一种靶向G蛋白偶联受体的化合物的筛选方法
文献发布时间:2023-06-19 12:14:58
技术领域
本发明涉及计算机辅助药物发现领域,特别是涉及一种靶向G蛋白偶联受体的化合物的筛选方法。
背景技术
G蛋白偶联受体(G protein-coupled receptors,GPCRs)是药物靶蛋白中数量最多的,介导了三分之一的药物作用。GPCRs对人体生理上的丰富调节在医学方面得到充分反映,34%的上市药物作用于GPCR。所以,大量的受体研究机构和药物发现机构对GPCRs进行了大量的研究。随着低温电子显微镜、深度突变扫描、基因组测序和信号蛋白分析等技术的进步,GPCR序列、结构和功能数据也迅速增加。最近的研究明确指出,药物和GPCR靶点之间的相互作用对候选药物的毒性或副作用有很大影响。此外,药物的重定位可以通过药物靶点的预测来实现,从而发现化合物和GPCR之间的相互作用,可以促使研究人员识别药物的潜在新用途。因此,筛选靶向GPCR的化合物是非常重要的。
虽然湿实验技术可以用来预测GPCR和化合物的相互作用,但是它们涉及到繁琐和耗时的工作。计算方法可以有效地筛选靶向GPCR的化合物,从而缩小靶向GPCR化合物的搜索空间,以供湿实验研究。在计算机上预测化合物和GPCR之间的相互作用是可取的,有效地补充了昂贵且费时的湿实验。新发现的靶向GPCR的化合物对于研发治疗某些疾病的新药是至关重要的。
目前,利用计算机筛选靶向GPCR的化合物的方法主要可以分为三类:基于结构的方法、基于配体的方法和基于化学基因组数据的方法。基于结构的方法主要采用化合物和GPCR的三维结构进行模拟,确定它们之间是否会有相互作用。然而,有些GPCR的三维结构尚不清楚,因此不能对其进行对接。基于配体的方法主要利用了相似化合物分子倾向于共享相似性质的概念,并且通常与相似的GPCR结合。也就是说,利用配体之间的相似性预测化合物和GPCR的相互作用。然而,当每个GPCR的已知配体数量不足时,基于配体的方法的预测结果可能变得不可靠。基于化学基因组数据的方法是同时使用化合物和GPCR的信息进行预测。基于化学基因组数据的方法的优点在于能够处理大量的生物数据进行预测。尽管目前基于化学基因组数据开展了靶向GPCR的化合物筛选研究,但是如何基于化学基因组数据利用深度学习进一步提升靶向GPCR的化合物筛选性能,是所属领域技术人员需要解决的技术问题。
发明内容
有鉴于此,本发明提供了一种靶向G蛋白偶联受体的化合物的筛选方法。本发明筛选方法能充分考虑化学基因组数据,包括化合物结构、GPCR氨基酸序列和已知的GPCR-化合物相互作用,对于筛选靶向G蛋白偶联受体的药物具有较好的性能和适用性。
为了实现上述发明目的,本发明提供以下技术方案:
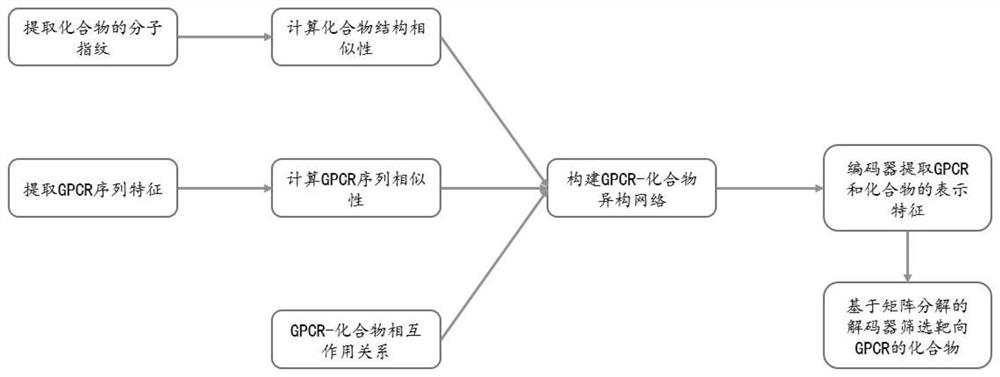
本发明提供一种靶向G蛋白偶联受体的化合物的筛选方法,包括:
步骤1:提取化合物的分子指纹,计算所述化合物结构相似性,获得化合物相似性矩阵;
步骤2:提取GPCR的序列特征向量;利用两个GPCR的序列特征向量的夹角来度量两个GPCR之间的相似性,构建GPCR相似性矩阵;
步骤3:利用化合物相似性矩阵、GPCR相似性矩阵和已知的化合物-GPCR关联关系,构建化合物-GPCR异构信息网络G=(V,E,R);利用编码器获得靶向GPCR的化合物的表示向量和GPCR表示向量;
步骤4:利用已知的化合物-GPCR关系训练解码器;根据靶向GPCR的化合物的表示向量和GPCR表示向量,利用训练好的解码器对靶向GPCR的化合物打分,预测GPCR和化合物之间的相互作用。
本发明步骤1中,所述分子指纹为MACCS密钥分子化学指纹fp。
本发明步骤1中,采用Jaccard相似系数计算所述化合物的两两相似性。其中,所述化合物是指所有候选的化合物,两两相似性是指这些化合物中每两个化合物之间的相似性。
本发明步骤1具体为:提取化合物的MACCS密钥分子化学指纹fp;采用Jaccard相似系数计算所述化合物的两两相似性sim(ci,cj),构建化合物相似性矩阵,所述sim(ci,cj)的计算公式如下:
其中,化合物ci和cj的二值分子指纹特征向量分别为fp(c
本发明步骤2中,所述提取GPCR的序列特征向量采用的方法为基于平行相关性伪氨基酸组成方法。
本发明步骤2中,所述提取GPCR的序列特征向量采用的方法具体为:
给定R表示GPCR的氨基酸序列,R
那么,氨基酸信息相关性θ
