一种利用HPLC法检测火麻油中5种大麻酚化合物的方法
文献发布时间:2023-06-19 10:48:02
技术领域
本发明属于分析化学领域,具体涉及一种利用高效液相色谱法(HPLC)检测火麻油中5种大麻酚类化合物的方法。
背景技术
大麻中主要的功效成分为大麻酚类物质,其中四氢大麻酚酸(THCA)、大麻二酚酸(CBDA)、大麻酚(CBN)、大麻二酚(CBD)、
五种大麻酚类物质中,四氢大麻酚酸、大麻二酚酸、大麻酚和大麻二酚均具有一定的药理活性,比如大麻二酚在医学上对癫痫、抑郁症、多发性硬化症等疾病有明显的治疗作用。而
火麻油是以工业大麻籽经冷榨产生的油脂。它含有高达80%的多不饱和脂肪酸,其中ω-6/ω-3的比例为3:1,这与欧洲食品安全局的建议(3-5:1)非常接近。长期食用火麻油可有效补充人体必需营养素α-亚麻酸,达到润燥滑肠、有益心脑血管健康的功效,还具有降低血脂、血压、血糖等功效。同时由于其含有CBD、CBN等大麻素类物质而具有额外的保健功效。然而,由于制备工艺及原料成熟度存在差异,导致火麻油中可能含有THC等影响中枢神经的活性物质。美国、德国、瑞士、比利时、欧洲工业大麻协会(EIHA)等许多国家相继发布了政策和法规,对火麻油中THC的含量上限进行了规定。而我国对存在的大量火麻油类产品还缺乏相应的管理规定,尤其是缺乏火麻油中THC、CBD等几种主要大麻素类物质的检测方法,使得生产商、监管部门无法确定其中THC、CBD等物质的含量,因而无法把控产品质量,造成市场上火麻油产品良莠不齐。所以非常有必要建立稳定、准确的火麻油中THC、THCA、CBD、CBDA、CBN的检测方法标准。
然而,由于大麻素的结构相似以及这些物质之间存在的转换关系,现有技术难以有效地分离出各种大麻素,这对分析方法的准确性提出了较高要求。Marinko Petrovic′等基于乙腈提取和GC-MS检测手段,发展了火麻油样品中CBD、THC、CBN含量的检测方法,同时探究了火麻油中大麻酚含量与脂肪酸组成的关系。
专利号为“CN107589203”,发明名称为“一种利用SPE-HPLC同时检测汉麻中三种大麻酚类化合物的方法”的专利,提供了一种基于固相萃取-高效液相色谱法来检测汉麻中三种大麻酚类化合物的方法,并对其进行定性定量分析。被检测的三种大麻酚类化合物为CBD、CBN和THC,不包含THCA和CBDA。
综上,目前尚未有报道同时分析检测火麻油中THC、THCA、CBD、CBDA、CBN的方法,同时已有的方法缺乏系统的结果验证过程。
发明内容
有鉴于此,本发明以同时检测5种大麻酚类化合物的方法为核心,包含两个子发明(即A发明和B发明),以及基于这两个发明的各自拓展。
本发明的目的之一在于提供一种基于高效液相色谱法来同时检测火麻油中5种大麻酚化合物的方法。具体技术方案如下。
一种利用HPLC法检测火麻油中5种大麻酚化合物的方法,所述方法采用C18色谱柱,以超纯水为流动相A、以乙腈为流动相B进行梯度洗脱,样品进入检测器检测;所述大麻酚类化合物为
进一步,所述C18色谱柱为Supersil-ODS2色谱柱(内径为4.6mm×250mm,填料颗粒粒径为3-5μm);柱温为25-35℃;检测波长为200-230nm。
进一步,所述流动相的流速为0.5-1.5mL/min。
进一步,所述样品的进样体积为1-10μL。
进一步,上述梯度洗脱的条件为:
0-20min,流动相A与流动相B的体积比为:30-10:70-90;
20-22min,流动相A与流动相B的体积比为:10:90;
22-24min,流动相A与流动相B的体积比为:10-30:90-70;
