一种快速获得脊椎动物线粒体基因组序列的方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于生物技术领域,具体涉及线粒体DNA的基因文库的建立方法和获取线粒体基因组序列的方法。
背景技术
动物线粒体基因序列具有母系遗传、拷贝数多、基因组序列数据量较核基因组小等特点,同时其整体上变异速率较高,但部分基因编码参与极为重要的生命活动而变异相对保守。基于此,动物线粒体基因序列成为了系统发育学与生态学等研究领域的良好工具。比如,目前环境DNA研究常以线粒体基因序列来评估环境中生物的种类和数量。然而不同研究者往往采用不同的线粒体基因标记,例如CO1,12S等基因序列,造成环境DNA数据库难以统一。同时,单个或数个分子标记的分析结果也往往不足以将一个具体生境情况研究透彻,而研究者们基于不同标记所开发的环境DNA采集提取、扩增富集和处理分析手段不尽相同,这又为科学家们选取适用于自身研究的方法设置了门槛,也增加了试错成本。而使用完整的线粒体基因组序列作为环境DNA数据库将解决上述问题。因为单位点线粒体基因或多位点线粒体基因的研究都可以使用线粒体基因组数据库作为参考数据库,而不受方法的限制,并且完整的线粒体基因组所提供的相应物种信息远大于数个位点基因序列,多位点结合分析后亦可解决因序列信息不足所导致的物种鉴定难题。
然而,用线粒体基因组作为环境DNA的数据库需要获得大量的线粒体基因组数据,而当前获得线粒体基因组获得方法要么耗时耗力,要么价格昂贵,如基于PCR和Sanger测序的引物步移法(Primer Walking)或长片段PCR扩增法(Long-range PCR Amplification),以及基于高通量测序的基因组概览(Genome Skimming)。
不同组织细胞中线粒体拷贝数具有显著差异,在含有线粒体的细胞中,线粒体的数量从数个到数百个不等。一般而言,动物肝脏和肌肉组织中的线粒体数量最多,相应的线粒体DNA的含量也更多。而核DNA与线粒体DNA在细胞裂解后也难以分开,因此目前没有专门用于提取线粒体的方法,而是直接提取全基因组DNA,随后用不同的手段将线粒体DNA纯化,便于后续的分析。
尽管线粒体DNA在细胞中拥有多个拷贝,但比起序列长度可达数十亿碱基的核DNA仍然是极其微小的一部分,因此,无论是哪一种测序方法,都需要对线粒体DNA进行一定程度的纯化,以提高其占比,从而增加测序的效率。而纯化某一类目标DNA通常采用两种思路:一种是将目标DNA通过PCR等手段进行扩增,增加有效数据量,从而提高其比例;另一种则是通过设计探针,杂交捕获目标DNA,同时洗去非目标DNA片段,使非目标DNA的数量下降,也能提高目标DNA的比例。
要想获得一个物种的完整线粒体基因组,除了上述DNA的提取、纯化与测序之外,还需要对测序的结果进行组装和校对。而在这个过程中,如果我们有目标物种的线粒体基因组参考序列,或是其近缘种的线粒体基因组参考序列,那么通过映射(mapping)的方式可以快速简便地获得较为准确的结果,但在研究过程中,研究者们往往会遇到不能确定目标物种的具体类型,或者通过形态学鉴定后发现没有目标物种的参考序列,那么组装的过程就会复杂许多。
靶序列捕获(Target Sequence Capture),也被称为基因捕获或基因富集,是一种用于捕获科学家感兴趣的目标序列的方法。它通常基于事先设计的RNA或DNA诱饵(探针),与已经建好的DNA文库中的目标序列杂交,再利用一系列手段将富集到的序列分离纯化以用于测序或下游实验。是利用探针杂交的原理将研究者们感兴趣的核酸片段富集纯化,从而提高有效数据的占比,降低测序和分析成本。该类方法的出现顺应了高通量测序技术的发展。此外也有利用核酸蛋白复合体如CRISPR/Cas系统对目标序列进行捕获的方法。