假设fi(i=1,2,…,20)表示归一化后的氨基酸频率;W∈(0,1)表示权重因子,那么GPCR特征向量的第u个值可表示为:
最终,GPCR特征向量可表示为:
ft=[ft
本发明的具体方案中,将参数λ设置为2。
本发明步骤3中,以g1和g2表示两个GPCR序列,所述两个GPCR序列基于余弦相似性的计算公式为:
其中,ft(g
本发明步骤3还包括利用下述公式更新异构信息网络中节点的表示向量的步骤:
其中,
其中,节点代表化合物或GPCR。
本发明步骤4中,根据化合物和GPCR的表示向量,利用解码器对靶向GPCR的化合物打分。
所述打分采用矩阵分解方法进行打分。具体地,所述采用矩阵分解方法进行打分具体为:假定D为可训练得到的对角矩阵,vgi和vcj分别为GPCR gi和化合物cj的表示向量,那么GPCR gi和化合物cj之间的分值为:
其中,编码器和解码器首先需要利用已知化合物-GPCR关系基于损失函数训练得到。
本发明靶向G蛋白偶联受体的化合物的筛选方法包括:采集化合物结构、GPCR序列以及化合物-GPCR相互作用关系的数据;基于所采集得到的结构序列数据提取化合物指纹和GPCR的特征向量,并对其进行计算,得到化合物结构相似性和GPCR序列相似性;根据化合物相似性、GPCR相似性和已知的化合物-GPCR相互作用构建异构图,再利用异构图神经网络对靶向G蛋白偶联受体的药物进行筛选。与现有技术相比,本发明具有以下有益效果:
1、本发明充分了考虑化学基因组数据,包括化合物结构、GPCR氨基酸序列和已知的GPCR-化合物相互作用,为进一步提升靶向GPCR的化合物的筛选性能提供了重要基础;
2、本发明利用编码器获得化合物和GPCR的表示向量,接着利用解码器预测GPCR和化合物之间的相互作用,通过AUC指标反应了本发明的预测性能,根据实验验证可知,本发明对于筛选靶向G蛋白偶联受体的药物具有较好的性能和适用性。
附图说明
图1示本发明筛选方法的流程示意图;
图2示各方法的AUC的测试结果。
具体实施方式
本发明提供了一种靶向G蛋白偶联受体的化合物的筛选方法。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明采用的试材皆为普通市售品,皆可于市场购得。
本发明提供一种靶向G蛋白偶联受体的化合物的筛选方法,包括:
步骤1:提取MACCS密钥分子化学指纹fp,MACCS密钥是166位结构密钥描述符,其中结构密钥描述符与SMARTS模式相关联;基于化合物的166位分子指纹采用Jaccard相似系数计算所有化合物两两相似性,从而构建化合物相似性矩阵。假定化合物ci和cj的二值分子指纹特征向量分别为fp(c
步骤2:采用基于平行相关性伪氨基酸组成方法提取GPCR的序列特征。给定R表示GPCR的氨基酸序列,R
那么,氨基酸信息相关性θ
假设fi(i=1,2,…,20)表示归一化后的氨基酸频率;w∈(0,1)表示权重因子(本发明中w设置为0.5),那么GPCR特征向量的第u个值可表示为:
最终,GPCR特征向量可表示为:
ft=[ft
利用两个序列特征向量的夹角来度量两个GPCR之间的相似性,从而构建GPCR相似性矩阵。假定GPCR g1和g2提取的特征向量分别为ft(g
步骤3:利用化合物相似性、GPCR相似性和已知的化合物-GPCR关联关系构建化合物-GPCR异构信息网络G=(V,E,R),为了更好地将网络拓扑结构与潜在向量融合起来,在图神经网络架构的驱动下,利用下述公式更新异构信息网络中节点(化合物和GPCR的表示向量h)的表示向量:
其中,
步骤4:利用解码器对靶向GPCR的化合物打分。解码器依赖于化合物和GPCR的表示向量对GPCR-化合物关系进行打分。
在本发明中,我们利用矩阵分解的方法作为打分函数(解码器),假定D为可训练得到的对角矩阵,vgi和vcj分别为GPCR gi和化合物cj的表示向量,那么GPCR gi和化合物cj之间的分值为:
本发明利用采用负采样方法构建负样本数据集,随机抽取与正样本个数相同多的中立样本作为负样本,然后利用交叉熵损失函数优化模型,如下:
其中,Τ表示所有的训练样本,sig()表示sigmoid函数,y为标签集合,对于正样本y=1,负样本y=0。
根据Adam优化算法求解编码器和解码器,设置epochs为1000,学习率lr=0.01,图卷积神经网络的层数layers=2,各隐含层的维度为[500,500,500],各层的激活函数均选择为ReLU函数。