24-30min,流动相A与流动相B的体积比为:30:70。
进一步,本发明所述的检测方法还包括在检测前对样品进行前处理;所述前处理包括将样品加入甲醇与异丙醇的混合溶剂中涡旋混匀,然后超声并离心,取上清液过滤的操作步骤。
进一步,所述混合溶剂中甲醇与异丙醇的体积比为:甲醇:异丙醇=50-80:50-20。
进一步,将样品加入甲醇与异丙醇的混合溶液中涡旋1-5min,然后超声提取10-20min,提取液以8000-12000rpm离心3-8min,然后取上清液过0.1-0.5μm的有机滤膜。
进一步,将样品加入甲醇与异丙醇的混合溶液中涡旋2-5min混匀,超声提取10-15min,提取液以10000rpm离心1-5min,取上清液过0.22μm有机滤膜后待测。
在本发明的其中一个实施例中,准确称取0.5g火麻油样品于10ml离心管中,加入2.5ml混合溶剂(甲醇:异丙醇=80:20,v/v),涡旋2min混匀,超声提取15min,提取液以10000rpm离心5min,取上清液过0.22μm有机滤膜后待测。
在本发明的其中一个实施例中,将过滤后的上清液移入m-PFC快速滤过型净化柱内继续过滤,缓慢推动注射杆,液相小瓶承接流出液,得到的滤液进行HPLC法检测。
本发明还提供一种分离检测火麻油中大麻酚类化合物的试剂组合物,所述试剂组合物采用C18色谱柱,以超纯水作为流动相A、乙腈为流动相B;所述大麻酚类化合物包括
本发明还可以提供一种基于高效液相色谱-串联质谱法联用同时检测5种大麻酚化合物的标准方法。具体技术方案如下。
一种利用HPLC-MS/MS同时检测5种大麻酚类化合物的方法,所述方法采用大麻酚专用色谱柱,以甲酸水溶液为流动相A、乙腈为流动相B进行梯度洗脱,进入检测器检测;所述大麻酚类化合物为四氢大麻酚酸(THCA)、大麻二酚酸(CBDA)、大麻酚(CBN)、大麻二酚(CBD)和
HPLC-MS/MS,即高效液相色谱-串联质谱法联用,是以高效液相色谱为分离手段,以质谱为鉴定工具的一种分离分析技术。HPLC方法在针对复杂样品进行分析时会受到一定要求的限制。而HPLC-MS/MS联用可以实现对复杂样品进行有效分离、定性和定量分析。
进一步,所述甲酸水溶液的质量浓度为0.1%-0.5%。
进一步,所述甲酸水溶液的质量浓度为0.1%、0.2%、0.3%、0.4%或0.5%。
进一步,所述大麻酚专用色谱柱内径为4.6mm×250mm,填料颗粒粒径为3-5μm;柱温为25-35℃。
进一步,所述流动相的流速为0.5-1.5mL/min。
进一步,所述方法采用多反应监控模式(MRM);所述5种大麻酚类化合物的离子化模式均为负离子模式。
进一步,采用上述方法进行梯度洗脱,所述梯度洗脱条件如下:0-15min,流动相A与流动相B的体积比为:30-5:70-95;15-15.1min,流动相A与流动相B的体积比为:5-30:95-70;15.1-25min,流动相A与流动相B的体积比为:30:70。
进一步,所述方法还包括在检测前进行样品的预处理操作;所述预处理操作包括使样品加热融化,然后溶于有机溶剂,离心后取上清液过有机滤膜。
进一步,所述样品为巧克力和/或糖果。
进一步,将融化后的样品加入甲醇溶液,涡旋1-5min后以8000-12000rpm离心3-10min,取上清液过0.1-0.5μm的有机滤膜。这一步操作的目的主要是为了提取大麻酚类化合物。由于巧克力中含有油脂,因此采用甲醇萃取,但很难不完全提取出脂类成分,而脂类的存在容易干扰大麻酚类的检测。因此后续又采用了离心,取上清液过有机滤膜来进一步纯化。得到的最终提取物适用于本发明的HPLC-MS/MS检测。
