一种不合成肠杆菌共同抗原和鞭毛的基因工程菌及其应用
文献发布时间:2023-06-19 11:50:46
技术领域
本发明涉及一种不合成肠杆菌共同抗原和鞭毛的基因工程菌及其应用,属于基因工程和发酵工程领域。
背景技术
在所有肠杆菌科菌株中发现的肠杆菌共同抗原(ECA)存在三种不同的形式:甘油磷脂结合型、脂多糖核结合型和环型。ECA的生物合成和组装由一个基因簇编码。ECA具有保守的重复单元结构,由三个氨基糖组成:N-乙酰氨基葡萄糖、N-乙酰-D-甘露糖醛酸和4-乙酰氨基-4,6-二脱氧-D-半乳糖。ECA在细胞膜完整性、鞭毛的生成、有机酸耐受性和细胞膜通透性等方面具有多种细胞功能。ECA也是一种毒力因子,在食品领域可能加速产品变质和病原菌群污染,增加医疗领域的治疗难度和感染风险。此外,ECA的生成可能会消耗细菌的能量和底物,这些能量和底物本应更好地用于促进细胞生长或代谢产物的产生。此外,位于外膜和周质间隙的ECA可阻碍物质交换和膜通透性,导致营养限制或工业发酵微生物生长缓慢。细菌鞭毛是另一种生物膜结构,既是运动细胞器,又是蛋白质输出/组装装置。鞭毛负责生物膜的形成、运动和趋化。此外,鞭毛介导的运动是导致发展中国家旅行者和儿童腹泻的一个关键毒性特征。
聚羟基丁酸酯(PHB)是一种广泛存在于微生物中的天然高分子生物材料。PHB在细胞内以不溶性球形包裹体或PHB颗粒的形式存在,作为生物体内的碳源和储能物质。PHB具有良好的生物相容性、生物降解性和热加工性,可作为生物医用材料和生物降解包装材料。据报道,截断脂多糖结构可以重新平衡碳和氮代谢,导致PHB的产量显著增加。目前,PHB的研究主要集中在生物合成途径的修饰、廉价原料的开发和利用以及PHB的再生和改性等方面。L-苏氨酸是一种必需氨基酸,广泛应用于医药、化学试剂、食品强化剂和饲料添加剂中。L-苏氨酸已通过微生物发酵工业化生产。L-苏氨酸的生物合成与TCA途径竞争,需要大量的能量来源,这是L-苏氨酸高产的瓶颈。L-苏氨酸的研究热点主要集中在代谢途径修饰上,如脂肪酸阻断、磷酸转移酶系统和底物再分配。
PHB和L-苏氨酸目前依然存在产量不高,生产工艺复杂,生产成本高的问题,如何解决这些问题,成为目前研究的热点和难点。
发明内容
技术问题:
本发明要解决的技术问题,是提供一种能够提高产物产量的底盘细胞的构建方法;同时提供一种能够提高产物产量的基因工程菌及其构建方法和应用。
技术方案
为了解决上述技术问题,发明人课题组考虑到ECA在大肠杆菌K-12亚群中不同领域的不良影响较多,MG1655菌株通过基因工程手段敲除所有12个参与ECA生物合成的基因,构建了ECA缺陷型WQM021,并随机选取了3种不同的产物:mCherry蛋白(蛋白质类)、PHB(碳源类)和L-苏氨酸(氨基酸类),考察WQM021对发酵产品生产的促进作用。结果表明,去除ECA可以促进细胞在LB和M9培养基中的增长,促进PHB和L-苏氨酸的生产,并促进mCherry蛋白的生产和胞外分泌。最后,根据WQM021的代谢分析,鞭毛合成基因被进一步从WQM021中敲除,获得ECA和鞭毛双缺失的突变株WQM022。研究发现,WQM022可以合成更多的PHB和L-苏氨酸,并进一步促进mCherry蛋白的生产和胞外分泌。因此,WQM021和WQM022具有作为优秀的底盘菌株的应用潜力,同时也表明,通过去除微生物非必需的膜壁结构,不仅能减少致病性风险,还可以将节约的底物和能量用于发酵产物的合成,这对其他工业微生物的优化和转化具有特殊的指导意义。
本发明提供了一种基因工程菌,所述基因工程菌以大肠杆菌为宿主,敲除大肠杆菌基因组上的肠杆菌共同抗原基因簇,所述肠杆菌共同抗原基因簇分别为wecA,wzzE,rffE,rffD,rffG,rffH,rffC,rffA,wzxE,rffT,wzyE and rffM,其序列的NCBI登录号依次为948789,944815,944789,948977,948300,948299,948298,948296,948294,2847677,948293和948301。