下面结合实施例,进一步阐述本发明:
实施例1
以Yamanishi’s study数据库(共含有224个化合物和95个GPCRs)为例采用本发明基于异构图神经网络方法筛选靶向GPCR化合物,步骤如下:
以采自Yamanishi’s study的GPCR-化合物相互作用,Drugbank数据集(https://go.drugbank.com)中的化合物SMILES描述符,KEGG GENES数据集中(https://www.genome.jp/kegg/genes.html)GPCR的氨基酸序列。数据集中包含224个化合物、95个GPCRs和635个化合物-GPCR相互作用。实验环境为:CPU:Intel Core i7-10875H,GPU:NVIDIAGeForce RTX 2070,内存:32G,系统:Windows 10,集成开发环境:PyCharm社区版,解释器:python3.7.6,pytorch:1.5.0+cu101,dgl:0.5.3。
1)提取化合物的分子指纹。
化学指纹是表征分子的二进制值(0和1)的列表,本发明采用广泛使用的MACCS密钥分子化学指纹fp。MACCS密钥是166位结构密钥描述符,其中结构密钥描述符与SMARTS模式相关联。
2)计算化合物结构相似性。
基于化合物的166位分子指纹采用Jaccard相似系数计算所有化合物两两相似性,从而构建化合物相似性矩阵。假定化合物ci和cj的二值分子指纹特征向量分别为fp(c
3)提取GPCR序列特征。
GPCR是由氨基酸组成的生物分子,考虑到GPCR在理化性质下的平行相关性,本发明采用基于平行相关性伪氨基酸组成方法提取GPCR的序列特征。给定R表示GPCR的氨基酸序列,R
那么,氨基酸信息相关性θ
假设fi(i=1,2,…,20)表示归一化后的氨基酸频率;w∈(0,1)表示权重因子(本发明中w设置为0.5),那么GPCR特征向量的第u个值可表示为:
最终,GPCR特征向量可表示为:
ft=[ft
4)计算GPCR序列相似性。
本文利用两个序列特征向量的夹角来度量两个GPCR之间的相似性,从而构建GPCR相似性矩阵。假定GPCR g1和g2提取的特征向量分别为ft(g
5)编码器获取药物表示向量
利用化合物相似性、GPCR相似性和已知的化合物-GPCR关联关系构建化合物-GPCR异构信息网络G=(V,E,R),为了更好地将网络拓扑结构与潜在向量融合起来,在图神经网络架构的驱动下,利用下述公式更新异构信息网络中节点(化合物和GPCR的特征向量h)的表示向量:
其中,
6)解码器筛选靶向GPCR的化合物
本发明利用解码器为靶向GPCR的化合物打分。相对于编码器将化合物和GPCR映射到向量不同的是,解码器依赖于化合物和GPCR的表示向量对GPCR-化合物关系进行打分。在本发明中,我们利用矩阵分解的方法作为打分函数,假定D为可训练得到的对角矩阵,vgi和vcj分别为GPCR gi和化合物cj的表示向量,那么GPCR gi和化合物cj之间的分值为:
7)模型训练
本发明中采用负采样方法构建负样本数据集,随机抽取与正样本个数相同多的中立样本作为负样本,然后利用交叉熵损失函数优化模型,如下:
其中,Τ表示所有的训练样本,sig()表示sigmoid函数,y为标签集合,对于正样本y=1,负样本y=0。
根据Adam优化算法求解模型,设置epochs为1000,学习率lr=0.01,图卷积神经网络的层数layers=2,各隐含层的维度为[500,500,500],各层的激活函数均选择为ReLU函数。
为了验证发明的有效性,我们采用了数据集中的百分之八十样本作为训练集,百分之十的样本作为校验集,百分之十的样本作为测试集。
对于上述验证方式采用AUC(接收者操作特征曲线(receiver operatingcharacteristic curve,ROC)下方的面积大小)作为评价指标。采用本发明实施例基于异构图神经网络对靶向GPCR的化合物进行筛选并且与RLSMDA和GRMF方法进行了对比。图2描述了对于AUC各方法的性能比较图,本发明的AUC值为0.9187,比其他两种方法的AUC值更大。
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 一种靶向G蛋白偶联受体的化合物的筛选方法
- 一种靶向抑制维生素K依赖性γ-谷氨酰羧化酶的小分子化合物的筛选方法及应用