具有改善的生物利用度的药物组合物
文献发布时间:2023-06-19 09:52:39
本申请是申请日为2014年1月20日、申请号为201480005646.7(国际申请号为PCT/EP2014/050974)、名称为“具有改善的生物利用度的药物组合物”的发明专利申请的分案申请。
发明领域
本发明涉及微沉淀块状粉末(Micro-precipitated Bulk Powder,MBP),或喷雾干燥产物的固体无定形分散体,其包含化合物4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸(化合物A)以改善所述化合物的生物利用度、安全性和耐受性。
本发明涉及包含稳定化固体无定形分散体、具有高载药量(例如50%-70%)的极其低溶解度化合物(化合物A)的药物组合物,其导致超过所述化合物晶形的显著提高的溶出度和生物利用度。化合物4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸(化合物A)及其制备方法公开在美国专利号8,354,444和WO2011/098398中。
4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸(C
以上确认的国际专利申请和美国专利描述了晶体形式的化合物A,且其在此通过参照整体并入。该化合物的晶体形式具有约277℃的起始熔点。该晶体形式在生理学pH(范围为pH1.5-8.0)下具有相对低的水溶性(水中<0.05μg/mL)并因此低于最佳生物利用度(高可变性)。因此,期望获得具有改善的溶解度/溶出速率和生物利用度的化合物形式。
本发明提供了化合物A的无定形形式,其实质上不含结晶化合物。以重量计,所述化合物以大于或等于30%复合物的量存在于化合物/聚合物复合物(complex)中。
本发明的另一方面是包含本发明复合物的药物组合物,其中化合物A以治疗有效量存在。
本发明的另一方面是无定形药物与聚合物的复合物在高载药量下是稳定的。
本发明的另一方面是含有稳定化无定形形式的药学活性化合物的本发明复合物的制备方法。
本发明的主要特征是:
a)化合物A的稳定化固体无定形分散体的制备,
b)最终产品中10-70%的载药量。
c)在无定形固体分散体在高载药量例如70%是稳定的情况,实现最佳暴露的最佳载药量是10–50%。
d)以30%至99%的水平使用聚合物例如醋酸羟丙甲纤维素琥珀酸酯(hypromellose acetate succinate)、聚维酮和共聚维酮,
本发明还包括如下项:
1.一种物理稳定的固体分散体,其包含化合物和稳定化聚合物,所述化合物具有小于1μg/ml的水溶性和>270℃的熔点。
2.根据项1所述的固体分散体,其中所述具有小于1μg/ml的水溶性的化合物是式(A)的4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸
3.根据项1或2所述的固体分散体,其中所述稳定化聚合物是醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)。
4.根据项1或2所述的固体分散体,其中所述稳定化聚合物是EUDRAGIT
5.根据项1或2所述的固体分散体,其中所述稳定化聚合物是共聚维酮(PVP VA64)。
6.根据项2至5中任一项所述的固体分散体,其中所述固体分散体中的根据项2的化合物A的以重量计的量与其中所述稳定化聚合物的以重量计的量的比率为5:95~70:30。
7.根据项5所述的固体分散体,其中所述固体分散体中的化合物A的以重量计的量与其中所述稳定化聚合物的以重量计的量的比率优选为30:70~50:50。
8.根据项7所述的固体分散体,其中所述固体分散体通过喷雾干燥包含化合物A和共聚维酮(PVP VA 64)的溶液来获得。
9.根据项7所述的固体分散体,其中所述固体分散体通过微量沉淀包含化合物A和醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)的溶液来获得。
10.单位剂量固体制剂,其包含项6至9中任一项所述固体分散体,和选自由以下构成的组的常用药学成分:崩解剂、稀释剂、润滑剂、助流剂和薄膜包衣剂。
11.根据项10所述的单位剂量固体制剂,其包含呈无定形固体分散体形式的约80%的项6至9中任一项所述的固体分散体,和约7%交联羧甲基纤维素钠、约6.8%甘露醇、约4%交聚维酮、约1.5%胶体二氧化硅和约0.7%硬脂酸镁,其然后装入胶囊或压制并包衣为片剂。
12.制备具有小于1μg/ml的水溶性的化合物和离子聚合物的固体分散体的方法,其包括形成所述化合物和聚合物在二甲基乙酰胺或任何其它适当溶剂中的溶液,并使用抗溶剂共沉淀所述药物与聚合物。
13.药物制剂,其含有项1至9中任一项所述的固体分散体,连同附加的药学上可接受的佐剂。
