一种通过竞争ELISA法进行标签蛋白定量的方法
文献发布时间:2023-06-19 19:38:38
技术领域
本发明涉及蛋白定量技术领域,尤其是一种通过竞争ELISA法进行标签蛋白定量的方法。
背景技术
标签蛋白是与目的蛋白一起融合表达的一种多肽或者蛋白,以便于目的蛋白的表达、检测、示踪和纯化等。标签是重组蛋白纯化的一个重要且有效的工具,它们不仅便于对融合蛋白进行检测与纯化,而且可能对目标蛋白的理化性质产生有利的影响,包括提高重组蛋白的产量、增强重组蛋白的可溶性以及促进重组蛋白的正确折叠等,同样的,标签对于重组蛋白定量也是非常重要的,这就需要和标签蛋白抗体一起发挥作用,标签蛋白抗体是一类经过亲和纯化的抗体,用于检测含有标签的目的蛋白的表达量、定位及其功能,标签序列可以直接设计在商品化的表达载体上。
高通量表达应用在进行细胞稳转株构建的过程中,加压筛选是必不可少的一个步骤。现在常规的操作是将转染后的细胞进行96孔板稀释接种,接种20块孔板,每孔约2000个细胞,然后进行加压筛选找出活性和表达量最佳的minipool。最后将筛选出的minipool进行手动有限稀释或者机器的自动化分选,找出活性和表达量最佳的单克隆进行方法培养,得到可以稳定表达的细胞株。随着分子相互作用技术的发展,近几年出现了以Octet分子相互作用技术为代表的各种分析蛋白质间相互作用的仪器,这些仪器大多以SPR技术为原理进行开发,能够全方面的对蛋白之间的相互作用,蛋白的浓度以及活性进行分析和把控,但是高昂的费用却让很多初创公司望而止步,为此如何在低成本的情况下还能够更准确的进行高通量定量是一个需要解决的问题。
对于一个已经纯化的蛋白,常用的定量方法有BCA法定量、Bradford 法定量、Lowry法定量、紫外分光光度法定量以及ELISA夹心法定量等,以上定量方法除了利用抗原抗体特异性结合的ELISA夹心法定量之外,几乎所有的方法都需要纯蛋白进行定量,同时还受到样本缓冲成分的影响,显然对培养基上清进行定量是行不通的,因为在细胞培养过程有一些宿主细胞内源性的蛋白会被释放出来以及培养基成分复杂从而影响测量结果,而ELISA夹心法定量则对抗体抗原原料是有相当高要求,特别是需要有特异性的抗体配对抗原,这种方法不具有普遍性。
发明内容
为了克服上述现有问题的不足,本发明提供了一种通过竞争ELISA进行标签蛋白定量的方法。
本发明解决其技术问题所采用的技术方案是:一种通过竞争ELISA法进行标签蛋白定量的方法,其特征是,包括如下步骤:
S1,标准品的制备,通过重组表达的方式生产阳性标准品,所述阳性标准品为带有标签的蛋白;
S2,HRP标记,将HRP标记至显色蛋白上;
S3,阳性标准品的检测;
包被100-1000ng/ml的标签抗体,每孔100ul,在4℃条件下包被18-24hr;
包被结束后进行3-5次的PBST洗板,每次200ul,洗板结束后甩干处理;
将阳性标准品与0.2-2ug/ml显色蛋白混合,随后再进行15个孔的稀释,稀释条件为利用封闭液对阳性标准进行对半稀释后加入EP管中,随后再将前一稀释的样本再进行对半稀释加入第二个EP管中,以此类推,最后将15个处理组与显色蛋白混合,混合样品的体积用PBST补至100ul加入酶标板的孔中,其中一个孔不加入阳性标准品,直接加入PBST做为对照组,数据处理过程中称为T0,然后在37℃条件下孵育1hr;
孵育结束后进行3-5次PBST洗板,每次200ul;
最后加入100ul的TMB显色液显色3-8min,显色结束后加入100ul 0.25M的HCl终止,在酶标仪上进行OD450读数;
S4,样本检测:
包被100-1000ng/ml的标签抗体,每孔100ul,在4℃条件下包被18-24hr;
包被结束后进行3-5次的PBST洗板,每次200ul,洗板结束后甩干处理;
根据预估的表达量取1-50ul的样品与0.2-2ug/ml显色蛋白混合,随后将阳性标准品2-3倍梯度进行稀释,混合样品的体积用PBST补至100ul,共计2-5个孔,在37℃条件下孵育1hr;
孵育结束后进行3-5次的PBST洗板,每次200ul;
最后加入100ul的TMB显色液显色3-8min,显色结束后加入100ul 0.25M的HCl终止,在酶标仪上进行OD450读数;
S5,绘制标准曲线并进行样品浓度分析,根据OD450的读数进行标准曲线的绘制和样品浓度的分析:
首先用阳性标准品检测出的OD450值除以对照组样品OD450值,将T/T0进行对数处理后的数据作为Y轴,加入阳性标准品的浓度做为X轴;
随后在Excel中进行XY散点图的绘制,找出线性范围;
最后将样品的T/T0数值的对数代入公式中即可得到蛋白稀释后的浓度,将数据乘以对应的稀释倍数即可得到实际样本中蛋白的表达量。
根据本发明专利的另一个实施例,进一步包括,S1中,所述标签为His、Flag、HA、c-Myc、GFP、人Fc等标签中的一种或多种。
根据本发明专利的另一个实施例,进一步包括,S2中,使用过碘酸钠法或Catcher-SPY体系进行定点标记。
根据本发明专利的另一个实施例,进一步包括,所述阳性标准品带有的标签包含样本带有的标签。
通过将对应标签的抗体包被在ElISA板中,将不同浓度梯度的阳性标准品和显色蛋白进行孵育后根据OD450值进行标准曲线的绘制,样本稀释不同的梯度与显色蛋白孵育后得出的OD450值代入绘制的标准曲线中,最终得到样本中含有标签的目的蛋白的含量。最终通过绘制标准曲线进行定量,批间差<5%,线性范围广,完全可以做到绝对定量。
本发明的有益效果是:
1、针对标签的融合蛋白进行绝对定量,将抗体直接包板至ELISA板中,将HRP直接标记在显色蛋白上,后续不需要添加其余的抗体进行信号的放大,避免了多加一步抗体带来的误差和繁琐的操作;
2、可直接对培养基上清中含有标签的蛋白进行定量,不需要对蛋白进行纯化,只要求培养基中的上清中不含降解蛋白的组分;
3、解决了ELISA夹心法需要抗体抗原配对的问题,只需要蛋白含有标签就可以进行定量,具有普遍性;
4、所用原料都可通过重组表达平台进行生产,产品可大量生产,成本低,批次可控,数据稳定;
5、操作步骤简单,只需要一台酶标仪即可完成,数据分析只需要用Excel进行,可以由生产人员进行操作;
6、可进行大量样本的同时定量,真正意义上做到了通量化定量;
7、应用多样灵活,也可以进行定性分析,不需要进行标准曲线的绘制。
附图说明
下面结合附图和实施例对本发明进一步说明。
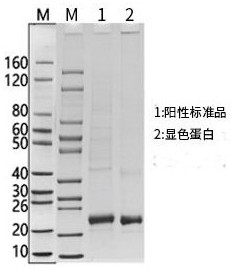
图1是His标签阳性标准品和His标签显色蛋白的SDS-PAGE电泳图;
图2是重组抗体的SDS-PAGE电泳图;
图3是实施例1数据处理后的曲线图;
图4是图3中曲线拟合后的线性范围;
图5是人Fc标签阳性标准品和人Fc标签显色蛋白的SDS-PAGE电泳图;
图6是重组抗体的SDS-PAGE电泳图;
图7是实施例2数据处理后的曲线图;
图8是图7中曲线拟合后的线性范围。
具体实施方式
实施例1:
S1,标准品的制备:
通过重组表达的方式生产阳性标准品,阳性标准品是带有His标签的蛋白,稳定性高;
1.1、全基因合成
全基因合成含有Anti-His mAb、His标签阳性标准品和His标签显色蛋白的序列,序列直接合成在真核表达载体上,载体上含有BamHI和XhoI酶切位点。
