光固化性氟聚醚类弹性体组合物的涂膜形成方法
文献发布时间:2023-06-19 11:57:35
技术领域
本发明为涉及一种通过用光、特别是用紫外线照射而进行固化的光固化性氟聚醚类弹性体组合物的涂膜形成方法的发明,且本发明提供一种在涂布组合物的基材的暗处或深处以及基材界面(特别是在树脂基材界面)上形成均匀的固化物涂膜的涂膜形成方法。
背景技术
以往,光固化性氟聚醚类凝胶组合物和橡胶组合物,作为通过照射光、特别是照射紫外线进而在常温且短时间的条件下能够得到耐热性、低温性、耐化学品性、耐溶剂性以及耐油性等综合性能优异的凝胶和橡胶固化物的材料一事已被公开(专利文献1和专利文献2)。
另外,光固化性氟聚醚类弹性体组合物可通过预先将硅烷类底漆涂布在基材上,进而作为具有低温固化性且粘接性的材料使用一事也被公开(专利文献3)。
在上述组合物的固化工序中,由于不需要加热,因此无需加热炉所带来的制造设备的省空间化以及引入制造设备的费用的降低、以及从间歇式生产向连续式生产的转换成为可能,因此,从制造效率的改善、以及伴随其所带来的制造成本降低等生产率提高的观点来看,上述组合物为有用的材料。
另外,上述组合物由于不受由使用丙烯酸类紫外线固化材料而容易产生问题的氧而引起的固化阻碍或不受由使用环氧类紫外线固化材料容易产生问题的水分而引起的固化阻碍,因此,不受来自安装操作和设备环境等的制约,进而能够容易地制造固化物。
另外,由于上述组合物即使对紫外线照射后的固化速度也能够通过调节紫外线照射量和环境温度进行控制,例如,即使在对基材上的组合物进行紫外线照射之后再与另一基材进行粘接操作的情况下,也能够通过调节固化速度来确保充足的操作时间。另外,上述组合物还能够通过在紫外线照射后进行加温来促进固化反应,进而缩短固化时间。
另外,由于上述组合物还具有暗处固化性和深处固化性,因此,即使在涂布组合物的基材的结构上具有部分光照射不到的(背阴)的部位、或用厚膜涂布也欲使其固化的情况下,也能够得到充分的固化物。
现有技术文献
专利文献
专利文献1:日本专利6020327号公报
专利文献2:日本特开2009-149782号公报
专利文献3:日本特开2017-039810号公报
发明内容
发明要解决的问题
但是,上述光固化性氟聚醚类凝胶组合物、橡胶组合物以及弹性体组合物虽然具有暗处固化性和深处固化性,但是存在下述问题:相对于明部其暗处的面积大或膜的厚度为厚的情况下,以及在组合物中含有二氧化硅等散射光的添加剂的情况下等,该组合物固化不充分;或者,在需要长时间固化、且要对基材进行粘接的情况下,在将组合物涂布在基材上并对该组合物进行紫外光照射后,必须将该基材与其他基材粘接。另外,还具有如下问题:在将组合物涂布在基材上、特别是将组合物涂布在树脂制的基材上并进行光固化的情况下,受到来自基材的固化阻碍,进而在基材界面(特别是在树脂基材界面)的固化变得不充分。
用于解决问题的方案
本发明者们为解决上述问题,汇集精心研究的结果,发现不是在将光固化性氟聚醚类弹性体组合物涂布基材上后再对其进行光照射,而是通过将预先用光照射过的光固化性氟聚醚类弹性体组合物涂布在基材上,即使在基材(特别是树脂基材)的暗处或深处以及界面处也能够得到充分固化了的均匀的固化物,进而完成了本发明。即,本发明为提供以下的光固化性氟聚醚类弹性体组合物的涂膜形成方法的发明。
[1]
一种光固化性氟聚醚类弹性体组合物的涂膜形成方法,其中,
包括将已被光照射的光固化性氟聚醚类弹性体组合物涂布在基材表面上的工序。
[2]
如[1]所述的光固化性氟聚醚类弹性体组合物的涂膜形成方法,其特征在于,
完成对光固化性氟聚醚类弹性体组合物进行光照射,然后,将所述光固化性氟聚醚类弹性体组合物涂布在基材表面上。
[3]
如[1]所述的光固化性氟聚醚类弹性体组合物的涂膜形成方法,其特征在于,
在对光固化性氟聚醚类弹性体组合物进行光照射的同时将所述光固化性氟聚醚类弹性体组合物涂布在基材表面上。
[4]
如[1]~[3]中任1项所述的涂膜形成方法,其特征在于,
将最大峰波长为300~400nm范围的紫外线,以累积光量为100mJ/cm
[5]
如[1]~[3]中任一项所述的涂膜形成方法,其中,
光固化性氟聚醚类弹性体组合物含有以下成分,
(A)成分:在一个分子中具有两个以上的烯基、且在主链中具有全氟聚醚结构的直链聚氟化合物,所述(A)成分的含量为100质量份;
(B)成分:在一个分子中具有两个以上已直接键合在硅原子上的氢原子(SiH基)的含氟有机氢硅氧烷,所述(B)成分的含量为相对于(A)成分的烯基1摩尔作为SiH基为0.5~3.0摩尔的量;
(C)成分:光活性型氢化硅烷化反应金属催化剂,所述(C)成分的含量为相对于(A)成分以金属原子的质量换算为0.1~500ppm的量。
[6]
如[5]所述的涂膜形成方法,其中,
光固化性氟聚醚类弹性体组合物进一步含有以下的(D)成分,
(D)成分:在一个分子中具有一个烯基、且在主链中具有全氟聚醚结构的聚氟单烯基化合物,相对于(A)成分100质量份,所述(D)成分的含量为1~300质量份。
[7]
如[5]或[6]所述的涂膜形成方法,其中,
光固化性氟聚醚类弹性体组合物进一步含有以下的(E)成分,
(E)成分:选自由以下记通式(1)~通式(3)表示的化合物组成的组的至少一种非官能性全氟聚醚化合物,相对于(A)成分和(B)成分的总量100质量份,所述(E)成分的含量为0~150质量份,
A-O-(CF
在式(1)中,A为以式C
A-O-(CF
在式(2)中,A与上述相同,c和d分别为1~300的整数,
在式(3)中,A与上述相同,e和f分别为1~300的整数。
[8]
如[5]~[7]中任一项所述的涂膜形成方法,其中,
所述光固化性氟聚醚类弹性体组合物进一步含有以下的(F)成分,
(F)成分:BET比表面积为0.1m
[9]
如[5]~[8]中任一项所述的涂膜形成方法,其中,
所述光固化性氟聚醚类弹性体组合物进一步含有以下的(G)成分,
(G)成分:氢化硅烷化反应的反应控制剂。
[10]
如[5]~[9]中任一项所述的涂膜形成方法,其中,
(A)成分为以下述式(I)所表示的直链聚氟化合物,