例如Sevigny J等根据所有后生动物的线粒体基因组序列设计探针,随后利用探针对任意后生动物的高通量测序文库进行富集,富集后测序。但由于探针设计采用全序列,大幅增加了合成成本,加上富集只针对文库短序列,有效数据占比不高,位点外信息不全,因此成本高,效率低。
因此,需要建立一种能够快速获得线粒体基因组序列的方法,提高有效数据占比,降低测序成本,同时能够获得更全面的序列信息。
发明内容
本发明旨在提供建立快速获得动物线粒体基因组序列的方法。
一种用于富集线粒体DNA的RNA探针,其核苷酸含有选自SEQ ID No.1-No.11中的任意一个或任意多个的序列。
优选的,所述RNA探针的核苷酸序列选自SEQ ID No.1-No.11中的任意一个或任意多个的序列。
更优选的,所述RNA探针的核苷酸序列如SEQ ID No.1-No.11所示。
进一步,所述的RNA探针用生物素修饰。
上述的RNA探针可用于富集动物线粒体、测定动物线粒体基因组序列、获取动物线粒体基因组序列或者建立动物环境DNA线粒体基因组数据库。尤其是用于富集脊椎动物线粒体、测定脊椎动物线粒体基因组序列、获取脊椎动物线粒体基因组序列或者建立脊椎动物环境DNA线粒体基因组数据库。
本发明另一个方案为,一种建立线粒体基因组文库的方法,包括以下步骤:
(1)样本总DNA与上述的RNA探针杂交后,捕获靶序列;
(2)收集靶序列并进行扩增,获得扩增后的线粒体DNA。
优选的,步骤(2)中,用修饰链霉亲和素的磁珠收集靶序列。
步骤(2)中,用多次退火环状循环扩增,然后清洗获得扩增后的线粒体DNA。
进一步,所述建立线粒体基因组文库的方法还包括以下步骤:
扩增后的线粒体DNA进行破碎、平末端修复、连接测序接头和索引PCR,构建文库。
本发明的另一个方案为,一种快速获得脊椎动物线粒体基因组序列的方法,其特征在于,包括以下步骤:
I.将上述方法获得的基因文库进行测序;
II.测序后的结果进行组装。
优选的,步骤II中,采用Trinity和NOVOPlasty进行组装。
进一步,用Trinity从头组装筛选得到的保守位点读序,组装出保守位点序列的重叠群(contig)后,由NOVOPlasty进行延伸,得到完整的线粒体基因组。
优选的,测序结果经过预处理,随后根据富集位点的保守序列利用BLAST筛选出最相似的序列,再用Trinity从头组装。组装结果再进一步利用BLAST筛选得到最相似的contig,并使用NOVOPlasty,对最终的contig进行延伸,得到完整的线粒体基因组。
本发明是基于长片段富集的“先富集后建库”的方法,与传统的富集较短片段文库DNA的方法不同,该方法在构建测序文库之前即对原始的长片段线粒体DNA进行富集,旨在获得更多线粒体DNA数据,从而提高富集效率,降低测序和后续组装成本。
以模式生物斑马鱼(Danio rerio)为例,结果表明,在相同测序数据量的基础上,该新方法比传统的基因组概览,线粒体基因组数据量的占比提高了180倍以上,即便是与传统富集方法相比,线粒体基因组数据量的占比也提高了3倍左右。更为重要的是,传统方法线粒体基因组数据的覆盖度不全,造成线粒体基因组组装不完整,而同等条件下,本方法数据的覆盖度达到100%,说明本方法降低了由于非富集位点序列的缺失而无法组装出完整线粒体基因组的可能性,能够实现完整的组装。
同时计算了使用该方法获得脊椎动物完整线粒体基因组的必要数据量,并与其他方法比较。结果表明改进后的方法仅需要50MB即可组装出完整线粒体基因组,所需的最低数据量不到5%,这大大降低了测序成本。
此外用中华鳖(Pelodiscus sinensis)和猪(Sus scrofa)进行了验证,同样成功地组装出了这两种不同脊椎动物的线粒体基因组,表明该方法也适用于其他纲的脊椎动物。采用本发明的探针和方法,尤其适用于检测和组装脊椎动物线粒体基因组。
