)。
对在上述过程中得到的Sub3-2-8(35.2g、110mmol)、Sub3-3-3(39.9g、132mmol)、碳酸钾(45.6g、330mmol)、四(三苯基膦)钯(7.6g、6.6mmol)通过与所述Sub3-4-1的试验方法相同的方法进行试验得到生成物Sub3-4-8(31.5g、69%)。
对在上述过程中得到的Sub3-4-8(20.8g、50mmol)、三苯基膦(32.8g、125mmol)、邻二氯苯(205mL)以与所述Sub3(1)的试验方法相同的方法进行试验得到生成物Sub3(66)(12.5g、65%)。
Sub3的示例如下,但不限定于此。
(表4)
Sub4示例
Sub4的示例如下,但不限定于此。
(表5)
最终成品2的合成例
将Sub3(1)(15.3g、47.3mmol)放入圆底烧瓶内用甲苯(500mL)溶解,之后添加Sub4-15(14.8g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.1g、5.2mmol)、叔丁醇钠(15g、156.1mmol)在100℃下进行搅拌。等反应结束后,用二氯甲烷和水提取,之后用硫酸镁干燥浓缩有机物层,之后对生成化合物进行硅胶柱层析(silicagelcolumn)及再结晶得到生成物17.0g(反应收率:68%)。
对Sub3(7)(18.1g、47.3mmol)、甲苯(500mL)、Sub4-1(8.2g、52.0mmol)、三(二亚苄基丙酮)二钯(2.0g、2.2mmol)、三叔丁基膦(0.9g、4.4mmol)、叔丁醇钠(12.7g、132mmol)利用所述3-6合成法得到最终生成物15.6g(反应收率:72%)。
对Sub3(1)(15.3g、47.3mmol)、甲苯(500mL)、Sub4-25(15.4g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物19.3g(反应收率:70%)。
对Sub3(1)(15.3g、47.3mmol)、甲苯(500mL)、Sub4-53(15.1g、52mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物19.4g(反应收率:71%)。
对Sub3(13)(16.9g、47.3mmol)、甲苯(500mL)、Sub4-55(16.1g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物20.2g(反应收率:73%)。
对Sub3(17)(18.1g、47.2mmol)、甲苯(500mL)、Sub4-56(16.7g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物19.8g(反应收率:67%)。
对Sub3(59)(20.5g、47.4mmol)、甲苯(500mL)、Sub4-57(13.7g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物20.1g(反应收率:69%)。
对Sub3(45)(16.9g、47.3mmol)、甲苯(500mL)、Sub4-58(14.6g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物20.5g(反应收率:72%)。
3-52合成例
Sub3(50)(15.8g、47.4mmol)、甲苯(500mL)、Sub4-12(20.2g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物20.0g(反应收率:66%)。
对Sub3(60)(17.7g、47.4mmol)、甲苯(500mL)、Sub4-59(17.8g、52.0mmol)、三(二亚苄基丙酮)二钯(2.4g、2.6mmol)、三叔丁基膦(1.05g、5.2mmol)、叔丁醇钠(15g、156mmol)利用所述3-6合成法得到最终生成物22.8g(反应收率:71%)。
(表6)