在本发明的一种实施方式中,所述基因工程菌还敲除了大肠杆菌基因组上的鞭毛基因簇;所述鞭毛基因簇基因分别为fliE,fliF,fliG,fliH,fliI,fliJ,fliK,fliL,fliM,fliN,fliO,fliP,fliQ,fliR,fliY,fliZ,fliA,fliC,fliD,fliS,fliT,flgN,flgM,flgA,flgB,flgC,flgD,flgE,flgF,flgG,flgH,flgI,flgJ,flgK,flgL,flhE,flhA,flhB,cheZ,cheY,cheB,cheR,tap,tar,cheW,cheA,motB,motA,flhC and flhD,其序列的NCBI登录号依次为946446,946448,946451,946456,946457,946454,946449,946443,946442,946423,946458,946462,946463,946464,948833,946833,948824,949101,946428,946429,946433,945634,946684,946300,945678,946687,945813,945636,945639,945647,946996,947534,947456,945648,945646,946094,946390,946391,946392,946393,946394,946396,946397,946399,946400,946401,946402,947564,947280和945442。
在本发明的一种实施方式中,以Escherichia coli str.K-12substr.MG1655为宿主细胞。
在本发明的一种实施方式中,所述基因工程菌的载体为:将β-酮基硫解酶、乙酰辅酶A还原酶和PHB合成酶的编码基因的PHB合成基因簇phaCAB连接到表达质粒上,构建得到载体。
在本发明的一种实施方式中,所述β-酮基硫解酶protein ID为QBK40993.1。
在本发明的一种实施方式中,所述乙酰辅酶A还原酶protein ID为QBK40994.1。
在本发明的一种实施方式中,所述PHB合成酶的protein ID为QBK40992.1。
在本发明的一种实施方式中,所述基因簇phaCAB的序列的NCBI登录号为QBK40992.1。
在本发明的一种实施方式中,所述重载体为:pBHR68。
在本发明的一种实施方式中,所述基因工程菌的载体为:将thrA*BC和rhtC基因连接到表达质粒上,构建得到载体。
在本发明的一种实施方式中,所述基因工程菌以pFW01-thrA*BC-rhtC为载体。
在本发明的一种实施方式中,所述基因工程菌的载体为:将mCherry基因连接到表达质粒上,构建得到载体。
在本发明的一种实施方式中,所述基因工程菌以pBBR1MCS2-Tac-mCherry为载体。
本发明还提供了一种利用葡萄糖发酵生产聚3-羟基丁酸酯(PHB)的方法,所述方法为,采用上述含有PHB合成基因的重组载体的基因工程菌发酵制备。
在本发明的一种实施方式中,所述方法为:菌株在LB固体培养基上活化培养10-15h,接种到培养基中,置于35-38℃,150-250rpm条件下培养5-10h得到种子液,将制备得到的种子液按照5%(v/v)的接种量接种于发酵培养基,于35-38℃,150-250rpm条件下培养40-50h。
在一种实施方式中,所述发酵培养基包括:葡萄糖15-20g/L,Na
本发明还提供了一种提高重组蛋白胞外分泌的方法,所述方法为,将上述含有mCherry基因的重组载体的基因工程菌发酵制备。
在一种实施方式中,菌株在LB固体培养基上活化培养10-15h,接种到培养基中,置于35-38℃,150-250rpm条件下培养5-10h得到种子液,将制备得到的种子液按照1%(v/v)的接种量接种于发酵培养基,于35-38℃,150-250rpm条件下培养12-14h。