对于单个样本而言,完整实验流程只需要三天,而获得完整线粒体基因组的总花费不到传统方法50%。最终结果表明,本方法在无需相应物种参考基因组的前提下,可以相对快速、经济地获得各种脊椎动物的线粒体全基因组,该方法为建立脊椎动物环境DNA线粒体基因组数据库提供了可行的技术路线。同时也可以用于获得未鉴定或无参考序列的脊椎动物的线粒体基因组组装,有助于完善脊椎动物线粒体基因组数据库。
附图说明
图1为实施例1三种方法的深度分布曲线
图2为实施例1三种方法不同数据量下的覆盖度情况
具体实施方式
主要试剂:
(1)Buffer Tango(10x)(Thermo,货号:BY5);
(2)dNTPs(10mM each)(Invitrogen,货号:18427088);
(3)ATP(100mM)(Thermo,货号:R0441);
(4)T4 polynucleotide kinase(10U/μL)(Thermo,货号:EK0031);
(5)T4 DNA polymerase(5U/μL)(Thermo,货号:EP0061);
(6)T4 DNA ligase(5U/μL)(Invitrogen,货号:15224041)及T 4DNA ligase试剂盒配套的T4 DNA ligase buffer、PEG-4000(50%);
(7)Bsm polymerase,large fragment(8U/μL)(Thermo,货号:EP0691)及配套的Bsm buffer;
(8)KAPA HiFi taq Ready Mix(2x)(KAPABIOSYSTEMS,货号:KK2602);
(9)UltraPure
(10)UltraPure
(11)Denhardt's Solution(50x)(Invitrogen,货号:750018);
(12)10%SDS溶液(生工,货号:B548118-0100);
(13)SUPERase·In
(14)DEPC处理水(生工,货号:B501005-0500);
(15)Human Cot-1 DNA(Invitrogen,货号:15279011);
(16)Dynabeads
(17)Tween-20(Amresco,货号:0777-1L);
(18)TE buffer(生工,货号:B548106);
所使用的磁珠用链霉亲和素修饰。
用于富集的RNA探针序列(SEQ ID No.1-11)如下:
1.ccgggtactacgagcactagcttaaaacccaaaggacttggcggtgctttagatccacctagaggagcctgttctagaaccgataacccccgttaaacctcaccctctcttgttcttcccg
2.gactataagtttaacggccgcggtattttgaccgtgcaaaggtagcgcaatcacttgtcttttaaatgaagacctgtatgaatggcataacgagggcttaactgtctcctttttccagtcaatgaaattgatctccccgtgca
3.tcgacaagagggtttacgacctcgatgttggatcaggacatcctaatggtgcagccgctattaagggttcgtttgttcaacgattaaagtcctacgtgatctgagttcagaccggagtaatccaggtcagtttctatctatgccacgatcttttct
4.gctcgaaccctacctgaagagatcaaaactcttagtgcttccactacaccacttccctagtaaagtcagctaaataagcttttgggcccataccccaaacatgttggttaaactccttcctttgct