CH
在式(I)中,X为-CH
Y为-CH
CH
在式(Z)中,R
在式(I)中,X’为-CH
在式(Z’)中,R
在式(I)中的g独立地为0或1,
在式(I)中的Rf
在式(i)中,p和q分别为0或1~150的整数,且p和q之和的平均值为2~200,r为0~6的整数,t为2或3,
在式(ii)中,u为1~200的整数,v为1~50的整数,且t为2或3。
[11]
如[5]~[10]中任一项所述的涂膜形成方法,其中,
(B)成分的含氟有机氢硅氧烷在一个分子中具有一个以上的一价全氟烷基、一价全氟氧烷基、二价全氟亚烷基和/或二价全氟氧亚烷基。
[12]
如[5]~[11]中任一项所述的涂膜形成方法,
其中,(C)成分的光活性型氢化硅烷化反应金属催化剂为(η
发明的效果
根据本发明的光固化性氟聚醚类弹性体组合物的涂膜形成方法,即使在基材(特别是树脂基材)的暗处、深处以及界面也能够形成均匀的该组合物的固化物的涂膜,且该固化物的涂膜其耐热性、低温性、耐化学品性、耐溶剂性以及耐油性等优异。
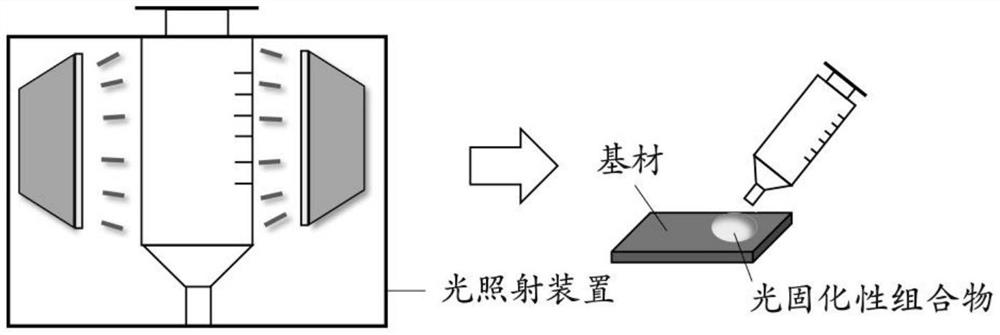
附图说明
图1为使用多个光源灯对在注射器中的组合物进行光照射后再涂布该组合物的方法的示意图。
图2为对在培养皿中的组合物进行光照射后再涂布该组合物的方法的示意图。
图3为在直线型的排出管部对组合物进行光照射的同时涂布该组合物的方法的示意图。
图4为在折叠型的排出管部对组合物进行光照射的同时涂布该组合物的方法的示意图。
图5为用铝箔覆盖铝制培养皿的一端、进而具有暗处的容器的示意图。
图6为在光照射通过排出管的组合物的同时将该组合物涂布在图5的铝制培养皿内的方法的示意图。
图7为将组合物涂布在铝制培养皿内,并从上方进行光照射的方法的示意图。
图8为在光照射通过排出管的组合物的同时,以厚度10mm的方式将该组合物涂布在不具有暗处的铝制培养皿内的方法的示意图。
图9为以厚度10mm的方式将组合物涂布在铝制培养皿内,并从上方进行光照射的方法的示意图。
图10为表示调查将参考例1~3的组合物密封在遮光瓶中,且在40℃条件下保存30天时的随时间推移的粘度变化的结果的图表。
图11为表示以厚度1.0mm的方式将组合物涂布在铝基材上进行光照射,且在25℃条件下随时间推移的测定弹性模量的结果的图表。
图12为表示以厚度1.0mm的方式将组合物(除参考例7之外)涂布在聚烯烃基材上,且在25℃条件下随时间推移的测定弹性模量的结果的图表。
图13为表示以厚度1.0mm的方式将组合物(除参考例9之外)涂布在PPS基材上,且在25℃条件下随时间推移的测定弹性模量的结果的图表。
图14为表示以厚度1.0mm的方式将组合物(除参考例10之外)涂布在PET基材上,且在25℃条件下随时间推移的测定弹性模量的结果的图表。
具体实施方式
以下,对本发明进行详细地说明,但本发明并不限于此。
[对光固化性氟聚醚类弹性体组合物的光照射条件]
首先,对用于本发明的光固化性氟聚醚类弹性体组合物的光照射条件进行说明。
光固化性氟聚醚类弹性体组合物可通过光照射而被固化。在固化时,所照射的光处在发射光谱中的最大峰值波长为300~400nm的区域,且处在短于300nm的波长区域中的各个波长的辐照度为所述最大峰值波长的辐照度的5%以下、优选为1%以下、更优选为0.1%以下,即越接近0越为优选。当将处于波长短于300nm的波长区域,且具有辐照度比所述最大峰值波长的辐照度的5%大的波长的光照射在所述组合物上时,有可能会发生聚合物末端基团的分解或者导致部分催化剂的分解,从而无法得到被充分固化的固化物。
使光固化性氟聚醚类弹性体组合物进行良好固化的紫外线照射量,作为累积光量为100mJ/cm
紫外线照射可以用具有多个发射光谱的光进行照射,也可用具有单一发射光谱的光进行照射。另外,单一的发射光谱也可为具有在300nm至400nm的区域中的宽光谱。具有单一的发射光谱的光为具有300nm至400nm、优选为350nm至380nm范围内的峰值(即,最大峰值波长)的光。作为用于照射这种光的光源,可列举例如,紫外线发光二极管(紫外线LED)或紫外线发光半导体激光器等的紫外线发光半导体元件光源。
作为照射具有多个发射光谱的光的光源,可列举为例如,金属卤化物灯、氙灯、碳弧灯、化学灯、钠灯、低压汞灯、高压汞灯、超高压汞灯等的灯等,氮气等的气体激光器,有机色素溶液的液体激光器,无机单晶中含有稀土类离子的固体激光器等。
当所述光源的光在发射光谱中于比300nm短的波长区域中具有峰值的情况下、或者存在当在比300nm短的波长区域中具有比在所述发射光谱中的最大峰值波长的辐照度的5%大的辐照度的波长的情况下(例如,当发射光谱在整个宽波长范围内很宽的情况下),可通过滤波器除去处于比300nm短的波长区域的波长的光。由此,将处于比300nm短的波长区域的各波长的辐照度调整为最大峰值波长的辐照度的5%以下、优选为1%以下、更优选为0.1%以下、进一步优选为0%。需要说明的是,当在发射光谱中于300nm至400nm的波长区域中存在多个峰值的情况下,将在其中显示最大吸光度的峰值波长作为最大峰值波长。滤波器没有特别限制,只要为能够除去比300nm短的波长的即可,也可以使用公知的滤波器。例如,可以使用365nm带通滤波器等。还需要说明的是,紫外线的照度和光谱分布可以用分光辐射度计、例如USR-45D(USHIO电机)进行测定。
作为光照射装置,没有特别限定,可以使用例如,点式照射装置、面式照射装置、条式照射装置以及传送式照射装置等的照射装置。
兼顾到光源照度,在使光固化性氟聚醚类弹性体组合物固化时的光照射时间的标准为,例如照射1~300秒、优选为10~200秒、更优选为20~150秒,且在上述光照射的1~60分钟后,特别为在上述光照射的5~30分钟后,光固化性组合物失去流动性,能够得到凝胶状或橡胶状的弹性体(固化物)。
[光固化性氟聚醚类弹性体组合物的涂膜形成方法]
接下来,对在本发明中的光固化性氟聚醚类弹性体组合物的涂膜形成方法进行说明。
本发明的涂膜形成方法的特征在于,将已光照射过的组合物涂布在基材的表面上,并示出以下两种方法。