在一种实施方式中,生产mCherry重组蛋白的发酵培养基,其组成包括:酵母粉1-5g/L,蛋白胨5-15g/L,NaCl 5-15g/L。
本发明还提供上述基因工程菌在表达目的蛋白中的应用。
本发明还提供了的基因工程菌在制备含有目的产物中的应用,所述目的产物为含有PHB的化学品,或含有L-苏氨酸的食品或化学品;所述基因工程菌为含有PHB合成基因的表达载体的基因工程菌或含有L-苏氨酸合成基因的表达载体的基因工程菌。
有益效果
(1)本发明在大肠杆菌中敲除大肠杆菌基因组上ECA基因簇的12个基因得到突变菌WQM021,WQM021菌株在营养丰富(LB)和营养缺乏(M9)培养基中生长变快,菌体总量增多,并且WQM021的细胞通透性明显增加。随后将mCherry重组蛋白、PHB和L-苏氨酸合成的三个质粒分别转化到ECA缺失菌株WQM021中,得到的重组菌WQM021/pBBR1MCS2-Tac-mCherry、WQM021/pBHR68和WQM021/pFW01-thrA*BC-rhtC可以高效合成PHB和L-苏氨酸,并促进mCherry重组蛋白的生产和胞外分泌。
(2)本发明构建得到的WQM021/pBHR026合成PHB的细胞干重(DCW)、PHB浓度和转化效率显著提高,分别达到2.60g/L、49.33%和8.02%,分别比MG1655/pBHR026提高了1.67倍、1.93倍和3.21倍,对照仅达到1.56g/L、25.54%和2.50%。
当WQM021用于合成苏氨酸时,WQM021/pFW01-thrA*BC-rhtC的L-苏氨酸产量、葡萄糖转化率和每小时L-苏氨酸产量分别达到1.44g/L、8.99%、0.50g/h。
(3)本发明在WQM021的基础上继续敲除鞭毛合成的50个基因,构建了WQM022,将pBHR68质粒导入WQM022中,PHB产量进一步增加;WQM022/pBHR68的DCW,PHB浓度和转化效率进一步提高,分别达到3.03g/L,78.95%,15.78%,比WQM021/pBHR68提高15.54%,60.04%和96.76%。
将含有L-苏氨酸生物合成和转运关键基因的质粒pFW01-thrA*BC-rhtC导入WQM022,得到重组菌株WQM022/pFW01-thrA*BC-rhtC;该菌株每小时L-苏氨酸产量,葡萄糖转化效率和L-苏氨酸产量分别达到1.83g/L,11.42%,0.63g/h。
附图说明
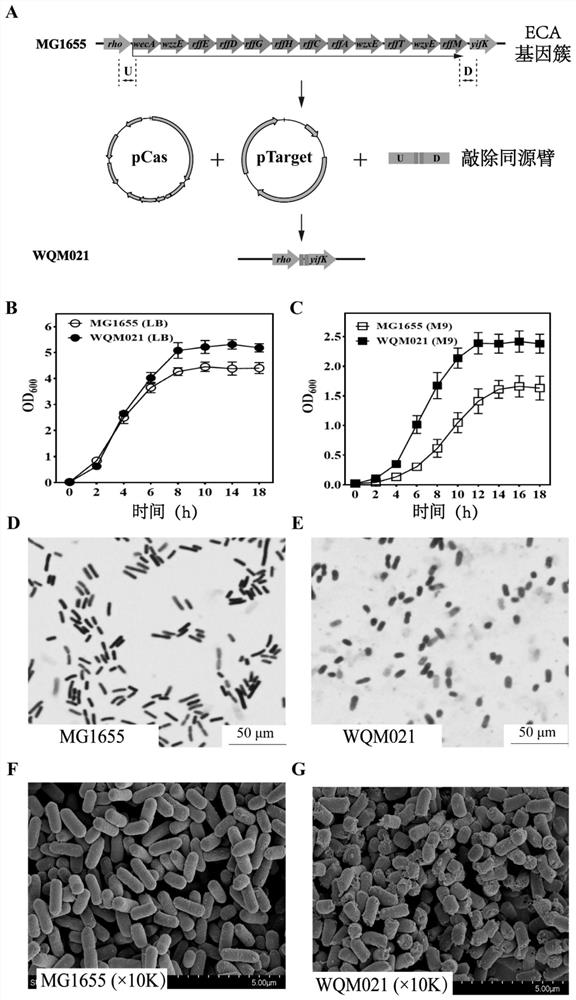
图1:ECA生物合成基因簇及MG1655、WQM021菌株的生长和形态变化;
其中,(A)ECA生物合成基因簇;(B)MG1655和WQM021在LB培养基中的生长曲线;(C)MG1655和WQM021在M9培养基中的生长曲线;(D)用光学显微镜观察MG1655的形态;(E)用光学显微镜观察WQM021的形态;(F)用扫描电镜观察MG1655的形态;(G)用扫描电镜观察WQM021的形态。
图2:MG1655和WQM021的膜通透性、重组蛋白分泌和自聚集分析;其中,(A)膜通透性分析;(B)Mcherry荧光蛋白的表达与分泌;(C)自聚集性分析。
图3:ECA缺失突变体WQM021用于PHB的生产;其中,(A)甲基酯化PHB的气相色谱保留时间;(B)GC-MS分析PHB的结构;(C)MG1655/pBHR68和WQM021/pBHR68生产PHB;(D)MG1655和WQM021胞内乙酰辅酶A含量。
图4:WQM022的构建及其在PHB和L-苏氨酸发酵中的应用;其中,(A)WQM022的构建;(B)利用WQM022进行PHB的生物合成;(C)利用WQM022生产L-苏氨酸。
具体实施方式
下述实施例中涉及的大肠杆菌JM109、pBSK质粒购自NEB。
(1)下述实施例中所涉及的质粒pBHR68的构建方法如下:
质粒pBHR68公开于SCI论文(Spiekermann,P.,Rehm,B.H.,Kalscheuer,R.,Baumeister,D.,Steinbuchel,A.,1999.Asensitive,viable-colony staining methodusing Nile red for direct screening of bacteria that accumulatepolyhydroxyalkanoic acids and other lipid storagecompounds.Arch.Microbiol.171(2),73-80,公开日:1999.01.17),其含有phaCAB基因簇并含有氨苄青霉素抗性标记,其中phaCAB基因簇含有β-酮基硫解酶、乙酰辅酶A还原酶和PHB合成酶的编码基因。
(2)基因的敲除方法
采用CRISPR/Cas9敲除系统对大肠杆菌进行“无痕”基因敲除。首先向大肠杆菌(Escherichia coli str.K-12substr.MG1655)中电转入pCas,通过L-阿拉伯糖诱导,使重组酶Gam、Bet和Exo表达。然后将同源臂片段和含有特定N20序列的pTargetF质粒同时电转入MG1655/Cas9感受态细胞。涂抗性平板,30℃培养18小时后,菌落PCR筛选突变菌株。
获得突变菌株后,向培养基中添加异丙基-β-D-硫代半乳糖苷(IPTG),诱导pCas转录形成sgRNA-pMB1,结合Cas9对pTargetF的pMB1复制子进行切割破坏,去除pTargetF。而含有pCas的菌株可直接进行下一轮的敲除,将含有pCas的菌株于42℃下过夜培养,去除温敏质粒pCas。
(3)生长曲线和细胞显微观察
通过每2小时监测600nm处的OD值来测量细胞生长曲线。为了观察细胞形态,大肠杆菌细胞在固体LB培养基上生长12小时,用1%结晶紫进行光学显微镜检查,并用油浸透镜(放大倍数100)和冷冻发射扫描电子显微镜(SEM,日立SU8220,日本东京)进行显微镜观察。
(4)PHB的提取、定性和定量分析
离心收集携带pBHR68的大肠杆菌细胞5mL,用pH 7.4的PBS缓冲液洗涤两次,离心收集后,将细胞转移到预先准确称重的离心管中,包上保鲜膜,并扎出多个小洞,使得水分可以蒸干,又避免菌液喷出,在真空冷冻干燥机中冻干48h至完全干燥,一般触摸管底为常温状态,表明已经完全干透,或者用手指轻弹管底,菌体可以散开并轻易脱离管壁,则视为菌体完全干燥。称重,计算出干重。
称取1-10mg完全干燥的菌体,同时称取PHB标准样品约10mg转移至预先准确称重的酯化管中,以便准确计算称得的样品重量。然后进行酯化操作,加入2mL甲醇(含有3%硫酸)和2mL氯仿,加上酯化管盖子并盖紧,期间可以超声,帮助样品散开,使得酯化更彻底,沸水浴6h以上。大约半小时后,即可观察到PHB标样一般以粉末形式迅速熔化。若未观察到半小时后标准品的形态变化与溶解,则应更换整套试剂,甲醇和氯仿的变质都会导致酯化无法正常进行,而更换新开封的试剂往往可以解决上述问题,经过六小时沸水浴后,在通风橱中冷却充分后,小心打开酯化管并加入1mL去离子水,这时需要分别将盖子旋紧并激烈震荡至体系完全充分混匀,此时将酯化管置于通风处中静置3h以上以分相,分相后,上层为水相,下层为有机相,取适量有机相加入到气相样品瓶中,盖紧盖子保持密封,保存在-80℃中。
气相色谱是定量PHB含量的准确灵敏的手段;采用Scion-SQ-456-GC模块,配备DB-5MS熔融石英毛细管柱(30m×0.25mm×0.25μm),对提取物进行分析,以确定其PHA组成和准确含量。气相色谱用70ev的电离能获得了正电子电离(EI),并用扫描间隔为0.5s的m/z50到m/z650的离子对质谱进行了编程。
PHB产量(wt%)以干细胞重量(DCW)百分比表示。气相定量采用岛津GC 2010气相色谱,使用安捷伦DB WAX 30m-0.32mm气相色谱柱和火焰离子化检测器,进样温度为250℃。以不同质量的商业化PHB作为标样绘制各标准样品的标准曲线。