5.cccacatcttctgcatgcaaaacagacattttaattaagctaaagccttactagacaggaaggcctcgatcctacaaactcttagttaacagctaagcgcttaaaccaacaagcatctgtctaacctttccccgc
6.ccatcttacctgtggcaatcacacgttgatttttctcaactaatcacaaagacatcggcaccctctatctagtatttggtgcttgagccggaatagtaggaactgcattaagcctcctaattcgggca
7.cacatgctttcgtaataattttctttatagtaatgccaattataattggaggttttggaaactgactagtgccactaatgattggtgcaccagacatggccttccctcgaataaataacatgagt
8.gccggcatcacaatacttctaacagaccgaaacctaaacacaaccttctttgaccctgccggaggaggagaccccatcctttaccaacacttattctgattctttggacaccctgaagtttatattct
9.agggtttattgtctgagcccatcacatgttcaccgtaggaatggacgtagatacacgggcttactttacttccgccacaataattattgccatcccaaccggagtaaaagtcttcagctg
10.cagtagccataattcaggcctatgtctttgttcttcttttaagcctttacctacaagaaaacgtttaatggcccatcaagcacacgcatatcacatagttgaccccagcccatgacccctaaca
11.tctttagccctcttctcccccaatctacttggtgatcctgacaacttcacccccgcaaaccctctagttacccctccccacattaaacccgaatggtacttcttatttgcctacgccatcctacgctcaat
用于MALBAC扩增的通用引物序列如下:
MALBAC+8N:gtgagtgatggttgaggtagtgtggagnnnnnnnn
MALBAC:gtgagtgatggttgaggtagtgtggag
实施例1
以斑马鱼为研究对象,从斑马鱼肌肉中提取总DNA,并建立线粒体基因组文库,测序后获得线粒体基因组。
(一)预杂交PCR
(1)在PCR仪上设置如下程序:95℃持续5分钟,65℃持续5分钟,65℃持续10分钟,60℃持续10小时,随后保持60℃;
(2)在冰盒上按照样品数和如下体系准备杂交混合液(Hyb Mix):
充分震荡混合均匀并将管壁上的试剂通过短暂离心的方式收集至管底,每个样品取5μL至事先准备好的空PCR管中;在冰盒或4℃冰箱内暂时保存至第(6)步再取用;
(3)在冰盒上按照样品数准备用于制备文库的DNA混合液(Lib Mix):
在写好相应样本编号的空PCR管中每管加入1.25μL的试剂,并加入6.25μL的样品DNA(100ng~500ng)。轻柔地吹打混匀,并短暂离心;在冰盒或4℃冰箱内暂时保存至第(5)步再取用;
(4)在冰盒上按照样品数和如下体系准备探针混合液(Baits Mix,SEQ ID No.1-11):
轻柔地吹打混匀混合液;在冰盒或4℃冰箱内暂时保存至第(6)步再取用;
(5)将Lib Mix转移至PCR仪中,开始执行(1)中设置的程序;DNA将在5分钟95℃下变性;
(6)当程序执行到第二步时,即温度从95℃降至65℃时,打开PCR仪,将Hyb Mix和Baits Mix放入(不要移动Lib Mix);所有混合液将在PCR仪内预热至65℃;
(7)当程序进入第三步时(65℃保持10分钟),将5μL的Hyb Mix和3μL的Baits Mix转移至Lib Mix,轻柔地吹打混匀;整个操作流程都在PCR仪上进行,不要将PCR管取出;