本发明还提供一种分离检测巧克力中大麻酚类化合物的试剂组合物,所述试剂组合物采用大麻酚专用色谱柱,以质量浓度为0.1%-0.5%的甲酸水溶液为流动相A、乙腈为流动相B;所述大麻酚类化合物包括四氢大麻酚酸(THCA)、大麻二酚酸(CBDA)、大麻酚(CBN)、大麻二酚(CBD)或
有益效果
本发明提供了一种基于高效液相色谱法同时检测火麻油中5种大麻酚类物质的分析方法。通过调节流动相配比,优化色谱条件实现被测组分色谱峰与基质中杂质峰的有效分离,无需使用样品净化柱,使得分析方法操作简便、耗时较短、经济。同时经过精密度实验、重复性实验以及加标回收率实验等对本方法的有效性进行验证,结果表明,本方法的检出限、定量限、精密度、回收率等结果均较好,可用于实际样品的分析确证。
此外,本发明通过对国内五个产地的火麻油进行检测,发现其中均含有THC,存在一定的食品安全风险。而本发明的技术方案可为相关检测方法标准的建立奠定方法学基础,同时对于杜绝大麻酚含量超标的火麻油流入市场以及促进国内外工业大麻食品有效监管具有重要意义。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍。显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。
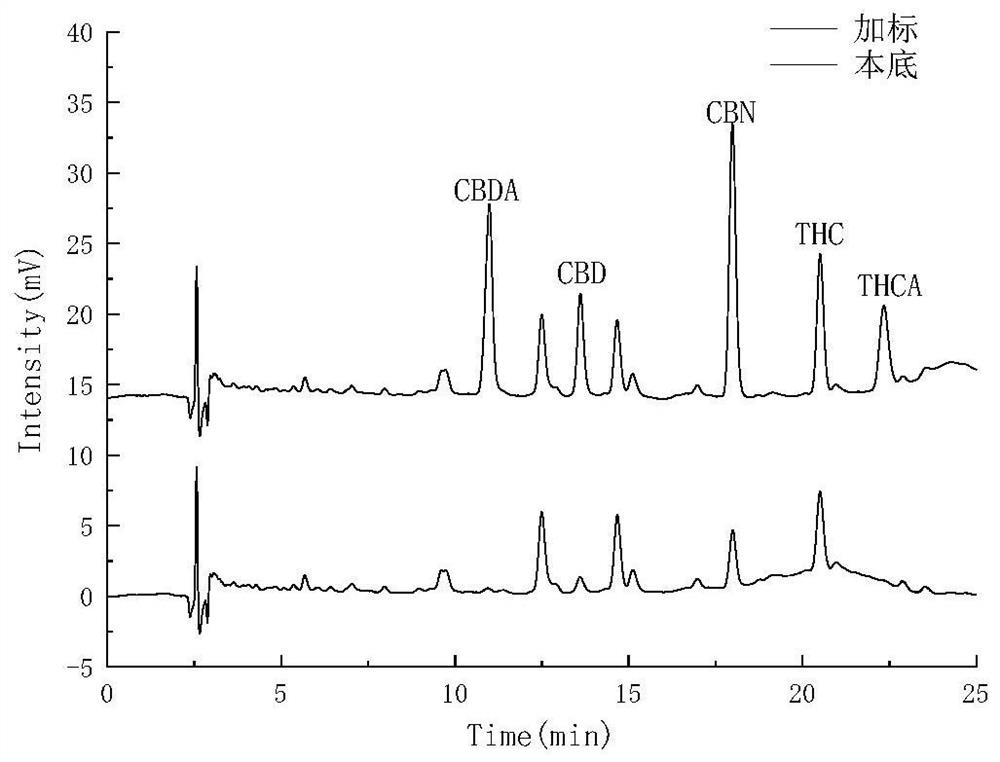
图1是两种不同色谱条件下的提取火麻油中5种大麻酚类物质分离图谱(A是梯度程序1;B是梯度程序2);
图2是火麻油样品净化前对比图(A是加标1μg/mL的混合标准品;B是加标5μg/mL的混合标准品);
图3是检测巧克力样品的高效液相色谱图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
需要说明的是,在本文中,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者装置不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者装置所固有的要素。在没有更多限制的情况下,由语句“包括一个……”限定的要素,并不排除在包括该要素的过程、方法、物品或者装置中还存在另外的相同要素。
如在本说明书中使用的,术语“大约”,典型地表示为所述值的+/-5%,更典型的是所述值的+/-4%,更典型的是所述值的+/-3%,更典型的是所述值的+/-2%,甚至更典型的是所述值的+/-1%,甚至更典型的是所述值的+/-0.5%。
在本说明书中,某些实施方式可能以一种处于某个范围的格式公开。应该理解,这种“处于某个范围”的描述仅仅是为了方便和简洁,且不应该被解释为对所公开范围的僵化限制。因此,范围的描述应该被认为是已经具体地公开了所有可能的子范围以及在此范围内的独立数字值。例如,范围
A发明:一种利用HPLC法检测火麻油中5种大麻酚化合物的方法
实施例一
检测方法
1)材料与仪器简介
大麻酚标准品:CBDA、CBD、CBN、THC、THCA标准品(1mg/mL,1mL,);甲醇、乙腈、异丙醇(色谱纯)。5个产地的火麻油,分别产于山西、陕西、广西、黑龙江和甘肃。
iChrom5100分析型液相色谱仪,配DAD检测器;Supersil-ODS2色谱柱(4.6mm×250mm,5μm);涡旋振荡器;离心机;分析天平;纯水仪;m-PFC快速过滤柱。
2)溶液配置
流动相:流动相A为纯水,为实验室纯水仪自制,电导率为18.2MΩ,使用前经水相滤膜抽滤,超声脱气。流动相B为乙腈(色谱纯),使用前经有机相滤膜抽滤,超声脱气。