(5)膜通透性和荧光蛋白分析
大肠杆菌细胞培养过夜,离心,洗涤2次,再悬液至1.92mL磷酸盐缓冲液中,使OD
(6)L-苏氨酸和乙酰辅酶A分析
通过离心获得的培养上清液适当稀释以分析细胞外氨基酸水平。配备Thermo250mm×4.0mm ODS-2HYPERSIL C18色谱柱的Agilent 1200或1260系列HPLC系统用于根据邻苯二甲醛柱前衍生化方法定量检测L-苏氨酸。
大肠杆菌细胞在M9培养基中培养至对数生长早期中期(OD600=0.8-1.0),用乙酰辅酶A测定试剂盒收集细胞内ATP和乙酰辅酶A水平。
(7)下述实施例中所涉及的培养基如下:
LB液体培养基(g/L):酵母粉5,蛋白胨10和NaCl 10。
M9液体培养基(g/L):葡萄糖4,Na
LBG培养基(g/L):葡萄糖20,酵母粉5,蛋白胨10和NaCl 10。
M9G培养基(g/L):葡萄糖20,Na
(8)下述实施例中所涉及的菌株或质粒的来源如表1所示:
表1在本发明构建中使用的菌株或质粒
实施例1:ECA和鞭毛缺失工程菌的构建
具体步骤如下:
(1)MG1655/pCas感受态细胞细胞制备:
接种带有pCas9质粒(Jiang,Y.,Chen,B.,Duan,C.,Sun,B.,Yang,J.,Yang,S.2015.Multigene editing in the Escherichia coli genome via the CRISPR-Cas9system.Appl Environ Microbiol,81(7),2506-14.)的Escherichia coli str.K-12substr.MG1655,于含100μg/mL卡那霉素的LB液体培养基中,30℃,200rpm培养12h,制备得到种子液;将制备得到的种子液按2%(v/v)接种量转接100mL LB液体培养基,30℃,200rpm培养至OD
(2)敲除质粒的构建
本发明中使用的质粒和引物分别列于表1和表2中。为了构建敲除质粒,使用引物F-sgRNA-wecA-rffM/R-sgRNA,F-sgRNA-fliE-R/R-sgRNA,F-sgRNA-fliY-T/R-sgRNA,F-sgRNA-flgN-L/R-sgRNA,和F-sgRNA-flhD-E/R-sgRNA以pTargetF为模板,通过反向PCR获得质粒片段pTargetF21,pTargetF21-1,pTargetF21-2,pTargetF21-3和pTargetF21-4。经过DpnI消化模板质粒,用T4 DNA连接酶连接环化载体片段,并分别转化到大肠杆菌JM109中,获得敲除质粒。引物T-wecA-rffM/T-sgRNA,T-fliE-R/T-sgRNA,T-fliY-T/T-sgRNA,T-flgN-L/T-sgRNA和T-flhD-E/T-sgRNA分别用于菌落PCR确认质粒的正确性。
(3)重组菌株的构建
通过CRISPR-Cas9双质粒无痕敲除系统,从Escherichia coli str.K-12substr.MG1655基因组中,敲除ECA生物合成相关的基因wecA,wzzE,rffE,rffD,rffG,rffH,rffC,rffA,wzxE,rffT,wzyE,rffM(图1A),构建了ECA缺陷型大肠杆菌WQM021。
使用引物对F1-wecA-rffM/R1-wecA-rffM and F2-wecA-rffM/R2-wecA-rffM扩增上游和下游臂片段。通过凝胶提取试剂盒纯化这两种PCR产物,并使用引物F1-wecA-rffM/R2-wecA-rffM重叠PCR,获得敲除同源臂DNA片段。将同源臂片段(400ng)和pTargetF21(100ng)混合并电转化到80μL MG1655/pCas感受态细胞中。将细胞在LB培养基,30℃培养下,培养1小时,使细胞愈合,然后将转化子涂布在含有壮观霉素和卡那霉素的LB琼脂平板上。30℃培养36h后,使用F1-wecA-rffM/R2-wecA-rffM引物,菌落PCR确认重组菌株的正确性。随后,正确的突变菌株,经IPTG诱导过夜后去除pTargetF21。接下来突变株在42℃下生长24h,去除pCas质粒。通过在含有壮观霉素或卡那霉素的平板上进行反向筛选来确认pTargetF21和pCas已经被去除。