另一方面,说明了由所述化学式1以及2表示的本发明的示例性合成例,但这都是基于布赫瓦尔德-哈特维希偶联(Buchwald-Hartwig cross coupling)反应、铃木-宫浦偶联(Suzuki cross-coupling)反应、分子内酸诱导环化(Intramolecular acid-inducedcyclization)反应(J.mater.Chem.1999,9,2095.)、Pd(II)催化氧化环化(Pd(II)-catalyzed oxidative cyclization)反应(Org.Lett.2011,13,5504)、格氏(Grignard)反应、循环脱水(Cyclic Dehydration)反应以及PPh3介导的还原环化(PPh3-mediatedreductive cyclization)反应(J.Org.Chem.2005,70,5014.)的,除了在具体合成例明示的取代基以外,结合由化学式1以及2定义的其他取代基(R
例如,反应式1中Sub1+Sub2->Final Products1反应、反应式3中合成Sub2的反应以及在反应式4中Sub3+Sub4->Final Products2的反应全部是基于布赫瓦尔德-哈特维希偶联(Buchwald-Hartwig cross coupling)反应,反应式2中Sub1-3+Sub1-4->Sub1反应以及反应式5中Sub3-2+Sub3-3->Sub3-4反应全部基于铃木-宫浦偶联(Suzuki cross-coupling)反应,反应式5中Sub3-4->Sub3反应是基于PPh3介导的还原环化(PPh3-mediatedreductive cyclization)反应(J.Org.Chem.2005,70,5014.)。具体地说,即使结合未明示的取代基,也可进行上述反应。
首先,在形成于玻璃基板的ITO层(阳极)上真空蒸镀N1-(naphthalen-2-yl)-N4,N4-bis(4-(naphthalen-2-yl(phenyl)amino)phenyl)-N1-phenylbenzene-1,4-diamine(以下称为“2-TNATA”)膜并且厚度为60nm。之后,真空蒸镀N,N'-Bis(1-naphthalenyl)-N,N'-bis-phenyl-(1,1'-biphenyl)-4,4'-d iamine(以下称为“NPB”)并且厚度为60nm来形成空穴传输层。
在空穴传输层上部使用由化学式1与化学式2表示的所述发明化合物以3:7的比例混合而成的混合物作为主体,作为掺杂物以95:5的重量比添加(piq)2Ir(acac)[bis-(1-phenylisoquinolyl)iridium(Ⅲ)acetylaceton ate],从而在所述空穴传输层蒸镀厚度30nm的发光层。将(1,1'-联苯)-4-油酸酯)双(2-甲基-8-羟基喹啉)铝(以下简称为“BAlq”)真空蒸镀10nm厚度作为空穴阻挡层,将三(8-羟基喹啉)铝(以下简称为“Alq3”)以40nm厚度进行成膜作为电子传输层。之后作为电子注入层将作为卤化碱金属的LiF蒸镀0.2nm厚度,接着以厚度为150nm来蒸镀Al而作为阴极来使用,从而制造出有机电致发光器件。
在如上述制造的实施例及比较例中的有机电致发光器件上施加正向偏压直流电压,利用photoresearch公司生产的PR-650测量电子发光(EL)特性,并在2500cd/m2的标准亮度下利用mcscience公司制造的寿命测量设备测量了T95的寿命。下列表显示器件制造及评估结果。
(比较例1至3)
单独使用化学式2表示的化合物作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例4)
单独使用比较化合物1作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例5)
单独使用比较化合物2作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例6)
单独使用比较化合物3作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例7)
单独使用比较化合物4作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例8)
混合使用比较化合物1与比较化合物2作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例9)
混合使用比较化合物3与比较化合物4作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(比较例10)
混合使用由化学式1表示的化合物与比较化合物4作为主体,除此之外通过与所述实施例1相同的方法制造有机电致发光器件。
(表7)
TRTP(时间分辨瞬态光致发光)测试试验
