一种检测亚硝酸盐自组装荧光探针的制备方法及其制备的荧光探针和应用
文献发布时间:2023-06-19 12:24:27
技术领域
本发明涉及荧光探针技术领域,尤其涉及一种检测亚硝酸盐自组装荧光探针的制备方法及其制备的荧光探针和应用。
背景技术
亚硝酸盐是一种致癌物质,是四大食品污染物之一,对人的生命造成威胁,因此对水中、食品和环境中亚硝酸盐的快速检测是极为重要的。目前开发的检测亚硝酸盐的方法很多种,比如:荧光光谱法、分光光度法、化学发光法、电化学检测法等。其中分光光度法具有较差的灵敏度,抗干扰能力较差,对检测环境要求较高;化学发光法由于不需要激发光源,因此没有关于光散射,具有光源不稳定或高背景的问题;电化学方法由于机械抛光效果不稳定,存在重现性较差的缺点。
荧光光谱法检测主要分为两类,一类是基于亚硝化反应检测NO
另一类是基于重氮化反应检测NO
公开号为CN110669026A的专利文件中,得到检测亚硝酸盐的荧光探针分子2-(2-氨基-4-R2-5-R1苯基)苯并噻唑,通过荧光信号强度变化检测亚硝酸盐含量,能够实现亚硝酸盐的快速检测,制备方法简单,但生物相容性差;
公开号为CN112280552A的专利文件中,公开的一种Dye-UCNPs 纳米探针的制备与亚硝酸盐的检测方法,但并不绿色环保;
公开号为CN108530459B的专利文件中,傅罗琴等公开的一种苯并噻唑-罗丹明类化合物,制备简单产率高,但检测体系为毒性环境;
公开号为CN111995573A的专利文件中公开了一类喹啉类化合物水溶性好,但制备时间过长;
公开号为CN110016336A的专利文件中以蒽酰亚胺为荧光团,邻苯二胺为识别基团的衍生物制备了一种荧光探针,制备成本低,但过程较繁琐;
公开号为CN110849856A的专利文件中提供了一种具有聚集诱导发光性能的水杨醛腙衍生物,但检出限较高。
综上,现存的荧光纳米团簇及荧光检测亚硝酸盐的方法具有检测速度快,灵敏度高等优点,但是这些荧光探针或荧光检测方法还存在一定的不足,例如生物相容性较低,合成较复杂,溶解性较差,和较高毒性等问题。
发明内容
本发明的目的是针对上述问题,提供一种检测亚硝酸盐自组装荧光探针的制备方法及其制备的荧光探针和应用。
为达到上述目的,本发明采用了下列技术方案:
一种检测亚硝酸盐自组装荧光探针的制备方法,包括以下步骤:
S1、制备不同粒径的纳米金,利用荧光光谱,以200nm为起点,以3~8nm为增长幅度连续改变激发光波长,直至最高发射峰的峰值不再升高,取1cm荧光比色皿,用移液枪取纳米金2mL于比色皿中;用荧光光谱仪进行扫描,观察其荧光光谱;
S2、取1cm荧光比色皿,用移液枪取纳米金2mL于比色皿中,以步骤S1测得的发射峰的最高峰值作为发射波长,观察其荧光光谱,测定纳米金的激发波长;
S3、取制备好的纳米金,利用荧光光谱仪在400~600nm波长下进行扫描,观察其出峰情况,确定最高吸收峰出现时的波长;再加入浓度为25μmol/LMb进行结合,每加0.02mL反应一段时间后进行1次荧光扫描,直至峰值猝灭,停止加入Mb,此时由Mb与AuNPs自组装制备的荧光纳米团簇Mb-AuNPs形成;
S4、纳米团簇制备好后,加入浓度为0.3×10-
进一步地,步骤S3中,上述纳米金的粒径为10~22nm,优选为 16.4nm。
进一步地,步骤S3中,每加0.02mLMb反应3~8min后进行1次荧光扫描。
进一步地,步骤S4中,上述避光搅拌的时间为18~32h,优选为 24h。