标准储备液:取CBDA(1mg/mL)、CBD(1mg/mL)、CBN(1mg/mL)、THC(1mg/mL)、THCA(1mg/mL)标准品转移至10mL容量瓶中,加入混合溶剂(甲醇:异丙醇=80:20,v/v)定容,配制成100μg/mL的5种大麻酚混和标准储备液,4℃冰箱冷藏保存,有效期为1个月。
标准工作液:准确吸取混合标准储备液0.05、0.10、0.25、0.50、1.00、2.50、5.00mL分别置于10mL容量瓶中,用混合溶剂(甲醇:异丙醇=80:20,v/v)定容,配制为质量浓度为0.5、1.0、2.5、5.0、10.0、25.0、50.0μg/mL的系列混合标准工作液,现用现配。
3)样品前处理:准确称取0.5g(精确到0.001g)火麻油于10ml离心管中,加入2.5ml混合溶剂(甲醇:异丙醇=80:20,v/v),涡旋2min混匀,超声提取15min,提取液以10000rpm离心5min,取上清液过0.22μm有机滤膜后待测。
4)样品前处理净化:取上述上清液2mL,过0.22μm有机滤膜后,移入m-PFC快速滤过型净化小柱上端,缓慢推动注射杆,液相小瓶承接流出液,待上机检测。
5)液相色谱条件:采用Supersil-ODS2色谱柱(4.6mm×250mm,5μm),进样体积为10μL,流速1mL/min,柱温:35℃,检测波长为220nm,流动相A相为超纯水,流动相B相为乙腈,梯度洗脱条件为0-20min,70%B-90%B;20-22min,90%B;22-24min,90%B-70%B;24-30min,70%B。
6)精密度试验:在相同仪器条件下,取1μg/mL浓度的标准工作溶液,按上述液相色谱条件连续进样6次,测得峰面积,代入标准曲线线性回归方程得出测定浓度值,再计算测定浓度的相对标准偏差RSD。
7)重复性试验:在相同的仪器条件下,精密称取0.5g(精确到0.001g)火麻油6份,按上述样品前处理下方法操作,将处理好的6份待测液按上述液相色谱条件进样分析,测得5种大麻酚峰面积,代入标准曲线方程,计算测定浓度的标准偏差RSD。
8)标准曲线:将配制好的0.5μg/mL、1μg/mL、2.5μg/mL、5μg/mL、10μg/mL、25μg/mL、50μg/mL标准工作溶液按上述液相色谱条件进样分析,分别以5种化合物的浓度为横坐标,峰面积响应值为纵坐标,绘制系列标准曲线,进行线性回归,得到5种大麻酚的标准曲线线性回归方程。
9)定量限、检出限:将上述标准储备液按上述液相色谱条件测定,测得5种大麻酚的峰面积,带入相应物质的标准曲线回归方程中。以最低水平标准溶液中目标组分的3倍平均信噪比为检出限(LOD),10倍平均信噪比为定量限(LOQ),计算得本方法的LOD和LOQ。
10)加标回收率:准确称取火麻油0.5g(精确到0.01g)12份,编号1-12,其中1-3号作为本底,4-6号加标配成0.5μg/mL待测液,7-9号加标配成1μg/mL待测液,10-12号加标配成5μg/mL待测液,按上述样品前处理下方法操作,将处理好的本底待测液以及低、中、高3种浓度的加标样品经HPLC分析,测得峰面积,计算回收率。
结果与分析
1)液相色谱条件优化