在WQM021的基础上,同样采用CRISPR-Cas9双质粒无痕敲除系统,及相应的引物,继续逐一敲除鞭毛合成的4个基因簇,构建了ECA和鞭毛双缺失的突变株WQM022(图4A),WQM022敲除的基因包括wecA,wzzE,rffE,rffD,rffG,rffH,rffC,rffA,wzxE,rffT,wzyE,rffM,fliE,fliF,fliG,fliH,fliI,fliJ,fliK,fliL,fliM,fliN,fliO,fliP,fliQ,fliR,fliY,fliZ,fliA,fliC,fliD,fliS,fliT,flgN,flgM,flgA,flgB,flgC,flgD,flgE,flgF,flgG,flgH,flgI,flgJ,flgK,flgL,flhE,flhA,flhB,cheZ,cheY,cheB,cheR,tap,tar,cheW,cheA,motB,motA,flhC and flhD,共计62个基因。
PBHR68质粒是从pBluescript SK-衍生而来,用于合成PHB。将pBHR68分别电转化到Escherichia coli str.K-12substr.MG1655、WQM021和WQM022中,得到重组基因工程菌MG1655/pBHR68、WQM021/pBHR68、WQM022/pBHR68,用于PHB合成。
同样,将质粒pFW01-thrA*BC-rhtC分别电转化到Escherichia coli str.K-12substr.MG1655、WQM021和WQM022中,制备得到菌株MG1655/pFW01-thrA*BC-rhtC,WQM021/pFW01-thrA*BC-rhtC和WQM022/pFW01-thrA*BC-rhtC,用于研究L-苏氨酸的合成。本部分涉及的菌株、质粒和引物如表1和2所示。
表2在本发明构建中使用的引物
实施例2:ECA缺失菌株WQM021的形态和生长变化
将实施例1构建得到的ECA缺失菌株WQM021和野生型菌株MG1655用于本实施例中菌株的形态和生长变化的研究。其中,通过Crispr-Cas9方法从Escherichia coli str.K-12substr.MG1655中敲除包含12个与ECA的生物合成和组装相关的基因的基因(wecA,wzzE,rffE,rffD,rffG,rffH,rffC,rffA,wzxE,rffT,wzyE,rffM),导致ECA缺失菌株WQM021(图1A)。
具体步骤如下:
将菌株MG1655和WQM021分别接种至LB液体培养基和M9液体培养基中,在37℃,200rpm条件下培养18h。生长状况如表3、图1B和1C所示:
表3:WQM021和MG1655的生长状况
结果显示:在LB和M9液体培养基中,WQM021生长更好,生物量大于MG1655(如图1B和1C所示),并且在M9培养基中,WQM021比MG1655生长的快。与LB培养基相比,M9液体培养基营养贫乏。当在M9液体培养基中生长时,MG1655细胞必须合成用于ECA合成的一些必需底物,而在WQM021中,可以节省用于ECA合成的底物和能量并用于促进细胞生长。
结果表明,去除大肠杆菌中的ECA有利于细胞生长,特别是在营养饥饿条件下,通过显微镜观察MG1655和WQM021细胞,MG1655细胞显示出长棒状(图1D);而WQM021细胞长度变短(图1E),SEM分析显示MG1655细胞具有完整的长杆形式(图1F),而WQM021细胞较短(图1G)。此外,在WQM021细胞表面观察到一些附着物(图1G),表明由于不存在ECA,WQM021的生物膜可能受损。