进一步地,步骤S4中,上述SH-β-CD的加入量为0.25~1.25mL 优选为1mL。
一种用于检测亚硝酸盐自组装荧光探针的制备方法与在亚硝酸盐检测中的应用,该应用包括以下步骤:
1)取制备好的SH-β-CD@Mb-AuNPs荧光探针于烧杯中,加入柠檬酸钠缓冲液,混合均匀后,测定荧光强度;
2)在SH-β-CD@Mb-AuNPs荧光探针中,分别加入柠檬酸钠缓冲液和已知不同浓度的亚硝酸盐溶液,溶液混合均匀后,测定荧光光谱,根据荧光数据绘制回归方程;亚硝酸盐溶液浓度c在0.01~1μmol/L和 1~5μmol/L范围内与荧光强度变化△F均呈现出可靠的线性关系,该荧光探针对亚硝酸盐的定量检测性能良好,线性方程为:
△F
3)需要测定未知浓度的亚硝酸盐溶液浓度时,在 SH-β-CD@Mb-AuNPs荧光探针中,分别加入柠檬酸钠缓冲液和未知浓度的亚硝酸盐溶液,溶液混合均匀后,测定荧光光谱,根据回归方程计算出亚硝酸盐溶液的浓度。
进一步地,步骤1)和步骤2)中,上述测定荧光强度的激发波长为200~300nm。
进一步地,步骤1)和步骤2)中,上述柠檬酸钠缓冲液的浓度为 0.1~0.5mol/L,优选为0.1mol/L,上述柠檬酸钠缓冲液的pH为4~8,优选pH为4。
本发明的有益效果是:
本发明的荧光探针相比于现存的亚硝酸盐检测的荧光探针具备绿色无毒、水溶性好、合成简单、制备时间短、生物相容性良好等优点。
本发明的亚硝酸盐检测方法具备成本低、制备简单、检测速度快、灵敏度高、检测限低的优点,检出限可低至0.13nmol/L。
本发明通过对荧光探针制备条件与检测体系的优化,得到了性能更好以及更稳定的荧光探针。
当然,实施本发明的任一产品并不一定需要同时达到以上的所有优点。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1是本发明的15.0nm的荧光光谱的激发波长规律图;
图2是本发明的15.0nm纳米金在发射波长552nm处的荧光强度图;
图3是本发明的Mb与AuNPs反应时间与荧光探针合成的关系图;
图4是本发明的Mb-AuNPs的荧光光谱表征图;
图5是本发明的Mb-AuNPs的紫外光谱表征图;
图6是本发明的Mb-AuNPs的红外光谱表征图;
图7是本发明的Mb-AuNPs的透射电镜扫描图;
图8是本发明的不同粒径纳米金与紫外吸收峰和吸光值的关系图;
图9是本发明不同粒径纳米金与紫外红移程度的关系图;
图10是本发明的不同粒径纳米金与荧光强度的关系图;
图11是本发明的Mb-AuNPs与SH-β-CD自组装过程中不同搅拌时间与紫外吸收峰和吸光值的关系图;
图12是本发明的Mb-AuNPs与SH-β-CD自组装过程中不同搅拌时间与紫外红移程度的关系图;
图13是本发明的Mb-AuNPs与SH-β-CD自组装过程中不同搅拌时间与荧光强度的关系图;
图14是本发明的SH-β-CD添加量与紫外吸收峰和吸光值的关系图;
图15是本发明的SH-β-CD添加量与紫外红移程度的关系图;
图16是本发明的SH-β-CD添加量与荧光强度的关系图;
图17是本发明的Mb-AuNPs、SH-β-CD@Mb-AuNPs的紫外光谱图;
图18是本发明的Mb-AuNPs、SH-β-CD@Mb-AuNPs荧光光谱图;
图19是本发明的SH-β-CD@Mb-AuNPs的透射电镜扫描图;
图20是本发明的不同缓冲液浓度与荧光强度的关系图;
图21是本发明的不同缓冲液浓度与荧光强度变化的关系图;
图22是本发明的不同缓冲液pH与荧光强度的关系图;