为尽可能的将待测的五种大麻酚物质与火麻油中的基质成分的分离,首先对流动相梯度程序进行了优化。以山西火麻油的甲醇提取液为本底溶液,向其中添加了5μg/mL混合标准品,采用两种不同的流动相梯度程序对本底液和加标液分别进行了色谱分离,两种流动相梯度程序分别为:(1)0-20min,80%B-90%B;20-22min,90%B;22-24min,90%B-80%B;24-30min,80%B。(2)0-20min,70%B-90%B;20-22min,90%B;22-24min,90%B-70%B;24-30min,70%B。分离结果如图1所示,并通过单标法对峰的归属进行了确证。结果表明在梯度程序(1)下CBD无法与火麻油基质成分分离,THC和THCA分离度较差(图1A)。而经过降低初始有机相的组成,在梯度程序(2)下,火麻油中各个峰分离情况较好,且基质中杂质峰和待测的五种大麻酚类物质的色谱峰没有相互掩盖(图1B),所以后续实验都采用梯度程序2进行实验优化和方法验证。
总结:从图1A和图1B的对比可以看出,通过调整流动相的比例,改变洗脱条件,能够实现将基质中杂质峰与被分析物色谱峰分离的效果,降低了杂质峰干扰的可能,从而省去对提取液进行净化处理的步骤,使得操作简便、耗时较短。
2)提取溶剂优化
根据5种大麻酚的理化性质,在仪器及实验条件相同情况下,本实验选择了(1)100%甲醇;(2)甲醇:异丙醇=80:20;(3)甲醇:异丙醇=50:50的3种溶剂,取相同火麻油样品,向其中添加了5μg/mL混合标准品,采用标准储备液的提取方式,分别用以上3种溶剂进行提取,并测定加标回收率,结果如下表1。从表中可以看出,添加了异丙醇后,样品的回收率可以控制在70%-100%之间,其中以甲醇:异丙醇=80:20进行提取,结果具有更好的稳定性,可以控制RSD<5%。所以在后续实验中采用甲醇:异丙醇=80:20的提取溶剂进行提取。
表1三种不同溶剂提取结果
3)提取液净化
由于火麻油基质中含有大量的脂肪酸,为了降低基质对待测物的影响,同时为了提高方法的灵敏度,通常需要采用样品基质净化的方法在保留待测物的前提下尽可能多的去除样品基质中的杂质。
滤过性净化法(m-PFC,multi-plug filtration cleanup)是一种基于QuEChERS法发展起来的对基质中干扰物质进行净化的方法。该方法将含有多壁碳纳米管、N-丙基乙二胺(PSA)、C18和无水硫酸镁的填料装于注射器中,使用时将提取液通过抽提或推送方式通过填料,使填料与基质中的色素、脂类、部分糖类、甾醇类、茶多酚、有机酸和生物碱等干扰物作用或吸附,而不与目标物反应,完成基质的净化。该方法与传统方法相比,无需称量吸附剂,大大缩短了净化时间,同时能够与LC-MS、IMS和拉曼等在线或离线联用,可大幅度提高前处理速度和效率。
本发明考察了m-PFC样品净化方式对检测方法的影响,本方法分别以添加了1μg/mL、5μg/mL的混合标品的山西火麻油甲醇提取物为样品,选用m-PFC快速净化柱为净化柱,采用上述样品前处理净化的方法对样品进行净化,并以上述样品前处理中的色谱条件进行了HPLC检测,结果如图2所示。其中,图2A为加标1μg/mL的混合标准品,图2B为加标5μg/mL的混合标准品。
总结:由图2可见,本实验选用的m-PFC净化柱净化效果并不佳,不仅引入了新的杂质,还对被检测组分THCA有严重吸附,导致THCA无检出,所以后续实验中不使用净化柱进行前处理。在之后的研究中可参考大麻酚的物理化学性质,对比多种净化柱净化效果,在不引入杂质的基础上,综合各方面因素,选择能对除被检测组分以外的其他物质有较好吸收的净化柱,以减小基质效应可能对检测结果产生的影响。
4)标准曲线、定量限、检出限
对5种大麻酚混合标准溶液系列进行测定,通过ichrom5100workstation绘制标准曲线回归方程,其线性范围、相关系数、LOD及LOQ如下表2所示。
表2五种大麻酚的线性范围、LOD和LOQ