(8)让混合好的溶液在PCR仪上完成程序的全部步骤;
(二)磁珠法富集和洗脱
(1)加入n×10μL(n为样品数)的MyOne(Invitrogen cat#65002)或M270(Invitrogen cat#:653-06)磁珠(链霉亲和素修饰)至新的PCR管中(单管不要超过180μL);
(2)将试管放到磁板上收集磁珠,待溶液澄清后吸去上清液;
(3)加入200μL的Binding Buffer(配方见附录)清洗磁珠;轻柔地吹打磁珠使其悬浮,随后再放回磁板收集磁珠并吸去上清液;
(4)重复步骤(3)两次;
(5)加入n×20μL Binding Buffer吹打使磁珠悬浮,加入1μL 10%Tween;
(6)取与样本数目对应的空PCR管,编号后每管加入180μL的Binding Buffer,随后加入20μL的磁珠悬浮液;
(7)60℃预热5分钟;
(8)将杂交溶液全部转移至磁珠溶液中,随后在杂交仪上以60℃孵育30分钟;随后用磁板收集磁珠,弃清液,过程尽可能迅速,注意不要结束PCR仪的程序,使其保持在60℃;
(9)在孵育过程中准备3×n个PCR管,分别加入190μL的Wash Buffer 2,并移至PCR仪中60℃预热至少10分钟;
(10)向磁珠管中加入186μL的Wash Buffer 2,轻柔地吹打磁珠使其悬浮,随后转移至PCR仪放置10分钟;随后用磁板收集磁珠,弃清液;
(11)重复步骤(10)两次,进行总计3次的60℃清洗;
(12)最后使用186μL的DEPC水在室温再进行一次清洗;
(13)在管中加入25μL的DEPC水;
(三)多次退火环状循环扩增(Multiple Annealing and Looping-BasedAmplification Cycles,MALBAC)
(1)在冰盒上按照样品数准备如下混合液:
准备与样品数对应的空PCR管,每管加入15μL的混合液,并加入10μL的带有磁珠的DNA,同时再增加一个负对照,采用DEPC水代替加入的10μL带有磁珠的DNA;充分混匀,短暂离心,最后在PCR仪上按照如下程序进行扩增:92℃持续3分钟,14-18个循环的:10℃持续45秒,20℃持续45秒,30℃持续45秒,40℃持续45秒,50℃持续45秒,68℃持续2分钟,92℃持续20秒,58℃持续20秒;完成循环后保持4℃;
上述PCR程序完成后,每管加入0.8μL的引物2:MALBAC,混合均匀,短暂离心,随后再执行如下程序:92℃持续2分钟,12-16个循环的:92℃持续20秒,58℃持续30秒,68℃持续1分钟;循环完成后4℃保存;
(2)采用磁珠法清洗DNA,最终加入30μL的水溶解DNA,并使用Nanodrop3300测定浓度,用于后续构建测序文库;
(四)DNA破碎
取0.3–1μg DNA到一个新的PCR管中,向管中加入可用于进行PCR反应级别的超纯水(以下简称PCR水)直至盖上后无气泡,使用Covaris M220 Focused-ultrasonicator(Covaris,Woburn,USA)超声破碎仪将DNA片段破碎至300bp左右(可使用琼脂糖凝胶电泳检测破碎后的片段长度);在文库构建开始前设置一个正对照和一个负对照;随后用Rohland等人开发的磁珠法清洗和收集DNA(在酒精清洗步骤后不加人新的溶液);
(五)平末端修复
(1)在冰盒上按照样品数和如下体系配置混合液:
(2)将混合液直接在前一步最后带有磁珠的PCR管中加入20μL混合液,混合均匀;
(3)在PCR仪上执行如下程序:25℃持续15分钟,12℃五分钟;
(4)用磁珠法清洗DNA(保持磁珠在管内,无需分离);
(六)连接测序接头
(1)在冰盒上按照样品数和如下体系配置混合液(不加接头,之后每个样品单独加入接头):
(2)每管加入38μL混合液,混合均匀;