14.根据项1至8中任一项所述固体分散体,用作用于治疗癌症,特别是AML或前列腺癌的药物。
15.实质上如本发明所述的新组合物、方法和用途。
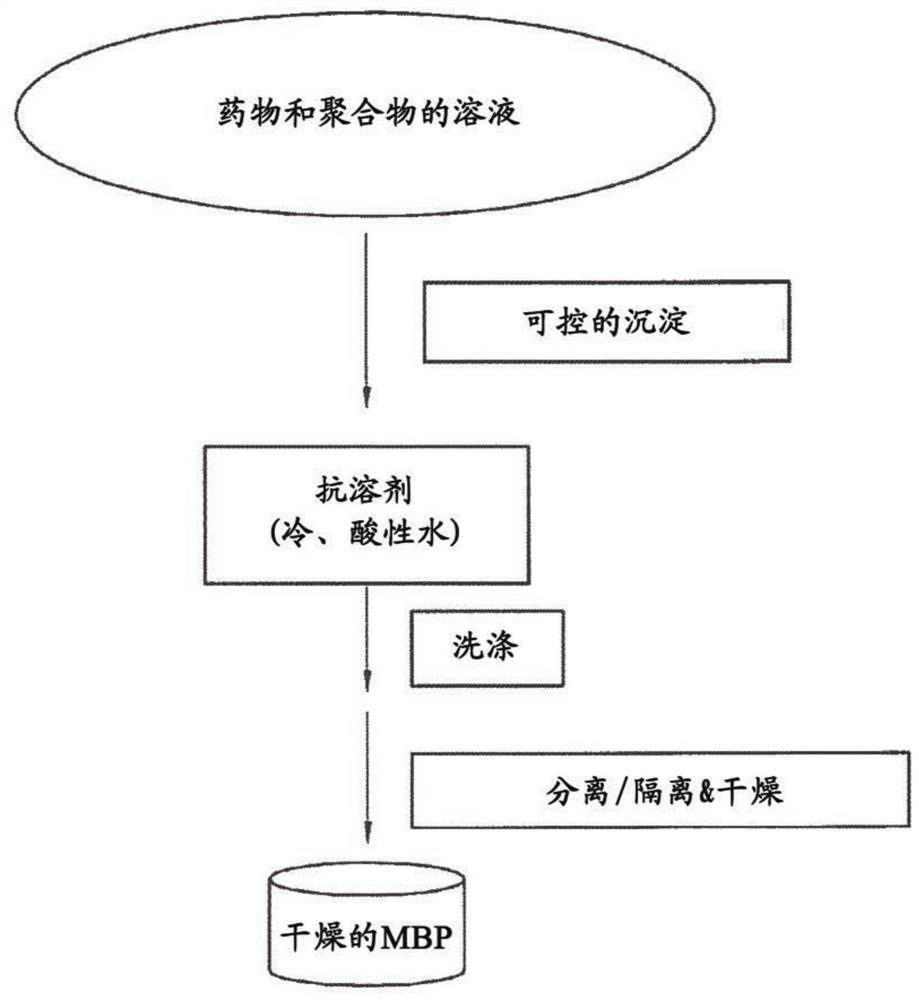
图1描述了微量沉淀方法。
图2描述了来自不同制剂策略的化合物A在猴中的暴露:晶体(原样)混悬液、晶体微米混悬液、采用Eudragit L100的固体分散体和采用HPMCAS的固体分散体。
图3描述了化合物A血浆浓度(plasma concentration)对比给药时间表:P1-30%载药量的采用HPMCAS的MBP形式的无定形固体分散体的混悬液剂型;P2-50%载药量的采用HPMCAS的MBP形式的无定形固体分散体的混悬液剂型;P3-30%载药量的采用PVPVA64通过HME制备的无定形的混悬液剂型;P4–晶体微米混悬液;P5-填充葡甲胺盐形式的化合物A颗粒的胶囊剂型。
图4描述了X-射线粉末衍射(XRPD)图中无定形固体分散体的稳定性。70℃处理a)4小时和b)8小时后固体分散体(50%载药量,采用HPMCAS聚合物)仍然是无定形的。
图5描述了X-射线粉末衍射(XRPD)图中无定形固体分散体的稳定性。无定形形式的50%载药量下的采用HPMCAS聚合物的MBP:a)起始、b)40℃/75%RH下6个月、c)25℃/60%RH下6个月。
图6A和B使用差示扫描量热法(DSC)加热循环法比较了MBP与相同比率的无定形API和聚合物的物理混合物的稳定性。A)物理混合物显示熔化。B)MBP显示玻璃转化(Tg)。
图7使用FTIR(傅里叶变换红外线)光谱描述了MBP中药物和聚合物之间的相互作用。a)物理混合物b)MBP。
图8a描述了根据实施例16获得的片剂(即,包含化合物A(50%wt/wt)与共聚维酮的喷雾干燥固体分散体)对比安慰剂(无固体分散体的片剂)的XRPD图案。片剂的XRPD图案对应于安慰剂的图案,表明没有可检测的结晶API(化合物A)。
图8b使用X-射线粉末衍射描述了根据实施例16的片剂的经时稳定性。对比片剂的初始测量(底物曲线)与在40℃和75相对湿度(RH)下在双泡(中间)和HDPE瓶(顶部)中贮存3个月后获得的曲线表明贮存期间没有可检测的结晶化合物A。
图9描述了包括通过1)微量沉淀(具有50%化合物A和HPMCAS),以及2)喷雾干燥(50%化合物A和PVP VA 64,实施例16)获得的固体分散体的化合物A的两层膜包衣的片剂制剂的体外溶出度。
治疗活性化合物生物利用度通常由(i)化合物的溶解度/溶出速率,和(ii)化合物通过受试者胃肠粘膜的分配系数/渗透率所决定。治疗活性化合物的不良生物利用度的主要原因通常是所述化合物的不良溶解度/溶出速率。由于患者对药物的不稳定吸收,不良生物利用度还经常伴随高度可变的患者血中浓度(blood level)以及不可预知的剂量/治疗作用。
如本发明使用的,当提及化学化合物涉及其在水或油中的溶解度时,术语"难溶的"可如美国药典与国家处方集(USP-NF)中所定义。根据此定义,溶解度的规定是根据溶解一份溶质所需的溶剂份数。在特定溶剂例如水中略溶(sparingly soluble)的化合物需要30-100份溶剂以溶解一份化合物。微溶性(slightly soluble)化合物需要100-1000份溶剂。十分微溶性(very slightly soluble)化合物需要1000-10,000份溶剂。不溶性(insoluble)化合物(例如化合物A)需要超过10,000份溶剂以溶解一份溶质。
此类药物的溶解度不足,以及不能在药学上可接受的载体中获得溶液中足够高的药物浓度,是配制这些药物的严重问题并因而限制此类化合物可实现的治疗益处。此外,溶解度不足在配制化合物用于需要显著高剂量并需要建立治疗有效剂量之上的非常高的安全界限的各种不同目标中是关注的问题。因此,存在对于增加这些药物溶解度的方法的显著需求。
为改善难溶性药物的期望特性,已开发了许多技术,其包括但不限于以下:
1.成盐:这是增加弱酸性或碱性NCE溶解度的最广泛使用的方法(Wadke,D.A.等人,Pharmaceutical Dosage Forms:Tablets,Vol.1,1989,pp 1-73)。盐的溶解度典型由抗衡离子影响且抗衡离子的选择是基于很多参数例如物理形态的溶解度、吸湿性和稳定性。虽然多种优点与成盐相关,但开发稳定的盐并不总是可行的。在许多情况下,难以实现增加的溶出速率,因为在生理环境中盐复原为其各自的酸或碱形式。
2.粒径减小:由于其不良溶解度,一些化合物的吸收/生物利用度受到溶出速率限制。粒径的减小显著改善溶出速率,其提供了更好的吸收潜能并潜在导致改善的治疗。湿磨(wet milling)(美国专利号5494683)和纳米技术(PCT国际申请WO 2004022100)是可应用于水难溶性药物的两种技术实例。虽然这些常规方法已通常使用以增加药物的溶出速率,但存在实际限制,因为期望的生物利用度增加不可能永远通过粒径减小而简单实现。而且,由于增加的表面能量导致的聚结或者润湿不良可推翻减小的粒径的任何益处。