实施例3:基因工程菌WQM021在重组蛋白生产中的应用
外膜透性分析通常采用NPN法。当NPN从上清液进入细胞时会变得非常强的荧光,而在水环境中的荧光很弱。分别对实施例1构建得到的ECA缺失菌株WQM021和野生型菌株Escherichia coli str.K-12substr.MG1655的膜通透性和荧光蛋白分析,结果表明:与对照野生型菌株MG1655(图2A)相比,WQM021的膜透性提高了69.71%,表明大肠杆菌中缺少ECA可以提高细胞通透性。
此外,通过表达和分泌胞内重组mCherry荧光蛋白进一步研究WQM021的膜通透性。
将pBBR1MCS2-Tac-mCherry质粒分别导入野生型菌株Escherichia coli str.K-12substr.MG1655和ECA缺失菌株WQM021;分别制备得到重组菌株MG1655/pBBR1MCS2-Tac-mCherry和WQM021/pBBR1MCS2-Tac-mCherry。
分别将上述制备得到的菌株接种至LB液体培养基中,在37℃,200rpm条件下培养12h后,分析了mCherry荧光蛋白在MG1655/pBBR1MCS2-Tac-mCherry和WQM021/pBBR1MCS2-Tac-mCherry中的表达水平(结果如图2B所示)。结果如表4所示:
表4:不同的菌株胞外和总荧光强度
结果显示:WQM021/pBBR1MCS2-Tac-mCherry的胞外荧光强度(EFI)高于MG1655/pBBR1MCS2-Tac-mCherry。并且,WQM021/pBBR1MCS2-Tac-mCherry的胞外和胞内总荧光强度(TFI)也高于MG1655/pBBR1MCS2-Tac-mCherry。
这表明大肠杆菌中ECA的缺失可以提高膜透性,WQM021从ECA合成中节省的资源可用于促进mCherry荧光蛋白的生产。此外,WQM021的自聚集性有显著提高,达到93.3%,比MG1655提高22.97%(图2C)。这可能是由于在没有ECA(图1G)的情况下产生的胞外絮凝物积累造成。
实施例4:基因工程菌在合成PHB的应用
1、简化大肠杆菌生物膜可以重新平衡碳和氮代谢,导致PHB产量显着增加。因此,对WQM021在PHB生产中的应用进行了研究。分别采用实施例1构建得到的WQM021/pBHR68和MG1655/pBHR68发酵制备PHB,具体步骤如下:
(1)种子液的培养
分别将上述菌株在LB固体培养基上,37℃培养10-15h后,制备得到单菌落,分别将制备得到的单菌落接种到LB液体培养基中,置于35-38℃,150-250rpm条件下培养5-10h,分别制备得到种子液。
(2)发酵生产PHB
培养方法:
将步骤(1)制备得到的种子液按5%(v/v)接种量接种于发酵培养基,置于35-38℃,150-250rpm条件下培养40-50h。
发酵培养基:葡萄糖15-20g/L,Na
将用于PHB生产的pBHR68引入大肠杆菌通常会生物合成PHB,但也有文献报道可以产生其他类型的聚羟基烷酸盐。因此,本发明采用气相色谱-质谱联用技术测定聚羟基烷酸酯的种类和数量。
分别对重组菌株MG1655/pBHR68和WQM021/pBHR68制备的聚羟基烷酸酯进行萃取、甲基酯化和分析。在GC谱图中,聚羟基烷酸的峰保留时间为5.508min,与PHB标准品的峰保留时间完全一致(图3A)。分别对WQM021/pBHR68、MG1655/pBHR68制备的PHB进行质谱分析,也显示出与标准PHB相同的图谱(图3B)。这证实了MG1655/pBHR68、WQM021/pBHR68合成的聚羟基烷酸酯为PHB。结果如表5、图3C、图3D所示。
如图3C所示,WQM021/pBHR026的DCW、PHB浓度和转化效率显著提高,分别达到2.60g/L、49.33%和8.02%,分别比MG1655/pBHR026提高了1.67倍、1.93倍和3.21倍,对照仅达到1.56g/L、25.54%和2.50%。