(3)每管再分别加入1μL的IS1和IS2(每个样品编号对应一组IS1+IS2用于在测序及后续分析区分不同样品,不同组IS1+IS2之间编号不能完全相同);
(4)在PCR仪上22℃孵育30分钟;
(5)用磁珠法清洗DNA(保持磁珠在管内,无需分离);
(七)填充
(1)在冰盒上按照样品数和如下体系配置混合液:
(2)每管加入40μL混合液,混合均匀;
(3)在PCR仪上37℃孵育20分钟;
(4)用磁珠法清洗DNA,最终使用35μL的TE Buffer溶解(保持磁珠在管内,无需分离);
(八)索引PCR(indexing PCR)
(1)在冰盒上按照样品数和如下体系配置混合液:
(2)每管加入13μL混合液,以及0.5μL的P7引物,并加入11μL的带有磁珠的DNA溶液,混合均匀,随后转移至PCR仪器;
(3)在PCR仪上执行如下程序:98℃持续45秒,12到16个循环的:98℃持续15秒,60℃持续30秒,72℃持续1分钟;循环完成后保持4℃;
(4)用磁珠法清洗DNA(加入新的磁珠),用25μL的TE Buffer溶解;
(5)取1μL使用琼脂糖凝胶电泳查看文库构建情况。
(九)测序和组装
文库采用Illumina Novaseq-PE150平台测序。测序结果进行预处理,包括解压缩、根据Inline Index对数据进行分类、去除接头序列和低质量序列。随后根据富集位点的保守序列利用BLAST筛选出最相似的序列并利用Trinity v2.11.0从头组装,组装结果再进一步利用BLAST筛选得到最相似的contig。并使用NOVOPlasty对最终的contig进行延伸,组装脚本见https://github.com/Checunmil y/mito_assemble。
测序数据(SRA:PRJNA796186)与组装序列结果(GenBank编号:OM236540,OM236541)均已上传至NCBI,与数据库中已有的序列并无显著不同。
从测序深度的分析可知,在经过富集和扩增后的,得到的线粒体DNA片段较长。
结果分析:
一、组装结果
对于每一个被测斑马鱼样本,配对的读序都使用BWA-MEM(v0.7.16a-r1181)默认参数比对至组装后的该斑马鱼样本线粒体基因组上,并使用Samtools(v1.10)的view、coverage和depth命令和默认参数读取比对结果的覆盖度、平均深度以及每个位点的深度。所有进行比较分析的数据在比对前都对读序数量进行了统一,截取了相同数量的读序(但由于读序长度不尽相同因此绝对数据量不同)用于分析,最终结果使用读序数量呈现。富集倍数使用每种富集策略所有样本平均深度的平均值除以标准建库策略的所有样本平均深度的平均值计算。
我们对用于测试方法效率的所有斑马鱼样本都要求了2GB的测序数据量,并且在数据预处理完成后每个样本获得了170MB~500MB的数据。以斑马鱼全基因组大小为参考(约1400MB),我们所测的数据在平均深度在1x以下,但对于斑马鱼线粒体基因组而言(约16k),富集后测序得到的数据量则远远大于其线粒体基因组大小。
除了本发明“先富集后建库”的方法,还采用了“直接建库”及“先建库后富集”的方法进行比较。
直接建库为,获得总DNA后,使用超声破碎仪进行破碎至300bp左右,进行平末端修复,添加接头;填充,以及indexing PCR完成后利用磁珠清洗法进行清洗。
先建库后富集的方法为:使用超声破碎仪将总DNA片段破碎至300bp左右;平末端修复;添加接头;填充;以及预杂交PCR。在以上每一步骤后都利用Rohland等人开发的磁珠法清洗和收集DNA。富集步骤基本参照外显子富集操作,但只进行一次富集步骤,包括:杂交;带有链霉素的磁珠捕获与洗脱;以及Indexing PCR。Indexing PCR完成后利用磁珠法进行清洗。