1.2、表达和纯化
采用polyscience的PEI max转染试剂进行质粒的转染。
将300μL PEI max加入至5ml optimem中,将100ug的真核表达质粒加入至5mLoptimem中,然后上下颠倒混匀后室温静置5min;然后将PEI max混合液缓慢加入含有质粒的混合液中室温静置10min,将其缓慢加入细胞中,20h后加入相应的添加剂,表达5天后进行离心,细胞离心后,取培养基上清进行纯化。
重组抗体采用重力柱纯化,将离心后的培养基上清进行纯化,100mL的Anti-HismAb培养基上清采用1mL rProteinA Beads 6FF 填料进行纯化。将1mL rProteinA Beads6FF填料进行重力柱装填,重力柱选用12ml的规格,首先用0.1M NaOH冲洗填料30min,接着用20倍柱体积的无内毒素水将NaOH冲洗干净,随后进行20倍柱体积的平衡液进行平衡冲洗,平衡液选用生理盐水,平衡冲洗后加入100mL培养基上清缓慢上样,上样的流速控制在1mL/min,上样结束后继续用20倍柱体积的生理盐水进行冲洗,冲洗后用0.1M Glycine(含有0.3M NaCl,pH=3.0)进行洗脱并分管收集,每管收集1mL,纯化后的蛋白进行SDS-PAGE电泳检测重组蛋白的纯度,最后过夜透析至生理盐水中。
重组的His标签阳性标准品和His标签显色蛋白采用重力柱纯化,将离心后的培养基上清进行纯化,100mL的His标签阳性标准品和His标签显色蛋白培养基上清采用1mL NiSmart Beads 6FF填料进行纯化。将1mL Ni Smart Beads 6FF填料进行重力柱装填,重力柱选用12ml的规格,首先用0.1M NaOH冲洗填料30min,接着用20倍柱体积的无内毒素水将NaOH冲洗干净,随后进行20倍柱体积的平衡液进行平衡冲洗,平衡液选用生理盐水,平衡冲洗后加入100mL培养基上清缓慢上样,上样的流速控制在1mL/min,上样结束后继续用20倍柱体积的生理盐水进行冲洗,冲洗后用3mM 咪唑(含有0.3MNaCl,pH=8.0)进行洗杂,最后用250mM咪唑(含有0.3M NaCl,pH=8.0)洗脱并分管收集,每管收集1mL,纯化后的蛋白进行SDS-PAGE电泳检测重组蛋白的纯度,最后过夜透析至生理盐水中。
电泳的具体条件如下:
上样2μg 的重组蛋白,4-20%的SDS-PAGE梯度胶采用MOPS缓冲液进行电泳,设置参数为160V电泳40min,最后进行考马斯亮蓝染色鉴定蛋白的大小和纯度。
实验结果如图1和图2所示,图1中阳性标准品(Lane 1)和显色蛋白(Lane 2)条带单一,纯度≥90%;图2中Anti-His mAb非还原电泳条带大小约160KDa(Lane 1),还原电泳呈现50KDa和25KDa两条带(Lane 2)。
S2,HRP标记:
将HRP标记至His标签显色蛋白上,可以用过碘酸钠法或Catcher-SPY体系进行定点标记;
取抗体10 mg,用0.01M Na
混旋过程中取装好的G-25脱盐柱用25 ml 平衡缓冲液(1mM CH
S3,阳性标准品的检测:
包被0.5ug/ml的His标签抗体,每孔100ul,在4℃条件下包被18-24hr;
包被结束后进行3-5次的PBST洗板,每次200ul,洗板结束后甩干处理;