i)在对光固化性氟聚醚类弹性体组合物进行光照射完了之后,将该光固化性氟聚醚类弹性体组合物涂布在基材表面上的方法
ii)在对光固化性氟聚醚类弹性体组合物进行光照射的同时,将该光固化性氟聚醚类弹性体组合物涂布在基材表面上的方法
以下,对上述两种涂布方法分别进行详细地说明。
i)在对光固化性氟聚醚类弹性体组合物进行光照射完了之后,将该光固化性氟聚醚类弹性体组合物涂布在基材表面上的方法
该方法为在对光固化性氟聚醚类弹性体组合物进行光照射完了之后,再将该光固化性氟聚醚类弹性体组合物涂布在基材表面上作为特征的方法。在该方法中,可以对容器中的组合物进行光照射,然后将已被光照射的组合物直接进行涂布、或者也可以将已被光照射的组合物转移到其它容器中并进行涂布。在任一种方法中,其光照射时的容器都选择无固化阻碍的影响或低固化阻碍的影响的容器。
作为无固化阻碍的影响的容器的例子,可列举玻璃制或金属制的容器。对于树脂制容器,虽存在固化阻碍的可能性,但依其树脂的种类、生产厂家以及等级等其影响也为各种各样。如果事先能够确认其为无影响或低影响的,则也可以适当地使用。
作为容器的形状,可以选择培养皿、盘、桶、瓶、罐、注射器以及药筒等易于使用的容器。特别是在所要涂布的位置为细节部分或者需要控制所涂布的量的情况下,可以适当地使用注射器或药筒。
在透过容器对组合物进行光照射的情况下,则对所要使用的容器要求具有透光性。作为透光性材质,可具体列举玻璃、丙烯酸类树脂、聚碳酸酯树脂、聚丙烯树脂以及聚对苯二甲酸乙二醇酯(PET)树脂等透明材料。
就光照射时的组合物的厚度而言,在厚度薄时,光会被无遗漏地照射在组合物上,光照射效率提高,因此优选为薄型。例如,在将组合物填充在培养皿、盘、桶等中并进行光照射的情况下(图2),其将组合物的厚度变薄并进行光照射时能够进一步提高固化速度。即使在将组合物填充在注射器或药筒中而进行使用的情况下(图1)也为同样,由于使用小直径的注射器、且对其中组合物进行光照射,或者将在培养皿中涂以薄层进行一次光照射后的组合物换装在注射器内再进行使用等的方法,由于光照射效率提高,进而能够得到良好的固化速度,因此为优选。
作为对组合物提高光照射效率的其它方法,可列举为对已被填充组合物的容器进行广范围的光照射的方法和多台设置光源灯进行光照射的方法以及使用反射镜的方法等。
作为在所述方法中的涂布方法,可表示为例如,图1和图2等。
ii)在对光固化性氟聚醚类弹性体组合物进行光照射的同时,将该光固化性氟聚醚类弹性体组合物涂布在基材表面上的方法
该方法为,在用光照射光固化性组合物的同时,将该光固化氟聚醚类弹性体组合物涂布在基材表面上的方法,其优选为在光照射已填充有组合物的容器的排出管部分的同时进行涂布的方法。与i)的方法的不同之处在于,对组合物进行光照射的部位仅限于马上用于涂布的部分。由此,例如在i)的方法中,如果容器中已经用光照射的组合物没有用完,则必须废弃该组合物。但在ii)的方法中,由于光照射的部位仅限于排出管部,因此,可以防止组合物的废弃。即使组合物在排出管部已被固化,如果更换排出管,仍然可以继续使用容器内的剩余组合物。
作为在被安装于容器的排出管部对组合物进行光照射的方法,可例举穿透排出管对组合物进行光照射的方法。在这种情况下,要求排出管的材质要具有透光性。作为透光性的材质,可例示在i)中所例举的材质,也可以根据需要进行加工和使用。
需要说明的是,在该方法中,上述容器优选为可推挤出方式的注射器等。
作为排出管的加工例,对排出管的粗度和长度以及形状的加工进行说明。关于粗度,因为细的一方光可容易无遗漏地照射到组合物,因此被优选。关于长度,也可以通过调整被光照射的排出管的路径长度来调整对组合物的光照射量。也就是说,在希望增加对组合物的光照射量的情况下,通过设计长排出管,且以在组合物通过该排出管期间进行光照射的方式进行设定,从而能够对组合物进行更为长时间的光照射。另外,关于形状,例如,由于通过使其形成为折叠形或环形,即使为相同的排出管长度也可以使光照射区域变窄,因此,其对涂布设备的小型化为有效。
作为光源灯,通过使用可在广范围内进行光照射的光源灯或已将数盏组合的光源灯以及使用反射镜的光源灯等,可以提高光照射效率且能够促进固化反应。
作为在所述方法中的涂布方法,可表示为例如,图3和图4等。
本发明的涂膜形成方法能够用在对在以往的涂膜形成方法中具有受到固化阻碍可能性的树脂基材(例如,聚苯硫醚(PPS)树脂基材、环氧树脂基材、酚醛树脂基材、聚对苯二甲酸乙二醇酯(PET)树脂基材、三聚氰胺树脂基材、脲醛树脂基材、聚酯树脂基材、聚氨酯树脂基材、聚酰亚胺树脂基材、聚酰胺树脂基材、聚烯烃树脂基材、聚氯乙烯树脂基材以及ABS树脂基材等有机树脂基材)上。
在本发明的涂膜形成方法中,作为将已被光照射的组合物涂布到基材上的方法,包括以喷涂、浸渍等进行涂布组合物的情况。此时,可以根据组合物的粘度适当地选择涂膜的形成方法,也可以根据涂膜的形成方法用溶剂适当地稀释来调节组合物的粘度。
作为用于稀释的溶剂,可例示如甲苯、乙酸乙酯、甲醇、2-丙醇、正己烷以及甲基乙基酮等常用的有机溶剂;1,2-双(三氟甲基)苯、1,3-双(三氟甲基)苯、1,4-双(三氟甲基)苯、1,3,5-三(三氟甲基)苯、1,3-双(三氟甲基)环己烷、1,1,1,2,2,3,3,4,4,5,5,6,6-十三氟辛烷、二氯五氟丙烷、乙基九氟丁基醚、乙基九氟异丁基醚、甲基全氟异丁基醚以及甲基全氟丁基醚等氟类有机溶剂。从对组合物的溶解性、溶剂的挥发性以及对基材的流平性等方面的考虑,可以适当地选择这些有机溶剂中的一种进行使用,或将由这些有机溶剂中的两种以上形成的混合物作为混合溶剂进行使用。
[光固化性氟聚醚类弹性体组合物]
接下来,以下对用于本发明的光固化性氟聚醚类弹性体组合物的各成分进行详细地说明。
〔(A)成分〕
在光固化性氟聚醚类弹性体组合物中的(A)成分为在一个分子中具有两个以上的烯基的直链聚氟化合物。其中,烯基数优选为在一分子中含有2~30个,更优选为2~10个,特别优选为2~6个。