(表8)
如表7的结果所示,混合由化学式1与化学式2表示的本发明的有机电致发光器件用材料作为磷光主体来使用的情况下(实施例1至100),相比于使用单一物质的器件(比较例1至7)显著改善驱动电压、效率以及寿命。
更详细地说,比较例1至比较例7为单独使用由化学式2表示的本发明的化合物、比较化合物1至比较化合物4作为磷光主体,其中相比于使用比较化合物的比较例4至比较例7,使用本发明比较化合物(3-6、3-61、3-74)的比较例1至比较例3体现出更高的效率与更长的寿命。
并且,可以确认到相比于使用所述单独物质的比较例1至比较例7,混合比较化合物1与比较化合物2或者比较化合物3与比较化合物4来作为磷光主体来使用的比较例8以及比较例9体现出更高的效率。
对比较例8与比较例9进行比较,可以确认到相比于混合具有同构氮原子的5环杂环化合物的比较例8,使用包括在5环环化合物中具有相互不同的杂原子(N、S)的异构多环环化合物的混合物的比较例9体现出更高的效率。
对比较例10与比较例9进行比较,可以确认到共同使用比较化合物3,并且将二咔唑材料(比较例9)与相当于本发明化学式1的化合物2-5分别作为主体材料来使用的情况下,使用本发明化合物2-5的比较例10体现出更高的效率与比较长的寿命,其中所述比较例10是使用在本发明化合物中相当于化学式1的化合物2-5与比较化合物3的混合物,所述比较例9是包括使用比较化合物3与比较化合物4的混合物。
另外,可以确认到相比于所述比较例1至比较例10的情况,混合化学式1与化学式2的混合物(本发明的化合物)作为主体材料来使用的实施例1至实施例100显著地体现出的高效率、高寿命以及低驱动电压。
本发明人根据上述实验结果,认为在混合化学式1的物质与化学式2的物质的情况下,除了各个物质的特性以外还具有其他新的特性,进而分别使用化学式1的(1-54)物质、化学式2的(3-6)物质、本发明混合物(1-54+3-6)如所述表8测试光致发光寿命(PLlifetime)。
如表8的结果所示,可以确认到在混合化学式1(1-54)与化学式2(3-6)(本发明的化合物)的情况下形成新的PL波长(556nm),并且可以确认到新形成的PL波长衰减和衰减时间比1-54以及3-6物质各个的衰减以及衰减时间分别增加62.6倍(3-6基准)至360倍(1-54基准)左右。据此,在混合使用本发明化合物的情况下,不仅通过各个物质具备的能级移动电子与空穴,还通过因为混合而形成的新能级的新区域(exciplex)而提高电子、空穴移动或者能量转移效率以及寿命。结果,使用所述本发明混合物的情况是混合薄膜体现出激光复合能量转移以及发光过程的重要示例。
另外,相比于使用混合比较化合物的磷光主体的比较例8至比较例10,本发明的组合更优秀的理由如下:在由不仅是电子(electron)而且还具有对空穴的稳定性、高T1等特征的化学式2表示的多环环化合物混合由空穴(hole)属性强的化学式1表示的化合物的情况下,因为高T1与高LUMO能量值而提高电子阻挡能力,并且更多的空穴(hole)快速且容易地移动于发光层。据此,增加空穴和电子的发光层中的电荷平衡(charge balance),进而在发光层内部实现良好的发光而不是在空穴传输层界面,因此还在HTL界面降低退化,进而最大限度地优化将器件整体的驱动电压、效率以及寿命。即,化学式1与化学式2的组合在电化学性方面起到协同作用,进而提高器件整体性能。
(表9)
如上述表9所示,对于本发明的化合物按照比例(2:8、3:7、4:6、5:5)分别制造元件并进行测试。
如上述结果所示,在化合物1-54与化合物3-6的混合物结构中,2:8、3:7的情况下,驱动电压、效率以及寿命的结果优秀,并且两者结果类似,而在4:6、5:5的情况,则确认到第一主体的比例增加的同时驱动电压、效率以及寿命的结果逐渐降低,据此化合物2-5与化合物3-61的混合物结果中也体现出相同的现象。这是因为如同2:8、3:7的情况,若将由空穴属性优秀的化学式1表示的化合物混合适当量,则可将发光层内电荷平衡最大化。
以上说明仅为本发明的一个示例,任何本发明所属技术领域中具有通常知识者可以在没有超出本发明本质特点的范围内进行各种变形。因此,在本说明书中所列出的实施例仅作为说明本发明,而并不限制本发明的思想和范围。本发明的权利保护范围应按照上述权利要求范围,而属于同等范围内的所有技术应被解释为包括在本发明的权利范围内。
- 有机电致器件用化合物、利用该化合物的有机电致器件及其电子装置
- 有机电致器件用化合物、利用该化合物的有机电致器件及其电子装置