总结:由表中数据可知,本方法在0.5~50μg/mL范围内有较好的线性关系,相关系数r2均>0.998。经计算,5种大麻酚类物质的检出限(LOD,S/N=3)均在0.10~0.25μg/g之间,定量限(LOQ,S/N=10)均在0.33~0.83μg/g之间。本发明的方法可以达到标准化,可以最低检测定量限附近的浓度。
5)精密度
按照上述精密度实验步骤,平行测定6次1μg/mL的混合标准溶液,测得的CBDA、CBD、CBN、THC、THCA五种大麻酚的浓度与RSD如下表3所示。
表3精密度实验结果
总结:由上表数据可知,精密度实验结果RSD在0.7%-3.9%之间,表明本方法稳定可靠,准确度高。
6)重复性
以陕西产的火麻油为样品,按上述重复性实验步骤,测得平行6组火麻油中CBDA、CBD、CBN、THC、THCA五种大麻酚的浓度及RSD如下表4所示。
表4重复性实验结果
总结:由上标数据可知,6次重复实验的RSD在1.9%-3.2%之间,表明本方法重复性较好。
7)加标回收率
准确移取一定量的混合标准溶液加入到样品中,用混合溶剂(甲醇:异丙醇=80:20,v/v)定容配制成0.5μg/mL、1μg/mL、5μg/mL的低、中、高三种浓度水平的加标样品,用本实验方法进行测定,并计算回收率,结果如表5所示。
表5加标回收率实验结果
总结:由表中数据可知,5种大麻酚的回收率在77.1%-103.3%,相对标准偏差RSD≤4.7%,表明本方法回收率好,结果准确可靠。
8)实际样品测定
按照本实验方对山西、广西、陕西、黑龙江、甘肃5个不同产地的火麻油进行分析测试,结果见表6。
表6不同产地火麻油中5种大麻酚含量
总结:实际样品分析结果表明,除山西火麻油中不含CBDA和THCA外,陕西、广西、黑龙江、甘肃等4个产地的火麻油中5种大麻酚都有检出,且具有成瘾性的THC的含量均>3.2μg/g,其中陕西火麻油中THC含量甚至高达11.9μg/g,这说明市售火麻油存在不同程度的大麻酚类物质污染,有一定的食品安全风险。
B发明:一种利用HPLC-MS/MS同时检测巧克力中5种大麻酚化合物的方法
实施例一
1)样品前处理:
称取0.5g巧克力样品于10ml离心管,盖好盖子,在45℃的水浴锅预热5分钟使其融化,然后加2.5ml甲醇,涡旋2min,1000rmp离心5min,取上清液然后过0.22μm有机滤膜,待用。
2)检测:
HPLC分离的色谱条件为:大麻酚专用柱(4.6mm×250I.D.,5.0μm),柱温为30℃。流动相A为0.1%甲酸水溶液,流动相B为乙腈,流速为1.0mL/min,流动相梯度洗脱。梯度洗脱程序为:0-15min,70%-95%B;15-15.1min,95%-70%B;15.1-25min,70%B。
3)质谱:
质谱部分采用多反应监控模式(MRM),五种大麻酚类物质的质谱参数如下表所示:
表7五种大麻酚类物质的质谱参数
4)结果
采用外标法测定糖果巧克力产品中5种大麻酚类含量。方法的检出限为10μg/kg,定量限为30μg/kg,5种大麻酚的回收率均在90%-110%之间,RSD<5%。
总离子流出图如图3所示。其中按出峰顺序,相应各峰依次为CBDA、CBD、CBN、THC、THCA-A。
上面结合附图对本发明的实施例进行了描述,但是本发明并不局限于上述的具体实施方式,上述的具体实施方式仅仅是示意性的,而不是限制性的,本领域的普通技术人员在本发明的启示下,在不脱离本发明宗旨和权利要求所保护的范围情况下,还可做出很多形式,这些均属于本发明的保护之内。
- 一种利用HPLC法检测火麻油中5种大麻酚化合物的方法
- 一种火麻油中四氢大麻酚检测的前处理方法