在(A)成分中,所说的“直链”是指由构成主链的全氟聚醚结构的全氟氧化亚烷基组成的重复单元彼此键合为直链的含义。每个重复单元本身也可含有支链状的全氟氧化亚烷基单元(例如,[-CF
作为(A)成分,特别优选为以由下述式(I)表示的在主链中的全氟氧化亚烷基重复单元中具有支链结构的直链聚氟化合物。
CH
[在式(I)中的X为-CH
Y为-CH
在式(Z)中,R
在式(I)中的X’为-CH
在式(Z’)中,R
在式(I)中的g独立地为0或1;
在式(I)中的Rf
在式(i)中,p和q分别为0或1~150的整数,且p和q之和的平均为2~200,r为0~6的整数,t为2或3,
在式(ii)中,u为1~200的整数,v为1~50的整数,且t为2或3。]
其中,作为R
其中,式(I)中的Rf
(在式(i)中,p和q分别为0或1~150的整数、优选为10~150的整数,且p和q之和的平均值为2~200、优选为20~160。另外,r为0~6的整数、优选为0~4的整数,t为2或3。)
(在式(ii)中,u为1~200的整数、优选为20~160的整数,v为1~50的整数、优选为5~40的整数,t为2或3。)
作为Rf
(在式(i-1)中,p1和q1分别为1~150的整数,并且p1+q1(平均)=2~200。L为2~6的整数。)
(式(i-2)中,p2和q2分别为1~150的整数,p2+q2(平均)=2~200。L为2~6的整数。)
(在式(ii-1)中,u1为1~200的整数,v1为1~50的整数。)
作为(A)成分的优选例,可列举以下述通式(II)表示的化合物。
[在式(II)中,X
作为以式(I)表示的直链聚氟化合物的具体例,可列举以下述式表示的化合物。
(其中p’和q’分别为1~150的整数,p’+q’(平均)=6~200)
(式中,p’和q’分别为1~150的整数,p’+q’(平均)=6~200。)
(式中,p”和q”分别表示为1~150的整数、且为满足p”+q”=2~200的数。)
在以式(I)表示的直链聚氟化合物中所含有的烯基的量,优选为0.00005~0.00050摩尔/g、更优选为0.00007~0.00040摩尔/g。如果在直链聚氟化合物中所含有的烯基的量过少,则有时固化物的物理强度降低或者得不到固化物。另外,如果烯基的量过多,则有时得到的固化物变脆,从而容易破裂。
需要说明的是,式(I)的直链聚氟化合物的粘度(23℃条件下)优选为在5~100000mPa·s的范围内、更优选为在500~50000mPa·s的范围内、进一步优选为在1000~20000mPa·s的范围内。如果将具有该范围内粘度的直链氟化合物用于光固化性组合物、且将该组合物用于密封、灌封、涂布、浸渍等方面,由于固化物具有适宜的物理特性,则为优选。式(I)的直链聚氟化合物可根据用途去选择最合适的粘度。此时,也可以将低粘度聚合物和高粘度聚合物进行混合,进而调整至所希望的使用粘度。需要说明的是,粘度可以用旋转粘度计(例如,BL型、BH型、BS型、锥板型、流变仪等)进行测定,特别是通过JIS K7117-1中所定义的粘度测定可以求出以上述通式(I)或通式(II)表示的直链聚氟化合物的粘度(23℃条件下)。
另外,以构成主链的全氟聚醚结构的全氟氧化亚烷基单元的重复数等所反映的直链聚氟化合物的聚合度(或分子量),例如,可以将氟类溶剂作为洗脱溶剂,且作为在凝胶渗透色谱(GPC)分析中的聚苯乙烯的数均聚合度(或数均分子量)等而求出。
进一步,在本发明中,为了将式(I)的直链聚氟化合物调整至根据目的所需要的平均分子量,可以预先在常规的方法和条件下,将直链聚氟化合物与在分子内含有二个氢甲硅烷基(Si-H基)的有机硅化合物进行氢化硅烷化反应所扩链而得到的生成物作为(A)成分使用。
这些直链聚氟化合物可以单独使用一种,或也可以组合使用二种以上。
在本发明的组合物中,(A)成分的含量优选为含有10~98质量%、更优选为20~95质量%、进一步优选为30~90质量%。
〔(B)成分〕
(B)成分为在一个分子中具有一个以上、优选为1~10个含氟有机基团、且具有2个以上、优选为3~50个直接键合到硅原子上的氢原子(即,以Si-H表示的氢甲硅烷基)的含氟有机氢硅氧烷。(B)成分为作为(A)成分的交联剂和/或扩链剂而发挥作用的成分。另外,从与(A)成分的相容性、分散性、固化后的均匀性等观点来看,作为含氟有机基团,(B)成分优选为在一个分子中具有一个以上的一价全氟烷基、一价全氟氧烷基、二价全氟亚烷基和/或二价全氟氧亚烷基等含氟基团的含氟有机氢硅氧烷。
作为该一价或二价的含氟有机基团,可列举例如,以下述式表示的基团等。
C
-C
(在式中,g为1~20的整数,优选为2~10的整数。)
(在式中,f为1~200的整数、优选为1~100的整数,h为1~3的整数。)
(在式中,i和j分别为1以上的整数、优选为1~100的整数,i+j的平均为2~200、优选为2~100。)
-(CF
(在式中,d和e分别为1~50的整数、优选为1~40的整数。)
另外,其优选为通过二价连接基团将这些全氟烷基、全氟氧烷基、全氟亚烷基或全氟氧亚烷基与(B)成分中的硅原子进行连接。作为该二价的连接基团,可列举为亚烷基、亚芳基以及它们的组合、或也可将醚键(氧原子)、酰胺键、羰基键、酯键、二有机亚甲硅基等介在于这些基团上者,例如,可列举下述的碳原子数2~13的二价的连接基团等。
-CH
-CH
-CH
-CH
-CH
-CH
-CH
-CH
-CH
-CH
(在式中,Ph为苯基,Ph’为亚苯基。)
另外,作为在该(B)成分的含氟有机氢硅氧烷中的上述一价或二价的含氟有机基团和已与硅原子键合的氢原予以外的、已与硅原子键合的一价取代基,可列举为例如,甲基、乙基、丙基、丁基、己基、环己基、辛基以及癸基等烷基;乙烯基、烯丙基等烯基;苯基、甲苯基以及萘基等芳基;苄基、苯乙基等芳烷基,以及这些基团的部分氢原子或全部氢原子被氯原子、氰基等取代的、例如,氯甲基、氯丙基以及氰乙基等的碳原子数为1~20、优选为碳原子数1~12的未经取代或经取代的一价烃基。
作为(B)成分的含氟有机氢硅氧烷,可以为环状、链状以及三维网状以及它们的组合中的任一者。该含氟有机氢硅氧烷中的硅原子数并无特别地限定,通常为2~60、优选为3~30左右。