3.脂质形成:难溶性药物可以远高于水性介质中的浓度溶于基于脂质的介质中。被给药后,脂质制剂分散于胃肠道流体中,其为药物提供了大表面积以使其从脂质中的溶液扩散至胃或肠道流体。药物在脂质制剂中的高溶解度提供了用于扩散的强推动力。自乳化药物递送系统(SEDDS)是一个实例。取决于脂质介质的选择,所得水性分散液可获得非常细或粗的乳液(参见例如美国专利号5969160、6057289、6555558和6638522)。这些配制技术的一些约束来自脂质介质中的不足药物溶解度、物理不稳定性(例如多晶型结晶伴随较小的溶解度)等。
4.固体分散体:近年来,固体分散体已在口服制剂领域中引起了注意,特别是对于难溶性化合物。固体分散体技术涉及在载体基质内稳定无定形形式的药物。无定形形式允许更快溶出药物并对于口服给药药物特别有前途(因为更广的载体基质选择)。然而,为有效使用此技术,需要确认与药物相容的适当载体。已开发了制备固体分散体的若干技术,包括共沉淀(参见例如美国专利号5985326和6350786)、熔融/喷雾干燥(参见例如美国专利号7008640)和热熔挤出(参见例如美国专利号7081255)。所有这些技术提供了聚合物基质中高度分散的药物分子,其改善了药物自分散体的溶出。制自不同方法的固体分散体可具有不同特性,例如多孔性、表面积、密度、稳定性、吸湿性、溶出度以及因此的生物利用度。然而,在文献中没有证据提示一种方法在另一方法之上的优势以实现期望的药代动力学模式,特别是更好的剂量比例性。
虽然这些技术中的一些是众所周知的,但它们大多数提供了许多独特挑战,并且不能适用于砖灰样化合物(brick dust like compound),即具有非常高的熔点并且实际上不溶于任何有机溶剂中。
此外,无定形固体分散体是存在额外挑战的高能制剂,因为它们天生是热力学上不稳定的。因此,它们的成功开发充分取决于对于负责其稳定化的特定相互作用的理解(Serajuddin,A.T.M.J.Pharm.Sci.1999,88,1058–1066;Janssens,S.和Van den Mooter,G.J.Pharm.Phamacol.2009,61,1571–1586.)。然而,没有通用或可靠的方法以选择工艺或聚合物以具有有保证的无定形稳定性和改善的生物利用度。已报道溶解度参数有助于选择聚合物。然而,如下表1中所示的,溶解度参数以及不同聚合物之间它们的排名在不同计算间是不一致的,因此基于不同计算可选择不同聚合物。因此,在提供稳定的无定形分散体方面,计算不能预测使用一种特定聚合物超过另一种的任何益处。
表1:化合物A与各种聚合物的溶解度参数计算
对于固体分散体制剂,若无定形重结晶,则可推测生物利用度由于来自无定形形式的溶解度改善的优势的损失而受到影响。然而,尚不清楚当无定形稳定性在宽的载药量范围内维持时,载药量或聚合物如何在生物利用度中发挥作用。
相关技术描述
WO2011/098398中已描述了化合物A、其合成方法以及含有该化合物的常规药物制剂。本专利申请描述了制备该化合物的热力学稳定形式的方法以及分子的作用机理。
美国专利号6350786公开了通过使用微沉淀块状粉末(MBP)技术获得的,包含各种不同化合物,即托卡朋、异维甲酸、沙奎那韦和数种其它化合物的无定形分散体的药物组合物。发现MBP技术是广泛适用的并发现数种不同聚合物,即
美国专利申请号US2010/0310659A1描述了使用MBP技术的丙烷-1-磺酸{3-[5-(4-氯-苯基)-1H-吡咯并[2,3-b]吡啶-3-羰基]-2,4-二氟-苯基}-酰胺与HPMC-AS的药物组合物。
美国专利申请号US2009/145999公开了丙烷-1-磺酸{3-[5-(4-氯-苯基)-1H-吡咯并[2,3-b]吡啶-3-羰基]-2,4-二氟-苯基}-酰胺与共聚维酮聚合物通过热熔挤出方法的无定形组合物。
美国专利申请号US12/902186详细说明了使用MBP和HME技术的低熔点药物HEP与HPMC-AS的药物组合物,其中无定形分散体通过HME方法显示出超过MBP制剂的略微改善的药代动力学行为。
在美国专利号6350786中,公开了使用具有大于80,000D的分子量的水不溶性离子聚合物的固体分散体,以提供稳定的无定形制剂。
美国专利号6548555描述了包括醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)的离子聚合物用于制备具有改善的溶解度和更好的生物利用度的固体分散体的用途。
WO2007/109605公开了喷雾干燥组合物,其包含药物和多种其它聚合物之中的PVP或PVP-VA。