将His标签阳性标准品与0.5ug/ml His标签显色蛋白混合,阳性标准品的起始浓度为200ug/ml,随后再进行15个孔的稀释,稀释条件为利用封闭液对阳性标准进行对半稀释后加入EP管中,随后再将前一稀释的样本再进行对半稀释加入第二个EP管中,以此类推,最后将15个处理组与显色蛋白混合,混合样品的体积用PBST补至100ul加入酶标板的孔中,其中一个孔不加入阳性标准品,直接加入PBST做为对照组,数据处理过程中称为T0,然后在37℃条件下孵育1hr;孵育结束后进行3-5次PBST洗板,每次200ul;
最后加入100ul TMB显色液显色3-8min,显色结束后加入100ul 0.25M HCl终止,在酶标仪上进行OD450读数;
S4,样本检测:
包被50ng/ml的His标签抗体,每孔100ul,4℃包被20hr;
第二天进行5次的PBST洗板,每次200ul,洗板结束后甩干处理;
取样品体积为25ul,随后再进行2个孔的稀释,稀释条件为将样品用PBST进行2倍体积的稀释,最后将3个处理组与His标签显色蛋白混合,混合样品的体积用PBST补至100ul。
实验过程中需要有一个孔不加入阳性标准品,直接加入PBS做为对照组,也就是T0组,然后37℃孵育1hr,孵育结束后进行5次PBST洗板,每次200ul,以上所有的孔都做复孔;最后加入100ul TMB显色液显色5min,显色结束后加入100ul0.25M HCl终止,在酶标仪上进行OD450读数。
S5,绘制标准曲线并进行样品浓度分析,根据OD450的读数进行标准曲线的绘制和样品浓度的分析:
根据OD450的读数进行标准曲线的绘制和样品浓度的分析。首先用检测出的OD450值(T值)除以T0(未加阳性标准品),将T/T0进行对数处理(log10)后的数据作为Y轴,加入阳性标准品的浓度做为X轴,随后在Excel中进行XY散点图的绘制,找出线性范围。
对线性范围的数据进行拟合,计算出对应的公式和R²。R²为回归平方和与总离差平方和的比值,R²介于0-1之间,越接近1,回归拟合效果越好。本实施例中R²≥0.98,证明实验数据可信。
最后将样品的T/T0数值的对数代入公式中即可得到蛋白稀释后的浓度,将数据乘以对应的稀释倍数即可得到实际样本中蛋白的表达量。
如图3为数据处理后的曲线图。
如图4为曲线拟合后的线性范围(0.8mg/L-25mg/L,R²=0.9855)。
样品稀释2倍后在线性范围内,将T/T0值代入曲线,得log(X)=0.3887左右,X=2.447ug,上清50ul,则计算上清的含量大约在47.6±3mg/L,而纯化后蛋白表达量是45±2mg/L,考虑到纯化过程中也会有一些损失,证明该方法计算得到的数据是可信的。
实施例2:
S1,标准品的制备:
通过重组表达的方式生产人Fc标签的阳性标准品,人Fc标签阳性标准品是带有人Fc标签的重组蛋白,纯度和稳定性高;
1.1、全基因合成
全基因合成含有mouse anti-human mAb、人Fc标签阳性标准品和人Fc标签显色蛋白的序列,序列直接合成在真核表达载体上,载体上含有BamHI和XhoI酶切位点。
1.2、表达和纯化
采用polyscience的PEI max转染试剂进行质粒的转染。具体方法参考实施例1中真核细胞瞬转实验。
重组抗体采用重力柱纯化,将离心后的培养基上清进行纯化,100mL的mouseanti-human mAb、人Fc标签阳性标准品和人Fc标签显色蛋白培养基上清采用1mLrProteinA Beads 6FF 填料进行纯化,步骤参考实施例1中对Anti-His mAb的纯化过程。纯化后的蛋白进行SDS-PAGE电泳检测重组蛋白的纯度,最后过夜透析至生理盐水中。
电泳的具体条件如下:
上样3μg 的重组蛋白,4-20%的SDS-PAGE梯度胶采用MOPS缓冲液进行电泳,设置参数为160V电泳40min,最后进行考马斯亮蓝染色鉴定蛋白的大小和纯度。