作为具有这种一价或二价的含氟有机基团和与硅原子键合的氢原子的(B)成分,可列举例如下述的化合物。这些化合物既可以单独使用一种,也可以并用两种以上。需要说明的是,在下述式中,Me表示甲基,Ph表示苯基。
在上述(B)成分的含氟有机氢硅氧烷中所含有的Si-H基团的量,优选为0.00050~0.01000摩尔/g、更优选为0.00100~0.00800摩尔/g。如果在含氟有机氢硅氧烷中所含有的Si-H基团的量过少,则有时导致交联密度不充分,所得到固化物的物理特性降低。如果Si-H基团的量过多,则有时在固化时产生发泡和/或所得到的固化物的物理特性会随时间的推移发生大的变化。
相对于(A)成分中所含有的乙烯基、烯丙基以及环烯基等的烯基1摩尔,上述(B)成分的配合量为(B)成分中的氢甲硅烷基,即Si-H基为0.5~3.0摩尔、优选为0.8~2.0摩尔的量。如果氢甲硅烷基(≡Si-H)过少,则会导致为交联密度变得不充分的后果,从而无法得到固化物。另外,如果氢甲硅烷基过多时,则会导致在固化时产生发泡。
〔(C)成分〕
(C)成分为光活性型氢化硅烷化反应金属催化剂。光活性型氢化硅烷化反应金属催化剂为通过光照射、特别是通过在300~400nm的近紫外线的照射使该催化剂中的金属活性化,进而促进(A)成分中的烯基与(B)成分中的氢甲硅烷基的加成反应的催化剂。该光活性型氢化硅烷化反应金属催化剂主要为铂族类金属催化剂或镍类金属催化剂。作为铂族类金属催化剂,有铂类、钯类以及铑类的金属络合化合物;作为镍类金属催化剂为,有镍类、铁类以及钴类的金属络合化合物。其中,由于铂类金属络合化合物较为容易入手,且表现出良好的催化活性,故被优选。
作为光活性型铂类金属络合化合物为例如,(η
当使用这些催化剂时,在它们为固体催化剂时虽可以以固态进行使用,但为了得到更均匀的固化物,优选为将催化剂溶解在适宜的溶剂中,然后再将得到的混合溶液与(A)成分的直链聚氟化合物相容而进行使用。
对所使用的溶剂的种类没有特别限制,只要为能够溶解催化剂的即可。但是,由于使用烃基的一部分氢被氟取代的溶剂、或者使用烃基的一部分氢被氟取代的溶剂和烃基的全部氢被氟取代的溶剂的混合溶剂具有可将催化剂更均匀地分散在组合物中的优点,因此更被优选。
作为烃基的一部分氢被氟取代的溶剂,可列举为,例如,1,3-双(三氟甲基)苯、1,2-双(三氟甲基)苯、1,4-双(三氟甲基)苯、1-甲基-3-(五氟乙基)苯、1,1,1-三氟-3-[3-(三氟甲基)苯基]丙-2-酮、4-氟-3-(三氟甲基)苯甲酸甲酯、七氟丁酸正丁酯、3,5-双(三氟甲基)苯甲酸乙酯、2-甲基-5-(三氟甲基)苯甲醛以及2,3-二甲氧基三氟甲苯等。
另外,作为烃基的全部氢被氟取代的溶剂,可列举为,例如,八氟甲苯、(1,1,2,3,3,3-六氟丙氧基)全氟苯、五氟乙基2,2,2-三氟乙醚,Fluorinert(3M公司制造)、PF5060(3M公司制造)、全氟聚醚低聚物等。
在将催化剂作为溶液使用的情况下,(A)成分的直链聚氟化合物和催化剂溶液的优选混合比为在100:0.01~1.00(质量比)的范围。在催化剂溶液的配合量大于该范围的情况下,有可能固化物的物理特性会受到影响;另一方面,在催化剂溶液的配合量小于该范围的情况下,可能导致对组合物产生分散不良。因此,以催化剂溶液的浓度控制在该混合比的范围内的方式进行适当地调整为宜。
相对于(A)成分的使用量,(C)成分的使用量为以该催化剂中的金属原子的质量换算为0.1~500ppm、优选为1~100ppm。如果(C)成分的使用量过少,则光固化组合物有可能不能得到足够的光固化性。如果(C)成分的使用量过多,则有可能导致产生发泡和/或对固化物的耐热性产生不良的影响。
〔(D)成分〕
在光固化性氟聚醚类弹性体组合物中,可以配合作为(D)成分的在一个分子中具有一个烯基、且在主链中具有全氟聚醚结构的聚氟单烯基化合物。
作为(D)成分,特别优选为以下述式(4)表示的聚氟单烯基化合物。
Rf
[在式(4)中,X’与上述式(I)中的X’相同,g为0或1。Rf
C
(式中s’为0~8的整数,h为1~6的整数,i为0~200的整数、优选为10~100的整数、更优选为20~50的整数,t’为1或2。)]
在上述式(4)中,作为Rf
(在式中,s为1~8的整数,i1为0~200的整数,i2+i3为0~200的整数。)
作为以上述通式(6)表示的聚氟单烯基化合物的具体例,可列举例如,下述的化合物。
(在上述式中,i’为0~200的整数。)
在上述聚氟单烯基化合物中含有的烯基的量优选为0.005~0.050摩尔/100g、更优选为0.010~0.040摩尔/100g。如果在聚氟单烯基化合物中含有的烯基的量过少,则伴随聚合物粘度的增加,其结果有时导致操作性降低;如果在聚氟单烯基化合物中含有的烯基的量过多,则有可能对组合物的溶解性降低且光固化性降低,或者有可能对固化物的物理特性带来不均匀等的不良影响。
需要说明的是,由于与(A)成分的直链聚氟化合物同样的理由,(D)成分的聚氟单烯基化合物的通过旋转粘度计所测定的粘度(23℃)优选为5~100000mPa·s的范围。
配合(D)成分的聚氟单烯基化合物可为任意。通过配合该成分,能够在不损害组合物的耐化学品性、耐溶剂性、低温特性等物性的情况下,调整组合物粘度、固化物的硬度以及断裂伸长率等的物性。
在光固化性氟聚醚类弹性体组合物中,相对于上述(A)成分的直链含氟聚合物100质量份,(D)成分的配合量为0~300质量份、优选为1~150质量份。由于配合(D)成分具有降低交联密度的效果,因此,依据固化产物的所需硬度是为橡胶状还是为凝胶状,能够适宜地调整其配合量来使用。即,如果(D)成分的配合量少,则固化后的交联密度增高,进而得到橡胶状的固化物。如果(D)成分的配合量多,则固化后的交联密度降低,进而得到凝胶状的固化物。如果(D)成分的配合量超过300质量份,则由于交联密度变得过低,在固化后也仍为液状,进而难以形成为固化物。
〔(E)成分〕
在本发明的组合物中,可配合作为(E)成分的、选自由以下通式(1)~(3)表示的化合物组成的组的至少一种非官能性全氟聚醚化合物。