而在组装流程结束后,三种策略中只有改进后的“先富集后建库”样本组装出了完整的线粒体基因组(GenBank编号:OM236539),直接建库与“先建库后富集”的样本均无法组装出完整的线粒体基因组。
在组装过程中,使用基因组组装软件如SOAPdenovo和SPAdes等,以及MITObim等专门用于线粒体的组装软件,都未能组装出完整的线粒体基因组。利用Trinity直接组装所有的测序结果也没有获得完整的线粒体基因组。
最终确定的组装策略为使用Trinity从头组装筛选得到的保守位点读序,组装出保守位点序列的contig之后将这些contig作为“种子”交由NOVOPlasty进行延伸,最终得到完整的线粒体基因组。
二、覆盖度及深度比较
在获得了斑马鱼样本完整线粒体基因组后,我们比较了不同建库策略的有效数据占比、平均深度、覆盖度和富集倍数。数据统计结果如表3-1所示。
表1.三种方法比对数据统计
从测序深度的分析可知,在经过富集和扩增后的,得到的线粒体DNA片段较长。
显著性检验对平均深度采用T检验,其中,先建库后富集与直接建库P<0.0025,先富集后建库与直接建库、先富集后建库与先建库后富集P<0.001。显著性检验结果表明,三种方法有效数据之间具有极其显著的差异。在直接建库的方法中,线粒体数据占比只有0.07%左右,考虑到斑马鱼核基因组与线粒体基因组大小的差异(约10万倍),以及线粒体DNA在单个细胞内的拷贝数区间(数个至数百个),该结果符合最初实验设计的预期。这一数据在不同物种以及不同组织之间会有较为显著的浮动,在核基因组相对较小的物种、线粒体含量较高的如肝脏、肌肉等组织测得的结果中,线粒体DNA的数据占比会更高;而对于那些核基因组较大、线粒体含量较少的组织样测得的结果中,这一占比则会更低。而通过与其他两种富集策略比较的结果显示,对线粒体DNA进行富集操作都能够显著提高线粒体DNA数据的占比,而其中效果最好的是改进后的“先富集后建库”方案,平均深度达到了直接建库数据占比的186倍,即便是比起传统的富集策略,也能有接近三倍的有效数据。尽管直接建库与先建库后富集的最低深度都为0,即存在没有序列信息的位点,无法组装出完整序列,但富集方法仍然显著提高了覆盖度。
若是提高数据量,先建库后富集的方法依然能够比较好地组装出完整的线粒体基因组。而在这样的低数据量下,“先富集后建库”方法仍然能够保持100%的覆盖度以及最低8的深度,也佐证了该方法所测得的数据确实涵盖了完整的线粒体基因组。
三、测序深度分布情况
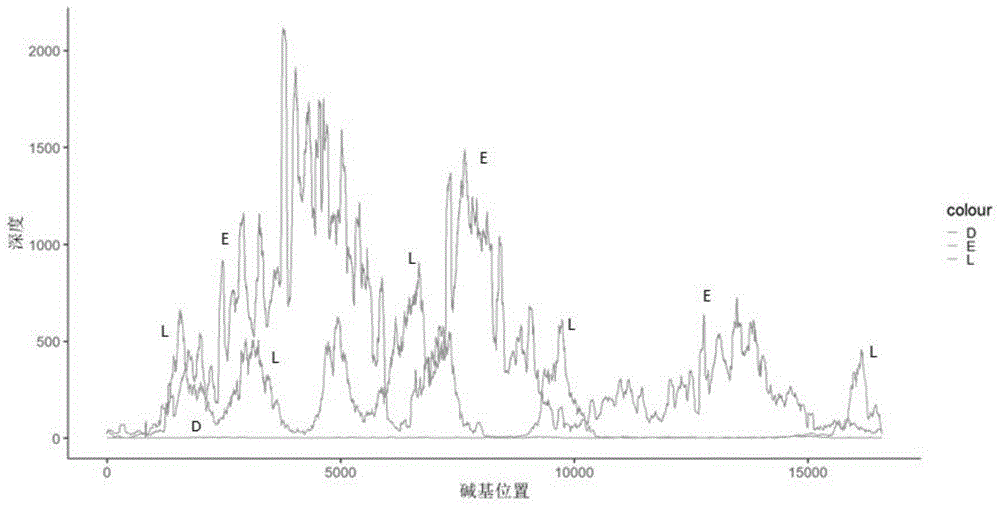
利用Samtools的depth命令和默认参数调出了所有测序样本每个碱基的深度,计算了不同策略每个碱基的平均深度,随后利用R对该数据进行了可视化,绘制了深度曲线图用于说明其深度分布情况,如图1所示,从上到下曲线依次为E(先富集后建库)、L(先建库后富集)和D(直接建库)。X轴代表线粒体基因组中的碱基顺序,Y轴代表每个碱基位点相应的深度。
深度曲线图同样显示了“先富集后建库”策略(E)有显著高于其他策略的最大深度和完整的覆盖度,然而,其在深度峰的位置却与“先建库后富集”策略(L)不同。“先建库后富集”的深度曲线与探针位点完全契合,在富集位点与其附近拥有较高的深度,而在富集位点之外的深度则很低,接近直接建库的深度。而“先富集后建库”的深度曲线则与富集位点不完全一致,甚至在某些本该出现峰值的位点很低(如7000和10000附近),同时在富集位点之外有一些地方出现了峰值(如11000和14000附近),在输出了单个样本的深度曲线后仍然表现出这一特征。我们推测出现这一现象的原因是在富集之后进行的MALBAC扩增步骤所用的随机引物被富集所用的探针争夺,并且RNA探针在复性过程中再次与目标位点结合,妨碍了扩增所用随机引物的结合,从而降低了该位点的扩增效率,而离富集位点较远的区域或是序列与富集位点差异较大的区域则不受影响,可以正常扩增,从而产生了这一现象。