HPMCAS是一种已被用于制造药物固体分散体的聚合物(例如,H.Konno、L.S.Taylor,Journal of Pharmaceutical Sciences,Vol.95,No.12,2006,2692-2705)。如本发明使用的其它聚合物,特别是聚维酮(PVP)和共聚维酮(PVP VA 64)是市售的,例如来自BASF SE(67056Ludwigshafen,德国)。
Kondo等人显示了难溶性药物在肠溶性共沉淀物中改善的口服吸收(例如J PharmSciences,83(4)1994)。此制剂中使用的聚合物是羟丙甲纤维素酞酸酯且该共沉淀物的制备是通过溶剂蒸发法,然后在80℃干燥。基于溶出数据,通过共溶剂或固体分散体途径增溶的此共沉淀物系统可复原回晶体形式,导致更高剂量下的生物利用度损失。
虽然已使用微共沉淀用于稳定数种固态形式的药物,但其可能并非必要于令人满意地调整此类难溶性化合物的药代动力学模式,特别是剂量依赖性暴露,而其对于管理该化合物的安全性和疗效而言是十分重要的。这些过饱和制剂可在贮存时或在压力条件下复原回晶体形式,导致生物利用度损失。发现针对无定形分散体的聚合物和方法的选择在该分散体的稳定化中发挥关键作用。然而,没有先验判断是否给定的聚合物或方法将提供无定形分散体的足够稳定性的绝对方法。
已发现固体无定形制剂中的载药量是关键的。通常载药量越低,稳定性越好。超过特定载药量,无定形固体分散体在保存期限期间具有高重结晶风险,并因而减少了改善的溶解度和生物利用度的益处。Lin和Cham(C.W.Lin,T.M.Cham.Effect of particle sizeon the available surface area of nifedipine from nifedipine-polyethyleneglycol 6000solid dispersions.Int.J.Pharm.,127(1996),pp.261–272)显示当使用5或10%萘普生载药(loading)时,相比于当使用20、30或50%载药时,PEG 6000中的萘普生固体分散体释放药物更快。这些结果可基于X-射线衍射结果解释,其表明具有低萘普生载药水平的分散体是无定形的而具有高载药量的那些是部分结晶的(DissolutionImprovement of High Drug-loaded Solid Dispersion.AAPS PharmSciTech 2006;7(2)Article 52)。药物产品开发中固体分散体技术的一个障碍是需要大量载体,即超过60%至90%wt/wt,以实现期望的溶出。此载体高百分率保证了生产时以及贮存期间的产品性能的一致性。
本发明涉及化合物A的稳定固体分散体,其特征在于加强的溶出速率和显著改善的生物利用度。在一个实施方式中,本发明固体分散体的制备是通过微沉淀,导致所述固体分散体为微沉淀块状粉末(MBP)。在另一实施方式中,本发明固体分散体的制备是通过喷雾干燥(SD)方法。取决于方法,可使用不同聚合物以在所述固体分散体中有效固定化合物A。
使用以下聚合物进行聚合物筛选:
·羟丙基甲基纤维素(HPMC 2910,E5)
·羟丙基纤维素HPC LF
·聚维酮K30(PVP K30)
·共聚维酮(PVP VA 64)
·Eudragit EPO(基于甲基丙烯酸二甲基氨乙酯、甲基丙烯酸丁酯和甲基丙烯酸甲酯的阳离子共聚物)
·Soluplus
·HPMCAS,LF
测试以下化合物A:聚合物的重量比:70%A:30%聚合物;50%A:50%聚合物;30%A:70%聚合物;以及50%A:45%聚合物:5%DOSS(琥珀磺酸二辛钠(Dioctyl sodiumsulfosuccinate)或多库酯钠)。
此外,以50%A:50%聚合物;以及30%A:70%聚合物的重量比测试以下聚合物:
·聚维酮12PF(PVP 12PF)
·聚维酮17PF(PVP 17PF)
·聚维酮K25(PVP K25)
·聚维酮K30(PVP K30)
·聚维酮K90(PVP K90)
·共聚维酮(PVP VA 64)
·Eudragit EPO(基于甲基丙烯酸二甲基氨乙酯、甲基丙烯酸丁酯和甲基丙烯酸甲酯的阳离子共聚物)
·Soluplus
·HPMCAS,LF
已证实在各种测试聚合物中,HPMCAS、聚维酮(PVP)和共聚维酮(PVP VA 64)显示出对于化合物A的改善的溶出模式。改善的溶出模式是指化合物A从由该化合物和各自聚合物形成的固体分散体的改善的释放。此外,聚维酮或共聚维酮的使用可导致独立于溶出环境中的pH值的溶出模式。因此,化合物A从由聚维酮或共聚维酮形成的固体分散体的溶出及因此的释放可在此固体分散体口服给药后早期就已发生,例如在胃中。这种化合物A的早期溶出/释放可因而显著改善化合物A的生物利用度。