A-O-(CF
[在式(1)中,A为以式:C
A-O-(CF
(在式(2)中,A为与上述相同,c和d分别为1~300的整数、特别为2~100的整数。)
(在式(3)中,A为与上述相同,e和f分别为1~300的整数、特别为2~100的整数。)
配合为(E)成分的非官能性全氟聚醚化合物可为任意。通过配合该成分,能够在不损害其它物性等的情况下,进一步改善组合物粘度的调整、提高固化物的耐化学品性或耐溶剂性以及低温特性。
由于与(A)成分的直链聚氟化合物同样的理由,(E)成分的通过旋转粘度计所测定的粘度(23℃条件下)优选为5~100000mPa·s的范围。
相对于(A)成分和(B)成分的总量100质量份,(E)成分的配合量优选为0~150质量份、更优选为0~100质量份、特别优选为0~80质量份。(E)成分的配合量如果超过150质量份,则随着时间的推移可能会从固化物中产生渗出。需要说明的是,(E)成分可以单独使用一种,也可以并用两种以上。
〔(F)成分〕
在本发明的组合物中,可以配合作为(F)成分的、BET比表面积为0.1m
该疏水性二氧化硅微粉末的BET比表面积优选为0.1~1000m
作为二氧化硅微粉末,可例示为干式二氧化硅、沉降法二氧化硅、凝胶法二氧化硅、胶体二氧化硅以及熔融球形二氧化硅等。其中,从对组合物中的分散稳定性或对固化物的增强效果等方面考虑,其最优选为干式二氧化硅。
上述疏水化二氧化硅微粉末,可以通过用疏水性处理剂处理已与二氧化硅表面的硅原子键合的羟基(硅烷醇基团)而得到。
作为疏水化处理剂,可例示为有机二硅氮烷、有机烷氧基硅烷、有机氯硅烷、环状有机聚硅氮烷以及线型有机聚硅氧烷等,其优选为有机二硅氮烷、有机烷氧基硅烷以及有机氯硅烷。更具体说来,可列举为六甲基二硅氮烷、六乙基二硅氮烷、六丙基二硅氮烷、1,3-二乙基-1,1,3,3-四甲基二硅氮烷、1,3-二甲基-1,1,3,3-四乙基二硅氮烷、1,3-二乙烯基四甲基二硅氮烷、1,3-二烯丙基四甲基二硅氮烷、1,3-二丁烯基四甲基二硅氮烷、1,3-二戊烯基四甲基二硅氮烷、1,3-二己烯基四甲基二硅氮烷、1,3-二庚烯基四甲基二硅氮烷、1,3-二辛烯基四甲基二硅氮烷、1,3-二壬基四甲基二硅氮烷、1,3-二癸基四甲基二硅氮烷、1,3-二乙烯基四乙基二硅氮烷、1,3-二甲基四乙烯基二硅氮烷等二硅氮烷化合物;甲基三甲氧基硅烷、乙基三甲氧基硅烷、丙基三甲氧基硅烷、乙烯基三甲氧基硅烷、烯丙基三甲氧基硅烷、丁烯基三甲氧基硅烷等烷氧基硅烷化合物;三甲基氯硅烷、三乙基氯硅烷、三丙基氯硅烷、二甲基二氯硅烷、二乙基二氯硅烷、二丙基二氯硅烷、二甲基乙烯基氯硅烷、烯丙基二甲基氯硅烷等氯硅烷化合物等。从操作性或与二氧化硅表面的硅烷醇基团的反应性等观点考虑,优选为使用硅氮烷化合物或氯硅烷化合物,特别优选为使用六甲基二硅氮烷、1,3-二乙烯基四甲基二硅氮烷以及二甲基二氯硅烷。
相对于(A)成分100质量份,该(F)成分的配合量为0.1~40质量份的范围、优选为1~30质量份的范围。在(F)成分的配合量低于0.1质量份的情况下,则不能调整所得到的固化物的物理性特性;另一方面,如果(F)成分的配合量高于40质量份,则组合物的流动性变差,另外,有时通过光进行的固化性也会显著降低。
疏水性二氧化硅细粉,可以使用组合上述疏水化处理剂进行了疏水处理的二氧化硅细粉,也可将分别使用疏水性处理剂进行了单独处理的二氧化硅细粉加以混合后进行使用。
〔(G)成分〕
在本发明的组合物中,在不损害本发明的效果的范围内,作为(G)成分,还可以配合以往所已公知的氢化硅烷化反应的反应控制剂。由此,组合物可以得到更好的保存性。作为反应控制剂,可列举例如,1-乙炔基-1-羟基环己烷、3-甲基-1-丁炔-3-醇、3,5-二甲基-1-己炔-3-醇、3-甲基-1-戊烯-3-醇、苯基丁炔醇等炔醇;3-甲基-3-戊烯-1-炔、3,5-二甲基-3-己烯-1-炔等炔属化合物等。另外,以下述的结构式表示的含氟炔醇化合物或聚甲基乙烯基硅氧烷环状化合物以及有机磷化合物等也可作为反应控制剂使用。
由于这些反应控制剂依其化学结构而具有不同的控制能力,因此,应该将配合量分别调整为最适合的量。通常,如果反应控制剂的配合量过少小,则不能在室温下得到长期保存性;如果反应控制剂的配合量过多,则固化速率变缓并且有可能无法得到足够的固化性。
〔其它成分〕
为了提高其实用性,在本发明的组合物中,除了上述的(A)成分~(G)成分之外,根据需要还可以配合无机质填充剂、染料、粘接促进剂、粘接助剂以及硅烷偶联剂等的各种添加剂。这些添加剂的配合量只要在不损害本发明的效果、且不损害光固化性组合物的特性和固化物的特性的范围内,可为任意的量。
作为无机填料,可列举例如,石英粉、熔融石英粉、硅藻土、碳酸钙等的增强性或半增强性填充剂;氧化铁、碳黑、铝酸钴等的无机颜料;氧化钛、氧化铁、碳黑、氧化铈、氢氧化铈、碳酸锌、碳酸镁、碳酸锰等的耐热改进剂;氧化铝、氮化硼、碳化硅、金属粉末等导热性赋予剂;碳黑、银粉、导电性锌白等导电性赋予剂等。
作为染料的例,可列举为以往公知的偶氮类化合物和醌类化合物等合成染料;胭脂红类化合物和靛蓝类化合物等天然染料。
另外,在本发明中,根据需要,也可配合羧酸酐、钛酸酯等粘接促进剂,粘接赋予剂和/或硅烷偶联剂。
〔光固化性氟聚醚类弹性体组合物的制造方法〕
光固化性氟聚醚类弹性体组合物可通过以下方法进行制造。
使用行星式搅拌机、ROSS搅拌机、霍巴特(Hobart)搅拌机等的混合装置、根据需要使用捏合机或三辊式滚轧机等的混捏装置,均匀地混合上述(A)成分、(B)成分以及(C)成分;优选为均匀地混合(A)成分、(B)成分、(C)成分以及(G)成分;(A)成分、(B)成分、(C)成分、(F)成分以及(G)成分;或者(A)成分、(B)成分、(C)成分、(D)成分、(E)成分、以及(G)成分和其它的任意成分。
对光固化性氟聚醚类弹性体组合物的制造方法没有特别地限制,可以通过将上述成分进行捏炼制造而成。另外,也可以用将上述成分事先制备成两种组合物,在使用时进行混合的方式进行制造。