结果显示,在最大序列深度和覆盖度上本方法显著优于原有方法。
四、获得完整线粒体基因组所需最低数据量检验
为了确定改进后方法获得完整线粒体基因组所需要的最少数据量,用于避免测序浪费,我们对“先富集后建库”的三个样本的数据进行了检验。在检验过程中,我们按照每次10000条序列(20000个读序)梯度减少了序列数,并将截取后的数据再次用BWA-MEM比对到组装得到的线粒体基因组上,并使用Samtools得到了覆盖度,输出每次的序列数与覆盖度,并用R绘制了相应的曲线图,不同数据量下覆盖度情况如图2所示。X轴代表读序数量,Y轴代表相应读序数量下的覆盖度。图中从上到下曲线依次为先富集后建库(E)、先建库后富集(L)和直接建库(D)。
从图中可以发现,“先富集后建库”方法覆盖度首次低于100%的读序数为140000,此后覆盖度开始降低,而其余两种方法的覆盖度在较少数据量的前提下则一开始就不满100%,并且下降趋势也显著快于“先富集后建库”方法。值得注意的是,即便是在极端低数据量的条件下(20000读序),改进后的新方法仍然能保持90%以上的覆盖度,说明我们富集长片段的思路确实能够获得离富集位点较远的DNA片段,从而保持了较高的覆盖度和较为均匀的深度。
五、“先富集后建库”方法与现有方法成本比较
根据上述实验结果可以得出,对于斑马鱼肌肉组织,该方法获得其完整线粒体基因组数据约为50MB。这一最低数据量同样需要考虑物种基因组大小及组织样差异,加上测序前扩增环节造成的PCR重复影响,因此推荐不少于200MB的测序数据量。而采用传统方法,例如先建库后富集的方案,进行高通量测序所需要的数据量在4G以上,因此大大节约了测序费用。而基于Sanger测序和PCR的方法由于单次扩增和测序所能获得的序列有限,因此需要重复进行PCR实验及测序,成本更高。
在时间成本方面,传统的基于Sanger测序方法需要反复地进行PCR扩增,该过程往往需要数天时间,如果算上每次扩增后的测序和分析时间则会更久。而本方法所采用的“先富集后建库”方案,从DNA提取到文库送测的全部实验流程仅需三天,且每个样只需要进行一次完整的实验流程,因此相较而言本方法在时间成本上也更为节约。但需要指出的是,基于高通量测序的“先建库后富集”方案在时间上要比本方法更快,这主要是由于省略了其中MALBAC全基因组随机扩增的步骤,同时先建库也能够减少一部分的实验操作,例如可以将多个样品混合后统一进行富集测序,在分析结果时再将不同样本分离,而“先富集后建库”由于没有在富集前事先加上测序接头和特异索引序列,故不能混样后富集,每一个富集管内只能有一种样品,尽管排除了富集偏好性的影响,但增加了很多实验操作。而“直接建库”方案由于没有富集和全基因组扩增的步骤,因此是基于高通量测序的方案中速度最快的,但该方法的效率也如前文结果所示是最低的。总而言之,尽管在富集效率和经济成本上有优势,但改进后的方法在时间上不如“先建库后富集”或是“直接建库”方案便捷,根据方案的选择所需时间差异在半天到一天。因此本方法在实验效率、经济成本和时间成本方面可以实现最优化。
实施例2
分别以爬行纲的中华鳖和哺乳纲的家猪的肌肉样本为研究对象,采用实施例1的方式,富集线粒体DNA后构建文库并测序,均要求测序公司提供4GB的数据量,并且也都组装出完整线粒体基因组。
测序数据(SRA:PRJNA796186)与组装序列结果(GenBank编号:OM236540,OM236541)均已上传至NCBI,与数据库中已有的序列并无显著不同。由此可见,本方法可以适用于脊椎动物线粒体基因组的测序,获得脊椎动物线粒体基因组,且无需目标物种的参考序列。
- 一种多鱗鯔鰕虎魚线粒体基因组全序列引物及设计、系统进化分析全序列扩增方法
- 一种多鱗鯔鰕虎魚线粒体基因组全序列引物及设计、系统进化分析全序列扩增方法