共有抗原
文献发布时间:2023-06-19 09:52:39
相关申请的交叉引用
本申请要求2018年5月23日提交的美国临时申请号62/675,649和2018年5月23日提交的美国临时申请号62/675,559的权益,出于所有目的,其中的每一种在此通过引用整体并入。
序列表
本申请含有序列表,其已经以ASCII格式电子提交,并且在此通过引用整体并入。创建于2019年5月22日的所述ASCII拷贝,命名为GSO-019_SL.txt,并且大小为6,925,585字节。
背景技术
基于肿瘤特异性抗原的治疗性疫苗作为新一代个性化癌症免疫疗法具有广阔的前景。
新抗原疫苗设计的一个问题是在受试者肿瘤内存在的众多编码突变中,哪种突变可以产生“最佳的”治疗性新抗原,例如能够引起抗肿瘤免疫并使肿瘤消退的抗原。
提出的初步方法并入了使用下一代测序的基于突变的分析、RNA基因表达及候选新抗原肽的MHC结合亲和力预测
事实上,多个团队所进行的关于由肿瘤细胞递呈的肽的分析显示,预计使用基因表达和MHC结合亲和力递呈的肽中不到5%可以在肿瘤表面MHC上发现
现有的递呈预测方法的这一低阳性预测值(PPV)提出了有关基于新抗原的疫苗设计的问题。如果使用PPV低的预测方法来设计疫苗,则大多数患者不太可能接受治疗性新抗原,且少数患者可能要接受一种以上新抗原(即使假设所有递呈的肽都具有免疫原性)。因此,用当前方法进行新抗原疫苗接种不太可能在众多具有肿瘤的受试者中取得成功。
此外,先前的方法仅使用顺式作用突变来产生候选新抗原,而在很大程度上忽视了考虑neo-ORF的其它来源,包括在多种肿瘤类型中出现且导致许多基因异常剪接的剪接因子的突变
最后,由于文库构建、外显子组和转录组捕捉、测序或数据分析的条件并非最佳条件,故肿瘤基因组和转录组分析的标准方法可能会遗漏产生候选新抗原的体细胞突变。同样,标准肿瘤分析方法可能会无意中促成序列伪影或生殖系多态性作为新抗原,而分别导致疫苗能力的低效使用或自身免疫的风险。
除了当前新抗原预测方法的挑战之外,可用于人中新抗原递送的可用载体系统也存在某些挑战,其中许多源自人。举例来说,许多人由于先前的自然暴露而对人病毒具有预先存在的免疫力,并且这种免疫力可能是使用重组人病毒进行新抗原递送以用于癌症治疗的主要障碍。
此外,靶向癌症患者之间共有的抗原作为疫苗策略具有广阔的前景,包括靶向具有突变的新抗原以及不具有突变的肿瘤抗原(例如,不适当表达的肿瘤抗原)。利用共有抗原疫苗策略的挑战至少包括以上讨论的那些。
发明内容
本文公开了一种用于递送抗原表达系统的组合物,其包含:所述抗原表达系统,其中所述抗原表达系统包含一个或多个载体,所述一个或多个载体包含:(a)载体主链,其中所述主链包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(聚(A))序列;和(b)抗原盒,其中所述抗原盒包含:(i)至少一个抗原编码核酸序列,其包含:(I)至少肿瘤特异性MHC I类抗原编码核酸序列,其包含:(A)MHC I类表位编码核酸序列,其中所述MHC I类表位编码核酸序列编码选自SEQ ID NO:57-29,357的MHC I类表位,(B)任选地,5′接头序列,和(C)任选地,3′接头序列;(ii)任选地,可操作地连接于所述抗原编码核酸序列的第二启动子核苷酸序列;和(iii)任选地,至少一个MHC II类抗原编码核酸序列;(iv)任选地,至少一个编码GPGPG氨基酸接头序列的核酸序列(SEQ ID NO:56);和(v)任选地,至少一个第二聚(A)序列,其中所述第二聚(A)序列是所述载体主链的原生聚(A)序列或外源聚(A)序列。