以下,示出在已配合(A)成分、(B)成分、(C)成分、(F)成分以及(G)成分的情况下的光固化性氟聚醚类弹性体组合物的制造方法的一例。
[基础化合物的制造方法]
首先,在加热或非加热条件下,相对于(A)成分100质量份,将在10~50质量份的范围的(F)成分配合在(A)成分内,且在加热-减压条件或加热-加压条件下进行捏炼,进而制作了(F)成分被高填充在(A)成分中的基础化合物。
在此,首先制作由(A)成分和(F)成分组成的基础化合物的目的在于,为了用为(A)成分的直链聚氟化合物去充分地覆盖为(F)成分的疏水性二氧化硅细粉的表面。由此,通过(B)成分和(G)成分难以被吸附在(F)成分的二氧化硅表面,或(B)成分和(F)成分彼此难以凝集在一起,以降低光固化性氟聚醚类弹性体组合物的粘度,从而提高光固化性。(A)成分和(F)成分的配合-捏炼可通过行星式搅拌机、门式搅拌机以及捏合机等的捏炼装置进行。
依据为(F)成分的疏水性二氧化硅粉末的种类,(A)成分与(F)成分的配合比会有所不同,但相对于(A)成分100质量份,(F)成分的含量优选为10~50重量份。相对于(A)成分100质量份,(F)成分在少于10质量份的情况下,则难以降低最终配合组合物的粘度,进而有可能导致粘度变得非常高。另外,相对于(A)成分100质量份,(F)成分在超过50质量份的情况下,则捏炼时的增稠和发热变得剧烈,此后,在配合剩余成分时,则不能均匀地混合,或者挥发性成分被蒸馏掉,有可能无法得到作为固化物所希望的特性。
有关基础化合物制作时的配合-捏炼的温度和时间,只要是适宜地决定就可,其优选为热处理温度为120~180℃,且以使成分均匀地分散的方式捏炼1小时以上。
有关配合-捏炼的压力,由于所使用的装置不同,因此,优选为根据该装置在加压或减压条件下进行。例如,在用行星式搅拌机或门式搅拌机进行捏炼的情况下,其压力条件优选为以表压为-0.05MPa以下的减压条件下进行。另外,当用捏合机进行捏炼的情况下,其压力条件优选为以表压0.4~0.6MPa的加压条件下进行。在该条件下进行操作的理由为,(A)成分容易沾湿(容易覆盖)在(F)成分的表面。
通过将上述(A)成分、(B)成分、(C)成分以及(G)成分配合在由以上述方法所得到的(A)成分和(F)成分组成的基础化合物中,进而能够得到光固化性氟聚醚类弹性体组合物。
需要说明的是,在使用光固化性氟聚醚类弹性体组合物时,根据其用途和目的,可以以所希望的浓度将该组合物溶解在适当的氟类溶剂中,例如,溶解在1,3-双(三氟甲基)苯、Fluorinert(3M公司制造)、全氟丁基甲基醚、全氟丁基乙基醚、全氟聚醚低聚物或它们的混合物等中进行使用。特别优选为将溶剂使用在薄膜涂布用途中。
[光固化性氟聚醚类弹性体固化物]
将上述(A)成分~(C)成分;优选为(A)成分、(B)成分、(C)成分以及(G)成分;(A)成分、(B)成分、(C)成分、(F)成分以及(G)成分;或者(A)成分、(B)成分、(C)成分、(D)成分、(E)成分、以及(G)成分作为主要成分的光固化性组合物,通过依照所述光照射方法对组合物进行光照射,进而能够形成耐化学品性和耐溶剂性优异且低透湿性的橡胶状或凝胶状的固化物。另外,通过依照上述涂布方法将组合物涂布在目标部位,即使在涂布组合物的基材的暗处和深处中也能够均匀地固化,并且能够得到不受来自基材的固化阻碍的固化物(涂膜)。
[实施例]
以下,通过所示参考例、比较参考例、实施例以及比较例,对本发明进行具体地说明,但本发明并不限于以下实施例。需要说明的是,在下述例中的份表示为质量份。另外,粘度表示为在23℃条件下的测定值(基于JIS K7117-1标准)。分子量表示为在将氟类溶剂作为洗脱液的GPC分析中的以聚苯乙烯换算的数均分子量。
用于光固化性氟聚醚类弹性体组合物的基础化合物的制造
[制造例1]
将用六甲基二硅氮烷对BET比表面积50m
光固化性氟聚醚类弹性体组合物的制造
[制备例1]
将以上述式(6)表示的69份聚合物连同在制造例1中已制造的20份基础化合物装入在行星式搅拌机中,并混合至均匀。在此,再依次配合0.05份(甲基环戊二烯基)三甲基铂(IV)的1,3-双(三氟甲基)苯溶液(铂浓度3.0质量%)、0.05份1-乙炔基-1-羟基环己烷的甲苯溶液(5.0质量%)、0.21份以下述式(7)表示的含氟有机氢硅氧烷(Si-H基含量为0.0050摩尔/g)、4.0份以下述式(8)表示的含氟有机氢硅氧烷(Si-H基含量:0.0026摩尔/g),并混合至均匀。然后,通过进行消泡操作进而制备了组合物。将制备例1的组成(单位:质量份)表示在表1。
[制备例2]
依次配合以下述式(9)表示的100份聚合物(粘度8500mPa·s、乙烯基含量0.00012摩尔/g、数均分子量15000)、0.05份(甲基环戊二烯基)三甲基铂(IV)的1,3-双(三氟甲基)苯溶液(铂浓度3.0质量%)、0.03份1-乙炔基-1-羟基环己烷的甲苯溶液(5.0质量%)、3.0份以下述式(10)表示的含氟有机氢硅氧烷,并混合至均匀。然后,通过进行消泡操作进而制备了组合物。将制备例2的组成(单位:质量份)表示在表1。
[制备例3]
将38份以上述式(6)表示的聚合物、12份以下述式(11)表示的聚合物(粘度900mPa·s、乙烯基含量0.023摩尔/100g、数均分子量4000)、50份以下述式(12)表示的全氟聚醚油(粘度770mPa·s、数均分子量4100)装入在行星式搅拌机中,并混合至均匀。在此,再依次配合0.05份(甲基环戊二烯基)三甲基铂(IV)的1,3-双(三氟甲基)苯溶液(铂浓度3.0质量%)、0.03份1-乙炔基-1-羟基环己烷的甲苯溶液(5.0质量%)、1.4份以上述式(7)表示的含氟有机氢硅氧烷并已混合至均匀。然后,通过进行消泡操作进而制备了组合物。将制备例3的组成(单位:质量份)表示在表1。
表1
[保存稳定性的评价]
[参考例1]
对将在上述制备例1所得的光固化性组合物在40℃条件下已密封的遮光瓶中保管30天时的粘度变化进行了评价。将其结果表示在图10。需要说明的是,其为根据JIS K7117-1的标准,使用日本东机产业股份有限公司制造的TV-10U型旋转粘度计(主轴No.H7、23℃、50rpm)而进行了粘度的测定。