图23是本发明的不同缓冲液pH与荧光峰变化的关系图;
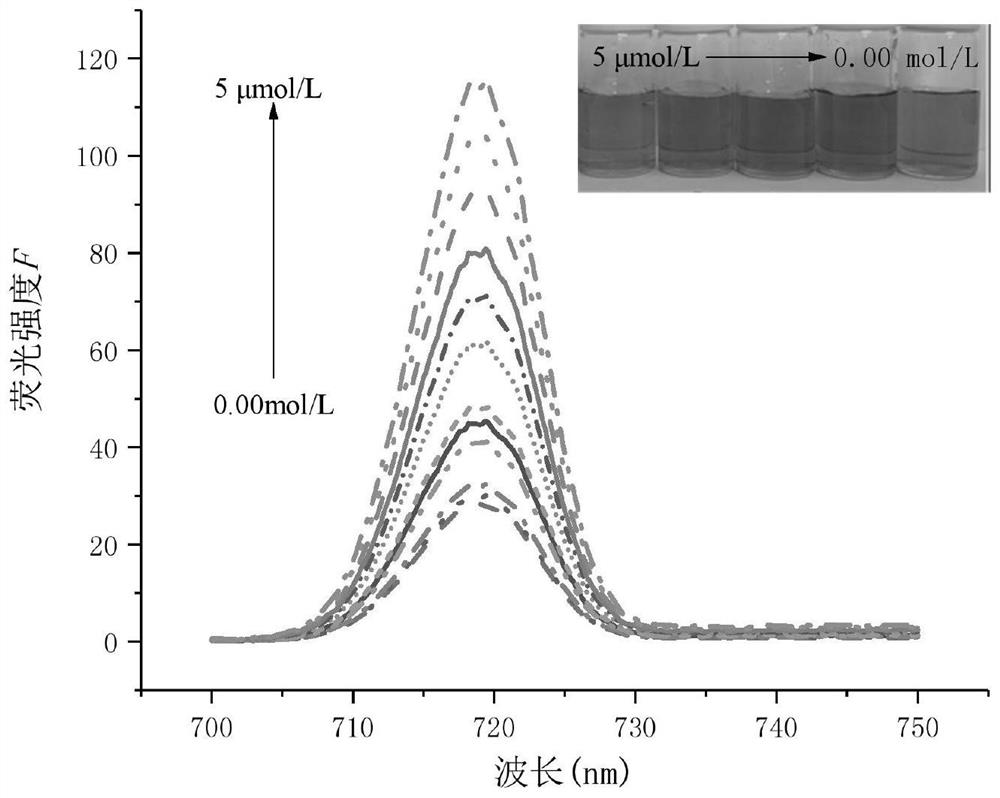
图24是本发明的SH-β-CD@Mb-AuNPs检测不同浓度亚硝酸盐荧光光谱图;
图25是本发明亚硝酸盐浓度与荧光强度变化的标准曲线图;
图26是本发明SH-β-CD@Mb-AuNPs的制备机理图;
图27是本发明Mb-AuNPs于不同浓度NO
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
实施例一
本实施例为一种测亚硝酸盐自组装荧光探针的制备方法,包括以下步骤:
S1、制备不同粒径的纳米金,利用荧光光谱,以200nm为起点,以3~8nm为增长幅度连续改变激发光波长,直至最高发射峰的峰值不再升高,取1cm荧光比色皿,用移液枪取纳米金2mL于比色皿中。用荧光光谱仪进行扫描,观察其荧光光谱;
S2、取1cm荧光比色皿,用移液枪取纳米金2mL于比色皿中,以步骤S1测得的发射峰的最高峰值作为发射波长,观察其荧光光谱,测定纳米金的激发波长;
S3、取制备好的粒径为16.4nm的纳米金,利用荧光光谱仪在 400~600nm波长下进行扫描,观察其出峰情况,在552nm附近出现最高吸收峰;再加入浓度为25μmol/LMb进行结合,每加0.02mL反应 3~8min后进行1次荧光扫描,直至峰值猝灭,停止加入Mb,此时由 Mb与AuNPs自组装制备的荧光纳米团簇(Mb-AuNPs)形成;
S4、纳米团簇制备好后,加入的1mL浓度为0.3×10-
一种用于检测亚硝酸盐自组装荧光探针的制备方法制备的荧光探针。
一种荧光探针在亚硝酸盐定量检测中的应用,该应用包括以下步骤:
1)取制备好的SH-β-CD@Mb-AuNPs荧光探针于烧杯中,加入pH 值为4的0.1mol/L的柠檬酸钠缓冲液1mL,混合均匀后,在激发波长 200~300nm测定荧光强度;
2)在SH-β-CD@Mb-AuNPs荧光探针中,分别加入pH值为4的 0.1mol/L的柠檬酸钠缓冲液1mL和已知不同浓度的亚硝酸盐溶液4mL,溶液混合均匀后,在激发波长200~300nm测定荧光光谱,根据荧光数据绘制回归方程;亚硝酸盐溶液浓度c在0.01~1μmol/L和1~5μmol/L 范围内与荧光强度变化△F均呈现出可靠的线性关系,该荧光探针对亚硝酸盐的定量检测性能良好,线性方程为:
△F
3)需要测定未知浓度的亚硝酸盐溶液浓度时,在 SH-β-CD@Mb-AuNPs荧光探针中,分别加入pH值为4的0.1mol/L的柠檬酸钠缓冲液1mL和未知浓度的亚硝酸盐溶液4mL,溶液混合均匀后,在激发波长200~300nm测其荧光光谱,根据回归方程计算出亚硝酸盐溶液的浓度。
实施例一中连续变化激发波长每增加5nm,发射峰波长红移10nm 左右,且荧光强度也有规律的增加。直至激发波长在275nm下,对应的发射峰为552nm时,此时纳米金的荧光强度峰值达到最大,继续变化激发波长,发射峰峰值出现下降,得出的荧光光谱图如图1所示。此种现象被称为非线性共振光散射。所以可以确定纳米金粒子的激发波长为275nm,发射峰为552nm。从图2中可以得出,固定552nm处的发射峰测得的激发波长为275nm。