如本发明使用的,术语“固体分散体”是指具有至少两种组分的任何固体组合物。在某些实施方式中,如本发明公开的固体分散体包含活性成分(例如化合物A);优选分散在至少一种其它组分(例如聚合物)中。在某些实施方式中,如本发明公开的固体分散体是包含至少一种药学或生物学活性成分(例如化合物A)的药物分散体。在一些实施方式中,固体分散体包含与聚合物分子分散的化合物A。优选该固体分散体是单相系统。一种特别优选的根据本发明的固体分散体是包含化合物A的微沉淀块状粉末(MBP)。在另一实施方式中,固体分散体是通过喷雾干燥获得并包含化合物A和作为聚合物的共聚维酮(PVP VA 64)。
如本发明使用的术语“分子分散的”是指化合物(例如,化合物A)与聚合物随机分布。在某些实施方式中,化合物以细分终态存在于聚合物中。参见,例如,M.G.Vachon等人,J.Microencapsulation,14:281-301(1997)和Vandelli等人,J.Microencapsulation,10:55-65(1993)。在一些实施方式中,化合物(例如,化合物A)可以其固态形式分散在由聚合物形成的基质内以使得该化合物以其无定形形式被固定。化合物是否分子分散于聚合物中可以各种方式证实,例如通过具有单一玻璃转化温度的所得固体分子复合物,或者通过X-射线衍射曲线中指示所述化合物(例如化合物A)的任何结晶量的信号的缺失。
如本发明使用的术语“固体分子复合物”是指包含分子分散于聚合物基质内的化合物A的固体分散体。
关于活性化合物在聚合物基质中的固定,如本发明使用的术语“固定”是指化合物分子与聚合物分子以这样一种方式相互作用以使得化合物维持在前述基质中并由于缺乏流动性而预防晶核形成。在一些实施方式中,聚合物可预防两个或多个化合物A药物分子间的分子间氢键或弱分散力。参见,例如,Matsumoro和Zografi,Pharmaceutical Research,Vo.16,No.11,p1722-1728,1999。
除非另有明确说明,否则如本发明使用的百分数(%)表示为重量百分数(重量%,wt/wt)。
因此,在第一方面中,提供了包含化合物A和聚合物的固体分散体。还提供了包含化合物A和聚合物的固体分子复合物。该聚合物可为非离子聚合物或离子聚合物。在某些实施方式中,聚合物选自由以下构成的组:醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)、羟丙甲纤维素、甲基丙烯酸共聚物等,以及其任意两种或多种的混合物。在一优选实施方式中,聚合物选自HPMCAS或聚维酮(PVP,
稳定的固体分散体包含分子分散于由聚合物形成的基质中的约10%至约90%,在某些实施方式中约30%至约70%,或约40%至约60%,或约20%至约50%,或约50%至约70%(wt/wt)的化合物A。在某些实施方式中,此聚合物是醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)或聚维酮(PVP)或共聚维酮(PVP VA 64)。最优选本发明化合物A的稳定化无定形分散体组合物包含非显著量的晶体化合物A,如通过所述组合物的无定形X-射线粉末衍射(XRPD)所证实的。
活性成分(即化合物A)具有化学名称4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸(化合物A)并可通过以下结构式表示:
化合物A(本发明有时称作“药物”,“API”)的晶体具有约277℃的熔点并在生理pH(pH 1.5-7.0)下具有非常低的水溶性(<0.05μg/ml),因此生物利用度非常低。如采用0.8x10
化合物A及其制备方法例如公开在美国专利号8,354,444和WO2011/098398中。更特别的,由于其抑制MDM2-p53相互作用的能力,化合物A具有治疗各种增殖性疾病例如癌症的潜力。如本发明使用的术语“癌症”是指实体瘤和血液肿瘤,其选自由以下构成的组:乳腺癌、前列腺癌、宫颈癌、卵巢癌、胃癌、结肠直肠癌(即,包括结肠癌和直肠癌)、胰腺癌、肝癌、脑癌、神经内分泌癌、肺癌、肾癌、恶性血液病(例如白血病)、黑素瘤和肉瘤。更特别优选的癌症选自由以下构成的组:恶性血液病、前列腺癌、乳腺癌、宫颈癌、卵巢癌、结肠直肠癌、黑素瘤和肺癌。在一特别优选实施方式中,癌症是急性髓性白血病(AML)或前列腺癌。
在一个实施方式中,本发明提供了物理稳定的固体分散体,其包含化合物和稳定化聚合物(stabilizing polymer),所述化合物具有小于1μg/ml的水溶性和>270℃的熔点。
在另一实施方式中,本发明提供了如上所述的固体分散体,其中具有小于1μg/ml的水溶性的化合物是4-{[(2R,3S,4R,5S)-4-(4-氯-2-氟-苯基)-3-(3-氯-2-氟-苯基)-4-氰基-5-(2,2-二甲基-丙基)-吡咯烷-2-羰基]-氨基}-3-甲氧基-苯甲酸(化合物A)。
在另一实施方式中,本发明提供了如上所述的固体分散体,其中稳定化聚合物是醋酸羟丙甲纤维素琥珀酸酯(HPMCAS)。