[参考例2]
除了将在上述参考例1中使用的光固化性组合物由制备例1的组合物变为制备例2的组合物之外,以同样的操作进行了评价。将其结果表示在图10。
[参考例3]
除了将在上述参考例1中使用的光固化性组合物由制备例1的组合物变为制备例3的组合物之外,以同样的操作进行了评价。将其结果表示在图10。
从图10的结果可以确认,光固化性氟聚醚类弹性体组合物均在遮光中显示出良好的保存稳定性。
[光固化性评价-光照射剂量检定-]
[参考例4]
将在上述制备例3中得到的光固化性组合物以厚度为2mm的量涂布在60mmφ×10mm的圆形铝制培养皿上并对其进行光照射。在此,所说的光照射为以使用表面照射型UV-LED照射器(CCS Co.,Ltd制造),365nm的照度为100mW/cm
[参考例5]
除了将光照射条件变更为100mW/cm
[参考例6]
除了将光照射条件变更为100mW/cm
[比较参考例1]
除了将光照射条件变更为50mW/cm
[比较参考例2]
除了将光照射条件变更为100mW/cm
表2
[光固化性评价-弹性模量评价-]
[参考例7]
将在上述制备例1中得到的光固化性组合物以在25℃的条件下且试料厚度1.0mm的方式涂布在铝基板上,然后进行光照射,并测定了在25℃条件下的弹性模量的时变。试料直径为8mmφ,光照射量为100mW/cm
[参考例8]
除了将在上述参考例7中使用的光固化性组合物由制备例1的组合物变更为制造例2的组合物之外,以与参考例7同样的操作进行了评价。将其结果表示在图11。
[参考例9]
除了将在上述参考例7中使用的光固化性组合物由制备例1的组合物改变为制备例3的组成之外,以与参考例7同样的操作进行了评价。将其结果表示在图11和图13。
从图11的结果可以确认,光固化性氟聚醚类弹性体组合物在25℃的条件下显示出良好的光固化性。
[暗处固化性的评价]
[实施例1]
将从60mmφ×10mm的圆形铝制培养皿的外端直至向内20mm处用铝箔覆盖,进而制作了如图5所示那样的具有暗处的容器。
将上述制备例3中得到的光固化性组合物注入在已连接玻璃制排出管(内径
涂布后立即遮光,且在23℃条件下放置2小时后,通过触摸对固化状态进行了确认。将其结果表示在表3。
[比较例1]
替代在实施例1中边进行光照射边将光固化性组合物涂布在铝制培养皿中的方法,而将其变为如图7所示那样,在以厚度为4mm的量的方式将组合物涂布在铝制培养皿内后,再从顶部进行光照射的方法。此时,光照射为使用与实施例1相同的表面照射型UV-LED照射器,且以365nm条件下的照度为100mW/cm
[深处固化性评价]
[实施例2]
将在上述制备例1中得到的光固化性组合物注入在具有排出管的聚丙烯制的注射器中,且如图8所示那样,在对排出管部的组合物进行照射的同时将该组合物涂布在8mmφ×15mm的圆形铝制培养皿内。涂布量为组合物的厚度为10mm的量,且以与实施例1同样,365nm的照度为100mW/cm
[比较例2]
替代在上述实施例2中的在对在排出管中的光固化性组合物进行光照射的同时将该光固化性组合物涂布在培养皿中的方法,将其变为如图9所示那样,在将制备例1的组合物以厚度为10mm的量的方式涂布在铝制培养皿内后,再从顶部进行光照射的方法。此时,以365nm条件下的照度为100mW/cm
表3
从表3的结果可知,在本发明的涂膜形成方法中,不论是在暗处还是在深处,均能够将组合物均匀地固化。
[在基材上的光固化性评价]
[实施例3]
将在制备例1中得到的光固化性组合物注入在具有排出管部的聚丙烯制的注射器中,且与实施例1同样,以365nm的照度为100mW/cm
[比较例3]
除了在以试料厚1.0mm的方式将在制备例1中得到的光固化性组合物涂布在聚烯烃基材上后再在聚烯烃基材上进行光照射以外,以与实施例3同样的操作对其弹性模量进行了测定。将其结果表示在图12。
从图12的结果可以确认,在本发明的涂膜形成方法中,未受到来自聚烯烃基材的固化阻碍;另一方面还可以确认,在比较例的方法中,由于受到来自聚烯烃基材的固化阻碍而导致弹性模量低,实际上,即使为弹性模量测定后的样品,其在基材界面也为固化不良。
[实施例4]
除了将实施例3中使用的光固化性组合物由制备例1的组合物变更为制备例3的组合物,且将其涂布组合物的基材由聚烯烃基材变更为聚苯硫醚(PPS)基材以外,与实施例3同样的操作对其弹性模量进行了测定。将其结果表示在图13。
[比较例4]
除了在以试料厚1.0mm的方式将在制备例3中得到的光固化性组合物涂布在PPS基材上后再在PPS基材上进行光照射以外,以与实施例4同样的操作对其弹性模量进行了测定。将其结果表示在图13。
从图13的结果可以确认,在本发明的涂膜形成方法中,未受到来自PPS基材的固化阻碍;另一方面还可以确认,在比较例的方法中,由于受到来自PPS基材的固化阻碍而导致弹性模量低,实际上,即使为弹性模量测定后的样品,其在基材界面也为固化不良。
[实施例5]
将在制备例3中得到的光固化性组合物以厚度为2mm的方式填充在8mmφ×15mm的圆形铝制培养皿内,并对其进行了365nm下的照度为100mW/cm
[比较例5]
除了在以试料厚1.0mm的方式将在制备例3中得到的光固化性组合物涂布在PET基材上后再在PET基材上进行光照射以外,以与实施例5同样的操作对其弹性模量进行了测定。将其结果表示在图14。
[参考例10]
除了在以试料厚1.0mm的方式将制备例3中得到的光固化性组合物涂布在铝基材上后再在铝基材上进行光照射以外,以与实施例5同样的操作对其弹性模量进行了测定。将其结果表示在图14。
从图14的结果可以确认,在本发明的涂膜形成方法中,未受到来自PET基材的固化阻碍;另一方面还可以确认,在比较例的方法中,由于受到来自PET基材的固化阻碍而导致弹性模量低,实际上,即使弹性模量测定后的样品也为固化不良。
- 光固化性氟聚醚类弹性体组合物的涂膜形成方法
- 水性涂料组合物以及使用该水性涂料组合物的涂膜形成方法及多层涂膜形成方法