实验结果如图5和图6所示,图5中人Fc标签阳性标准品重组蛋白的非还原电泳(Lane 1)和人Fc标签显色蛋白(Lane 3)条带单一,纯度≥95%,还原电泳呈现50KDa和25KDa两条带(Lane 2和Lane 4);图6中mouse anti-human mAb非还原电泳条带大小约160KDa(Lane1),还原电泳呈现50KDa和25KDa两条带(Lane 2)。
S2,HRP标记:
参考实施例1中对His标签显色蛋白的HRP标记方法。
S3,阳性标准品的检测;
包被100ng/ml的mouse anti-human mAb抗体,每孔100ul,在4℃条件下包被18-24hr;
包被结束后进行3-5次的PBST洗板,每次200ul,洗板结束后甩干处理;
将人Fc标签的阳性标准品与1ug/ml人Fc标签的显色蛋白混合,阳性标准品的起始浓度为250ug/ml,随后再进行15个孔的稀释,稀释条件为利用封闭液对阳性标准进行对半稀释后加入EP管中,随后再将前一稀释的样本再进行对半稀释加入第二个EP管中,以此类推,最后将15个处理组与显色蛋白混合,混合样品的体积用PBST补至100ul加入酶标板的孔中,其中一个孔不加入阳性标准品,直接加入PBST做为对照组,数据处理过程中称为T0,然后在37℃条件下孵育1hr;
孵育结束后进行3-5次PBST洗板,每次200ul;
最后加入100ul TMB显色液显色3-8min,显色结束后加入100ul 0.25M HCl终止,在酶标仪上进行OD450读数;
S4,样本检测:
包被100ng/ml的mouse anti-human mAb抗体,每孔100ul,4℃包被20hr;
包被结束后进行5次的PBST洗板,每次200ul,洗板结束后甩干处理;
取样品体积为20ul,随后再进行2个孔的稀释,稀释条件为将样品用PBST进行2倍体积的稀释,最后将3个处理组与显色蛋白混合,混合样品的体积用PBST补至100ul。
实验过程中需要有一个孔不加入阳性标准品,直接加入PBS做为T0,然后37℃孵育1hr,
孵育结束后进行5次PBST洗板,每次200ul,以上所有的孔都做复孔;
最后加入100ul TMB显色液显色5min,显色结束后加入100ul 0.25M HCl终止,在酶标仪上进行OD450读数。
S5,绘制标准曲线并进行样品浓度分析,根据OD450的读数进行标准曲线的绘制和样品浓度的分析:
数据处理方式请参考实施例1对His标签蛋白的数据处理。
如图7为数据处理后的曲线图。
如图8为曲线拟合后的线性范围(0.1mg/L-7.8mg/L,R²=0.9969)。
样品稀释20倍后在线性范围内,将T/T0值代入曲线,得log(X)=-1.15,X=7ug/ml,乘以稀释倍数计算上清的含量大约在140±5mg/L,而纯化后蛋白表达量是135±2mg/L,考虑到纯化过程中也会有一些损失,证明该方法计算得到的数据是可信的。
以上说明对本发明而言只是说明性的,而非限制性的,本领域普通技术人员理解,在不脱离所附权利要求所限定的精神和范围的情况下,可做出许多修改、变化或等效,但都将落入本发明的保护范围内。
- 肝型脂肪酸结合蛋白质制剂、对其评价的方法、抑制使用其的测定中的肝型脂肪酸结合蛋白质引起的测定值的变动幅度的方法、肝型脂肪酸结合蛋白质、其的制造方法、编码其的DNA、由该DNA转化得到的细胞、制作肝型脂肪酸结合蛋白质的校准曲线的方法及对该蛋白质进行定量的方法
- 一种定量检测重组人表皮生长因子直接竞争ELISA法试剂盒及其应用
- 采用间接竞争ELISA法检测动物源性过敏原牛血清白蛋白