在另一实施方式中,本发明提供了如上所述的固体分散体,其中稳定化聚合物是EUDRAGIT
在另一实施方式中,本发明提供了如上所述的固体分散体,其中稳定化聚合物是聚维酮(PVP)或共聚维酮(PVP VA 64)。
在另一实施方式中,本发明提供了如上所述的任一固体分散体,其中固体分散体中的化合物A的以重量计的量与其中稳定化聚合物的以重量计的量的比率为5:95~70:30。
在另一实施方式中,本发明提供了如上所述的任一固体分散体,其中固体分散体中的化合物A的以重量计的量与其中稳定化聚合物的以重量计的量的比率优选为30:70~50:50。
在又另一实施方式中,根据本发明的固体分散体是通过喷雾干燥共聚维酮(PVPVA 64)和化合物A的溶液而获得。可使用其中共聚维酮和化合物A二者均可溶的任何溶剂。优选地,将50%(以重量计)共聚维酮和50%(以重量计)化合物A溶于丙酮中。合并量的共聚维酮+化合物A占丙酮溶液的3-7%,优选5%(以重量计)。通过常规喷雾干燥方法喷雾干燥此溶液,然后是二次干燥过程。可使用所有常规二次干燥方法,优选盘式干燥器、螺旋干燥器或流化床干燥器。如此获得的喷雾干燥粉末的进一步特征在于粒径分布为约d
固体分散体,特别是根据提供的方法可获得的MBP和/或喷雾干燥产物,可以用于给药水难溶性药物(例如化合物A)的各种形式使用,特别是用于口服剂型。示例性剂型包括可干燥或通过加水重构以形成糊剂、浆体、混悬液或溶液而口服摄入的粉末或颗粒;片剂、胶囊或丸剂。可将各种添加剂与如本发明所述的固体分散体混合、磨碎或制粒以形成适于以上剂型的材料。潜在有益的添加剂可通常落入以下类:其它基质材料或稀释剂、表面活性剂、药物络合剂或增溶剂、填充剂、崩解剂、粘合剂、润滑剂和pH改性剂(例如,酸、碱或缓冲剂)。其它基质材料、填充剂或稀释剂的实例包括乳糖、甘露醇、木糖醇、微晶纤维素、焦磷酸钙和淀粉。表面活性剂的实例包括十二烷基硫酸钠和聚山梨酯80。药物络合剂或增溶剂的实例包括聚乙二醇、咖啡因、氧杂蒽、龙胆酸和环糊精。崩解剂的实例包括羧甲淀粉钠、海藻酸钠、羧甲基纤维素钠、甲基纤维素和交联羧甲基纤维素钠。粘合剂的实例包括甲基纤维素、微晶纤维素、淀粉和胶类例如瓜尔豆胶和黄芪胶。润滑剂的实例包括硬脂酸镁和硬脂酸钙。pH改性剂的实例包括酸例如柠檬酸、醋酸、抗坏血酸、乳酸、天冬氨酸、琥珀酸、磷酸等;碱例如醋酸钠、醋酸钾、氧化钙、氧化镁、磷酸三钠、氢氧化钠、氢氧化钙、氢氧化铝等,以及通常包含酸和所述酸的盐的混合物的缓冲剂。包含此pH改性剂的至少一种作用是控制药物、基质聚合物或二者的溶出速率,从而控制溶出期间的局部药物浓度。
可在固体无定形分散体形成期间或之后并入添加剂。除了以上添加剂或赋形剂以外,本领域技术人员已知的用于配制和制备使用本发明公开的组合物的口服剂型的任何常规材料和方法的使用是潜在有用的。
因此,在另一实施方式中,提供了单位剂量固体制剂,优选片剂,其包含根据本发明的固体分散体和选自由以下构成的组的常用药学成分:崩解剂、稀释剂、润滑剂、助流剂和薄膜包衣剂(film coat)。
在另一实施方式中,本发明提供了单位剂量固体制剂,其包含约80%的根据本发明的任一固体分散体,和约7%交联羧甲基纤维素钠、约6.8%甘露醇、约4%交聚维酮、约1.5%胶体二氧化硅和约0.7%硬脂酸镁,其然后装入胶囊或压制并包衣为片剂。
在又另一实施方式中,本发明提供了单位剂量固体制剂,其特征在于通过喷雾干燥化合物A和共聚维酮(PVP VA 64)获得的固体分散体占内核重量(kernel weight)的约80%wt/wt,使用滚筒搅拌器将其进一步与优选自甘露醇、微晶纤维素、一水乳糖或二氧化硅的填充剂(内核重量的6.8%直至10.8%);选自交联羧甲基纤维素钠或交聚维酮的一或两种崩解剂(内核重量的4%wt/wt);优选胶体二氧化硅的助流剂(内核重量的1%wt/wt);和润滑剂(内核重量的0.2%wt/wt)硬脂酸镁混合。
在又另一实施方式中,提供了根据实施例16的具体片剂制剂。
在另一实施方式中,本发明提供了制备具有小于1μg/ml的水溶性的化合物(优选化合物A)和离子聚合物的固体分散体的方法,其包括形成化合物和聚合物在二甲基乙酰胺或任何其它适当溶剂中的溶液,并使用抗溶剂(anti-solvent)共沉淀该药物与聚合物。优选此实施方式中的聚合物是HPMCAS。
在另一实施方式中,本发明提供了制备具有小于1μg/ml的水溶性的化合物(优选化合物A)和离子聚合物的固体分散体的方法,其包括形成化合物和聚合物在丙酮或任何其它适当溶剂中的溶液,并喷雾干燥该药物与聚合物。优选此实施方式中的聚合物选自聚维酮(PVP)或共聚维酮(PVP VA 64)。
在另一实施方式中,本发明提供了药物制剂,其包含根据本发明的固体分散体连同附加的药学上可接受的佐剂。