乙酰辅酶A是PHB合成的直接前体。为探讨ECA缺失导致大肠杆菌产生更多PHB的原因,测定了WQM021细胞内乙酰辅酶A的浓度。WQM021/pBHR026中的细胞内乙酰辅酶A水平显著高于MG1655/pBHR026,达到4.44nmol/g,比MG1655/pBHR026提高1.52倍,仅达到1.76nmol/g(图3D)。这很好地解释了为什么去除ECA有利于大肠杆菌中PHB的产生,并表明这种ECA缺失的WQM021可能是高效PHB生产的潜在宿主。
2、通过敲除ECA和鞭毛合成的62个基因构建WQM022(图4A)。然后,将pBHR68质粒导入WQM022中以研究它是否可以进一步促进PHB合成。
具体制备过程同步骤1,区别在于,将菌株更换为实施例1制备得到的WQM022/pBHR68,结果如表5、图4B所示:
表5:不同菌株制备得到的干重、PHB、转化率
如图4B所示,通过过表达编码PHB的必需基因,与WQM021/pBHR68相比,PHB产生进一步增强。WQM022/pBHR68的DCW,PHB浓度和转化效率进一步提高,分别达到3.03g/L,78.95%,15.78%,比WQM021/pBHR68提高15.54%,60.04%和96.76%。
实施例5:基因工程菌在合成L-苏氨酸中的应用
由于大肠杆菌已被开发用于生产L-苏氨酸,WQM021和WQM022高效生产L-苏氨酸的潜力也被进一步研究。分别采用实施例1制备得到的基因工程菌株MG1655/pFW01-thrA*BC-rhtC、WQM021/pFW01-thrA*BC-rhtC和WQM022/pFW01-thrA*BC-rhtC发酵制备L-苏氨酸。
(1)种子液的培养
菌株在LB固体培养基上在37℃培养10-15h,制备得到单菌落,分别将制备得到的单菌落接种到LB培养基中,置于35-38℃,150-250rpm条件下培养5-10h得到种子液。
(2)发酵生产L-苏氨酸
培养方法:
将步骤(1)制备得到的种子液按10%(v/v)接种量接种于发酵培养基,置于35-38℃,150-250rpm条件下培养30-40h。
发酵培养基,其组成包括:酵母粉1-5g/L,柠檬酸1-5g/L,(NH
分别对基因工程菌株MG1655/pFW01-thrA*BC-rhtC、WQM021/pFW01-thrA*BC-rhtC和WQM022/pFW01-thrA*BC-rhtC的发酵液进行L-苏氨酸检测,其结果如表6所示:
表6:不同的菌株对苏氨酸的产量、葡萄糖转化率、苏氨酸生产效率
如图4C所示,WQM021/pFW01-thrA*BC-rhtC的L-苏氨酸产量、葡萄糖转化率和每小时L-苏氨酸产量分别达到1.44g/L、9.00%、0.50g/h,比MG1655/pFW01-thrA*BC-rhtC分别提高了39.81%、39.97%和85.19%,仅达到1.03g/L、6.43%和0.27g/h,WQM022/pFW01-thrA*BC-rhtC比WQM021/pFW01-thrA*BC-rhtC合成更多的L-苏氨酸。
如图4C所示,WQM021/pFW01-thrA*BC-rhtC的L-苏氨酸产量、葡萄糖转化率和每小时L-苏氨酸产量分别达到1.44g/L、9.00%、0.50g/h,比MG1655/pFW01-thrA*BC-rhtC分别提高了39.81%、39.97%和85.19%,仅达到1.03g/L、6.43%和0.27g/h,WQM022/pFW01-thrA*BC-rhtC比WQM021/pFW01-thrA*BC-rhtC合成更多的L-苏氨酸。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 一种不合成肠杆菌共同抗原和鞭毛的基因工程菌及其应用
- 一种猪源产肠毒素大肠杆菌鞭毛蛋白3FliCon融合蛋白及其应用