实施例一中在向纳米金中加入Mb后的前五分钟的反应时间中,荧光强度变化△F逐渐上升。在1~2min时,荧光强度变化△F为25 左右,△F稍有增大,但没有明显的差异(P>0.05)。在2~5min时,荧光强度变化△F由25.9增至60.9,△F显著增大(P<0.05),即荧光强度持续下降;当结合反应时间在5~6min时,荧光强度变化△F由 60.9急剧下降至44.8,即荧光强度开始增强;6~10min时候,荧光强度变化△F一直保持在43~44左右无明显差异,得到反应时间与荧光强度之间的关系如图3。如此表明结合时间为5min时,结合体系达到稳定,荧光强度变化△F保持平稳,荧光猝灭最明显,因此选取Mb与 AuNPs最佳反应时间为5min。
实施例一中在AuNPs激发波长275nm下加入Mb结合后,如图4 所示AuNPs在552nm附近的荧光强度降低,加入足够量后,荧光强度发生猝灭荧光探针形成。当只有AuNPs进行荧光扫描时峰值达到了 589.3,随着每次Mb的加入,荧光峰值F在逐渐降低,直到Mb的加入量达到0.16mL时,荧光峰值F降到了最低。说明此时荧光比色皿中 15.0nm的2mLAuNPs与0.16mL25μmol/L的Mb发生了结合,致使荧光强度发生了猝灭。这可能是因为当足够的Mb将AuNPs表面的柠檬酸钠分子全部置换掉,溶液中的AuNPs的静电斥力作用消失,发生了团聚,使得荧光强度降低。
从图5中可以看出,实施例一中利用柠檬酸钠还原法制备的AuNPs 在520nm处有明显的紫外吸收峰,经过公式计算得出粒径为15.0nm,此时的AuNPs在溶液中的状态为分散。形成Mb-AuNPs后,紫外吸收峰由520nm红移至524nm,峰形变宽、峰值降低。根据电磁增强理论,当粒子之间的间距小于入射光的波长时,相邻粒子的表面等离子体波就会发生耦合,且随着粒子间距减小,耦合增强,并表现出协同等离子体共振吸收行为,表面等离子体共振吸收带出现红移、峰型变宽。 Mb-AuNPs在520nm处吸收峰较小,并在524nm附近有最大吸收峰,300~400nm间出现了Mb的吸收峰,说明Mb与AuNPs进行结合后发生了一定的结构变化,证明Mb中的巯基与AuNPs发生配位,形成Au-S 键,通过键的结合使得金溶胶由分散变为聚集,吸收峰向长波方向移动发生了红移的现象。
1020cm
制得的纳米金粒子呈球形结构,且各粒子之间因静电斥力而稳定存在,由图7的Mb-AuNPs透射电镜图可以看出大量的Mb-AuNPs通过形成的Au-S键聚集在一起,纳米粒子的聚集程度增加。这可能是由于Mb中S-H键与纳米金中的Au有很强的相互作用,通过形成Au-S键,使得含有静电斥力的AuNPs粒子混乱,从而发生聚集。
由图8可知,当纳米金粒径为10.3nm时,紫外吸收峰和吸光值相较于纳米金无明显变化;当纳米金粒径为13.2~21.7nm时,吸光值相较于纳米金有着明显的降低,结合图9可知,紫外吸收峰发生了明显的红移,且随纳米金粒径增大,紫外红移程度越明显(P<0.05),峰值降低越多。当纳米金粒径超过16.4nm时,峰值变化虽为明显,但是特征峰几乎消失。由图10可知,随着纳米金粒径的变化,荧光强度F变化显著(P<0.05),当纳米金粒径为16.4nm,荧光强度最低,显著低于其他粒径(P<0.05)。因此选取16.4nm为自组装 SH-β-CD@Mb-AuNPs最佳粒径。