在另一实施方式中,本发明提供了根据本发明的固体分散体,用作用于治疗癌症,特别是AML或前列腺癌的药物。
在另一实施方式中,本发明提供了根据本发明的固体分散体用于制造用于治疗癌症,特别是AML或前列腺癌的药物的用途。
极低的溶解度/生物利用度向获得针对化合物A的期望的暴露和安全界限提出了挑战。由于具有极低水溶性的的疏水性药物的低生物利用度可为严重问题,已开发了不同方法以实现期望的高水平药物溶解度和溶出速率。现将通过以下实施例进一步阐明这些方法,但其无意于限制本发明的范围。
实施例
A.:晶体配制方法
以下是采用化合物的晶体形式或盐形式的各种不同配制方法的细节(实施例1)。表1阐明了采用那些配制方法获得的相对生物利用度。
如下制备晶体制剂:
晶体混悬液的制备是通过将晶体化合物A分散在水基介质中,所述介质组成为2%羟丙基纤维素、0.15%聚山梨酯80、0.09%尼泊金甲酯和0.01%尼泊金丙酯。磨碎该混悬液以实现<10μm的中值粒径(d
表2.猴中的生物利用度改善
盐筛选已确认了化合物A的数种潜在盐类(参见下表3)。其中,葡甲胺是有前途的盐类,其具有最大改善的水溶性,因此在动物药代动力学研究中在含有颗粒形式的化合物A葡甲胺盐与泊洛沙姆188、交聚维酮、胶体二氧化硅和硬脂酸镁的颗粒的胶囊固体剂型中对其进行测试。生物利用度(暴露)未改善(图3)。
表3.化合物A各种盐的溶解度
B.:无定形固体分散体配制方法
发现化合物A的无定形固体分散体相比于该化合物的晶体或盐形式呈现出显著更高的生物利用度。
如实施例3-10中所示,评估各种可用技术生成适当的无定形制剂,即喷雾干燥、热熔挤出和微沉淀块状粉末技术。
考察的各种载体包括羟丙甲纤维素、醋酸羟丙甲纤维素琥珀酸酯、Kollidon PVP、Kollidon PVP VA64、Soluplus、丙烯酸和甲基丙烯酸共聚物,例如Eudragit L100-55、Eudragit L100、Eudragit EPO等。在5%-70%的各种载药范围内进行实验。
在30%至50%的载药量、在120℃至180℃使用热熔挤出机熔化化合物A、共聚维酮(或
通过在室温下搅拌将药物和聚合物(HPMCAS或Eugragit L100)溶于二甲基乙酰胺(DMA)中。然后将过滤或未过滤的溶液加至冷的、温度控制的抗溶剂水性介质(稀HCl,约pH3.0,温度为1℃至10℃),其可快速共沉淀该药物和聚合物。用冷酸水和冷水频繁洗涤萃取残留DMA,然后分离洗液和湿沉淀物并干燥该沉淀物。将所谓MBP的干燥粉末进一步加工为给药混悬液或片剂。以下所有制剂显示无定形XRD图案(图1)。
表4:来自采用HPMCAS的无定形固体分散体制剂的大鼠和猴中的生物利用度改善。
化合物A为无定形固体分散体(MBP)形式,组成含有30%化合物A和70%EudragitL 100聚合物的粉末。最终给药浓度等于水性介质中的6mg/mL化合物A,该介质含有2%w/w羟丙基纤维素、0.1%聚山梨酯80和0.09%尼泊金甲酯和0.01%尼泊金丙酯。(图2)
通过以30%载药量与70%HPMC-AS聚合物共沉淀(MBP)得到无定形固体分散体形式的化合物A。最终构建的给药混悬液浓度为水性介质中的1mg/mL化合物A,该介质含有2%w/w羟丙基纤维素、0.1%聚山梨酯80和0.09%尼泊金甲酯和0.01%尼泊金丙酯。
化合物A以含有50%化合物A和50%HPMC-AS聚合物的无定形固体分散体(MBP)的形式。然后将MBP构建为水性介质中浓度为1mg/mL化合物A的给药混悬液,该介质含有2%w/w羟丙基纤维素、0.1%聚山梨酯80和0.09%尼泊金甲酯和0.01%尼泊金丙酯(图3)。
化合物A呈无定形固体分散体(MBP)形式,组成含有70%化合物A和30%HPMC-AS聚合物的粉末。构建时混悬液浓度为4mg/mL。
将含有50%化合物A和50%HPMC-AS的化合物A的无定形MBP固体分散体进一步加工为片剂。组成为92.8%的无定形固体分散体(MBP)形式的化合物A,与5%交联羧甲基纤维素钠、1.5%胶体二氧化硅和0.7%硬脂酸镁。
将含有30%化合物A和70%HPMC-AS的化合物A的无定形固体分散体进一步加工为片剂。片剂组成为94%无定形固体分散体(MBP)形式的化合物A(30%化合物A和70%HPMC-AS),与3.7%交联羧甲基纤维素钠、1.2%胶体二氧化硅、0.5%羟丙基纤维素和0.6%硬脂酸镁。
将含有50%化合物A和50%HPMC-AS的化合物A的无定形固体分散体进一步加工为片剂。片剂组成为80%无定形固体分散体形式的MBP(50%化合物A和50%HPMC-AS),与7%交联羧甲基纤维素钠、6.8%甘露醇、4%交聚维酮、1.5%胶体二氧化硅和0.7%硬脂酸镁。然后可用传统的水性薄膜包衣混合物包衣片剂内核。
将约5mg MBP形式的化合物A置于20mL 37℃生物相关性流体(禁食状态模拟肠液和进食状态模拟肠液)中并经时过滤通过0.2μm滤器。然后通过HPLC分析滤液。