由图11可知,随着搅拌时间的不同,紫外光谱扫描得出的吸光值以及峰位置均有所变化。当搅拌时间为8h,相较于纳米金,紫外吸收峰位置无明显差异(P>0.05),结合图12可知,波长红移了2nm;当搅拌时间为16h,吸光值明显降低,紫外波长由520nm红移至527.5nm;当搅拌时间达到24h后,峰值降低,并发生明显的红移,紫外吸收峰由520nm红移至575nm。这可能是因为加入SH-β-CD后,Mb-AuNPs 与SH-β-CD通过Au-S键自组装形成了SH-β-CD@Mb-AuNPs,随着粒子尺寸的增大,能级差减小,吸收光的波长向长波方向移动即所谓的红移,红移程度越大粒子越大。当搅拌时间超过24h后,峰值无明显红移,这可能是因为时间搅拌过长,破坏了SH-β-CD@Mb-AuNPs自组装的过程及结构。由图13可知,当搅拌时间为8h、32h,荧光强度 F无明显变化,当搅拌时间为16h、24h荧光强度F小幅度降低,但两者差距不明显,选取24h为自组装SH-β-CD@Mb-AuNPs的最佳搅拌时间。
由图14可知,随着0.3×10-
由图17可知,在16.4nm下制备的Mb-AuNPs、 SH-β-CD@Mb-AuNPs相较于同一粒径的AuNPs,紫外波长发生了不同程度的红移,SH-β-CD@Mb-AuNPs红移的程度最大且峰值下降的最多。这可能是因为形成的荧光探针介电常数相较于AuNPs、Mb-AuNPs增加,使得紫外红移,峰值降低。因此,由SH-β-CD@Mb-AuNPs在紫外光谱中的红移程度可以得知,其粒子尺寸以及介电常数大于AuNPs 和Mb-AuNPs。
由图18可知,在275nm激发波长下,Mb-AuNPs的荧光发射峰位置在550nm,SH-β-CD@Mb-AuNPs的荧光发射峰位置在720nm,荧光发射峰出现了明显的红移。说明加入SH-β-CD后化合物的结构发生了变化,引起基态与激发态和第一激发态之间能级差的改变,从而使得发射光子的能量改变致使荧光峰值红移。在相同的激发波长下, Mb-AuNPs、SH-β-CD@Mb-AuNPs的荧光发射峰位置不同,这可能是因为,加入SH-β-CD后,通过Au-S键的形成,纳米团簇表面积变大,表面活化能高,使得纳米粒子更容易发生聚集成为粒子尺寸较大的荧光探针-SH-β-CD@Mb-AuNPs。
因静电斥力的作用,AuNPs粒子在溶液中稳定存在。图19中 SH-β-CD@Mb-AuNPs的粒子聚集程度相较于图7中Mb-AuNPs更为紧密,纳米粒子的聚集程度增加。说明SH-β-CD与Mb-AuNPs通过Au-S 键的形成,将Mb-AuNPs中多余的结合位点占据,使得 SH-β-CD@Mb-AuNPs相较于Mb-AuNPs在溶液中的状态聚集的更为紧密、范围更广,形成了更为稳定的荧光探针SH-β-CD@Mb-AuNPs。
控制其他条件不变时,加入检测体系的缓冲溶液的浓度对检测结果的影响较为显著(P<0.05)。由图20、21可知,随着缓冲液浓度增大,荧光峰值先是小幅度的降低,当缓冲液浓度超过0.3mol/L,荧光峰值变化发生骤降。这可能是因为SH-β-CD对酸不稳定,当缓冲液浓度过高破坏了SH-β-CD@Mb-AuNPs中Au-S键的结合,导致荧光探针不稳定。为了保持检测体系的稳定,因此选取0.1mol/L为检测亚硝酸盐最佳的缓冲溶液浓度。
控制其他条件不变时,加入检测体系的缓冲液的pH值对检测结果的影响较为显著(P<0.05)。固定缓冲液浓度为0.1mol/L,控制其他条件不变,由图22可知,当pH值超过4或小于4溶液体系中的荧光强度变化显著(P<0.05),结合图23可知,pH越高荧光峰值下降的更明显。因此缓冲溶液的pH值为4时为最佳。
由图24、25可知,在0.01~1μmol/L和1~5μmol/L范围内荧光强度变化与亚硝酸盐浓度成线性关系;标准曲线方程分别为△F
由于SH-β-CD的独特分子囊结构,能够包络各种客体分子。未修饰Mb时,AuNP在溶液中因静电斥力的作用而稳定存在,修饰Mb后因Mb中的巯基以及AuNPs中的Au以Au-S键相连,破坏了AuNPs 的静电斥力,使得AuNPs发生了聚集。由于AuNPs粒子体积较大,结合位点较多,因此通过SH-β-CD的加入,使Mb包络在β-CD的腔穴中,占据了AuNPs上多余的位点,形成了更为稳定的SH-β-CD@Mb-AuNPs 荧光探针,SH-β-CD@Mb-AuNPs荧光探针的制备机理如图26所示。
在酸性条件下向制备好的Mb-AuNPs中加入不同浓度的NO
实施例二
称取通过超市购买的某品牌火腿肠5g搅碎混匀后,置于烧杯中,加入12.5mL的饱和硼砂溶液。以65℃左右的水,将搅碎混匀的火腿肠洗至500mL容量瓶中。沸水浴加热15min,冷却至20℃左右,加入 5mL106g/L亚铁氰化钾溶液,混合均匀后加入5mL220g/L乙酸锌溶液。冰箱中封口保存备用。
在制备好的火腿样品液中加入不同浓度的亚硝酸钠标准液,以确定此检测体系对样品中不同浓度亚硝酸钠的加标回收率和精密度,每组实验重复测定3次,结果见表1。
表1火腿肠中亚硝酸盐含量的加标回收实验
结果表明,加标回收率在96.39%~102.12%之间,且组内相对标准偏差皆在5%以下,说明该荧光探针检测体系具有良好的加标回收率和精密度。经计算火腿肠中亚硝酸盐含量为8.56mg/kg,低于国家限量标准30mg/kg。
实施例三
将通过超市购买的某品牌榨菜于榨汁机中搅拌成糊状后,取100g 榨菜糊置于烧杯中,加入6mL的饱和硼砂溶液。以65℃左右的水,将榨菜糊洗至500mL容量瓶中。向容量瓶中加入2g活性炭粉,然后加入2mL106g/L亚铁氰化钾溶液,混合均匀后加入2mL220g/L乙酸锌溶液。冰箱中封口保存备用。
在制备好的榨菜样品液中加入不同浓度的亚硝酸钠标准液,来确定此方法检测亚硝酸盐的加标回收率和精密度,每组实验重复测定3 次,结果见表2。
表2榨菜中亚硝酸盐含量的加标回收实验
结果表明,加标回收率在95.31%~100.23%之间,且组内相对标准偏差皆在5%以下,说明该荧光探针检测体系具有良好的加标回收率和精密度。经计算榨菜中亚硝酸盐含量为2.85mg/kg,低于国家限量标准4mg/kg。
实施例四
取90g通过超市购买的某品牌纯牛奶于容量瓶中,依次加入24mL 的硫酸锌溶液、24mL的亚铁氰化钾溶液、40mL缓冲液,边加边摇,静置15~30min,用滤纸过滤,利用锥形瓶收集。冰箱中封口保存备用。
在制备牛奶中加入不同浓度的亚硝酸钠标准液,来确定此方法检测亚硝酸盐的加标回收率和精密度,每组实验重复测定3次,结果见表3。
表3牛奶中亚硝酸盐含量的加标回收实验
结果表明,加标回收率在93.33%~95.31%之间,且组内相对标准偏差皆在5%以下,说明该荧光探针检测体系具有良好的加标回收率和精密度。经计算牛奶中亚硝酸盐含量为0.0926mg/kg,低于国家限量标准0.2mg/kg。
以上公开的本发明优选实施例只是用于帮助阐述本发明。优选实施例并没有详尽叙述所有的细节,也不限制该发明仅为具体实施方式。显然,根据本说明书的内容,可作很多的修改和变化。本说明书选取并具体描述这些实施例,是为了更好地解释本发明的原理和实际应用,从而使所属技术领域技术人员能很好地理解和利用本发明。本发明仅受权利要求书及其全部范围和等效物的限制。
- 一种检测亚硝酸盐自组装荧光探针的制备方法及其制备的荧光探针和应用
- 一种过氧亚硝酸盐近红外荧光探针ONP及其制备方法和应用