表5.溶解度增加
(图4)
如X-射线粉末衍射(XRPD)中显示的,MBP在25℃/60%RH下贮存6个月后以及在40℃/75%RH下6个月后仍然是无定形的(图5)。
使用差示扫描量热法(DSC)加热循环法比较无定形MBP固体分散体与相同组分的物理混合物,显示物理混合物结晶而MBP仍然是无定形的。(图6)
FTIR(傅里叶变换红外线)光谱显示(图7)在MBP中药物和聚合物是分子分散的,提供较大的稳定性且不易于结晶。另一方面,物理混合物中的无定形API不是分子分散的,并因而易于结晶。因此,均匀分子分散是甚至在高载药量下的优异稳定性的主要因素。
该片剂含有800mg化合物A和共聚维酮(PVP VA 64)的喷雾干燥粉末(SDP),相当于400mg化合物A(游离碱)。
表6:片剂组成:
方法步骤:
1)将化合物A(喷雾干燥粉末的50%w/w)和共聚维酮(喷雾干燥粉末的50%w/w)溶于丙酮中以获得5%(w/w)的固体浓度。
2)将1)中获得的溶液进料通过4μm滤器至喷雾干燥器单元并使用干燥室中的转轮喷雾器、或两液喷嘴、或压力喷嘴雾化。由喷雾器创造的细雾与热氮流(110至150℃)混合并开始从液滴蒸发溶剂。调节溶液进料率以实现期望的出气口温度(60至90℃)。干燥气体携带细粉通过干燥室出至旋风分离器。旋风分离器自干燥气体分离粉末并通过重力将粉末收集入鼓桶中。实质上无粉末的气流进入滤袋壳体,其中超细颗粒保留在滤袋中。在冷凝器中冷却无粉末气体,其中发生溶剂凝结且再加热后的干燥气体再循环至干燥室干燥。
3)对2)获得的喷雾干燥材料进行二次干燥以减少产品中丙酮的存在量。使用盘式干燥器进行二次干燥。
4)使用滚筒搅拌器进一步混合3)获得的喷雾干燥粉末与作为填充剂的甘露醇;交联羧甲基纤维素钠,并且交联羧甲基纤维素钠作为崩解剂;作为助流剂的胶体二氧化硅。
5)在滚筒搅拌器中进一步混合4)获得的混合物与作为润滑剂的硬脂酸镁(内核重量的0.2%)。
6)使用配有口袋制粒机或星轮以及0.8mm开口筛网的Gerteis碾压机干燥制粒5)获得的混合物。
可使用星轮替代口袋制粒机。开口可为0.5mm至0.8mm。
7)在滚筒搅拌器中混合6)获得的颗粒与作为助流剂的胶体二氧化硅以及作为崩解剂的交联羧甲基纤维素钠。
8)在滚筒搅拌器中进一步混合7)获得的混合物与作为润滑剂的硬脂酸镁(内核重量的0.5%)以获得用于压片的最终混合物。
9)使用旋转压片机例如Korsch XL 100WipCon将最终混合物压制成片。调节片重至最终所需的剂量强度,即400mg。这些400mg内核具有20.1mm至9.5mm的尺寸。
10)使用穿孔鼓包衣机例如Glatt包衣机,使用含有PVA的薄膜包衣系统薄膜包衣9)获得的片剂。薄膜包衣的涂覆量为内核重量的3%。薄膜包衣的组成为1.2%部分水解的聚乙烯醇、0.6234%二氧化钛、0.606%聚乙二醇、0.444%滑石、0.099%氧化铁黄、0.024%氧化铁红和0.0036%氧化铁黑。薄膜包衣量可高达内核重量的5%。还可使用替代的薄膜包衣系统例如用于防潮。
在环境条件下透射几何中采用STOE STADI P衍射仪记录X-射线衍射图(Cu Kα辐射,初级单色仪,硅条探测器,角范围3°至42°2θ,总测量时间约30分钟)。样品的制备和分析未经进一步物质处理(例如碾磨或过筛g)(图8a、b)。
根据实施例16的固体分散体(即,包含50%化合物A和共聚维酮(PVP VA64)的喷雾干燥固体分散体)的XRPD图案,对应于安慰剂的图案,证实最初没有可检测的晶体API(化合物A)(图8a)。
所述固体分散体在双泡和HDPE瓶中贮存3个月(贮存条件:40℃和75%相对湿度(RH))后的XRPD图案对应于最初测量的固体分散体的图案,证实没有可检测的晶体API(化合物A)(图8b)。
针对包括通过1)微量沉淀(具有50%化合物A和HPMCAS的MBP),以及2)喷雾干燥(50%化合物A和PVP VA 64,实施例16)获得的固体分散体的膜包衣片剂形式的化合物A的两种制剂进行体外溶出度检测。使用的介质为0.01摩尔(0.01N)的盐酸(HCl)、模拟禁食的胃以及禁食状态模拟肠液(FaSSIF),模拟禁食的小肠。具体的设置总结在下表7中。
表7:用于溶出研究的参数
在预填充有500ml蒸馏水(dest)的1000ml容量瓶中称量(0.83ml)沸腾的HCL37%。添加水至1000ml。充分混合,并在使用前使其冷却至室温。
步骤1
为了制备缓冲液,在约2.700L的纯化水中溶解1.260g的NaOH(颗粒),13.410gNaH
步骤2
向约1.5L缓冲液中添加6.720g SIF粉末原料。搅拌直至粉末充分溶解。在室温下利用缓冲液补充至体积(3.000L)。
图9中显示了根据该方法获得的溶出度数据。
- 具有改善的生物利用度的药物组合物
- 具有改善的生物利用度的药物组合物