本文还公开了一种用于递送抗原表达系统的组合物,其包含:所述抗原表达系统,其中所述抗原表达系统包含一个或多个载体,所述一个或多个载体包含:(a)载体主链,其中所述主链包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(聚(A))序列;和(b)抗原盒,其中所述抗原盒包含:(i)至少一个抗原编码核酸序列,其包含:(I)至少10、11、12、13、14、15、16、17、18、19或20个彼此线性连接的肿瘤特异性MHC I类抗原编码核酸序列,其包含:(A)KRAS_G12A MHC I类表位编码核酸序列,其中所述KRAS_G12A MHC I类表位编码核酸序列编码包含SEQ ID NO:19,831的序列的MHC I类,(B)KRAS_G12C MHC I类表位编码核酸序列,其中所述KRAS_G12C MHC I类表位编码核酸序列编码包含SEQ ID NO:14,954的序列的MHC I类表位,(C)KRAS_G12D MHC I类表位编码核酸序列,其中所述KRAS_G12D MHC I类表位编码核酸序列编码选自SEQ ID NO:19,749和19,865的MHC I类表位,和(D)KRAS_G12V MHC I类表位编码核酸序列,其中所述KRAS_G12V MHC I类表位编码核酸序列编码选自SEQ ID NO:19,976、19,979、19,779、11,495和19,974的MHC I类表位,其中所述肿瘤特异性MHC I类抗原编码核酸序列中的每一个包含I类表位编码核酸序列,任选地其中每个MHC I表位编码核酸序列编码选自SEQ ID NO:57-29,357的MHC I表位,并且其中所述肿瘤特异性MHC I类抗原编码核酸序列中的每一个包含:(A)任选地,5'接头序列,和(B)任选地,3'接头序列;(ii)任选地,可操作地连接于所述抗原编码核酸序列的第二启动子核苷酸序列;和(iii)任选地,至少一个MHC II类抗原编码核酸序列;(iv)任选地,至少一个编码GPGPG氨基酸接头序列的核酸序列(SEQ ID NO:56);和(v)任选地,至少一个第二聚(A)序列,其中所述第二聚(A)序列是所述载体主链的原生聚(A)序列或外源聚(A)序列。
本文还公开了一种用于递送抗原表达系统的组合物,其包含:所述抗原表达系统,其中所述抗原表达系统包含一个或多个载体,所述一个或多个载体包含:(a)载体主链,其中所述主链包含:(i)至少一个启动子核苷酸序列,和(ii)至少一个聚腺苷酸化(聚(A))序列;和(b)抗原盒,其中所述抗原盒包含:(i)至少一个抗原编码核酸序列,其包含:(I)至少20个彼此线性连接的肿瘤特异性MHC I类抗原编码核酸序列,其包含:(A)KRAS_G12A MHC I类表位编码核酸序列,其中所述KRAS_G12A MHC I类表位编码核酸序列编码包含SEQ IDNO:19,831的序列的MHC I类,(B)KRAS_G12C MHC I类表位编码核酸序列,其中所述KRAS_G12C MHC I类表位编码核酸序列编码包含SEQ ID NO:14,954的序列的MHC I类表位,(C)KRAS_G12D MHC I类表位编码核酸序列,其中所述KRAS_G12D MHC I类表位编码核酸序列编码选自SEQ ID NO:19,749和19,865的MHC I类表位,和(D)KRAS_G12V MHC I类表位编码核酸序列,其中所述KRAS_G12V MHC I类表位编码核酸序列编码选自SEQ ID NO:19,976、19,979、19,779、11,495和19,974的MHC I类表位,(E)KRAS_G13D MHC I类表位编码核酸序列,(F)KRAS_Q61K MHC I类表位编码核酸序列,(G)TP53_R249M MHC I类表位编码核酸序列,(H)CTNNB1_S45P MHC I类表位编码核酸序列,(I)CTNNB1_S45F MHC I类表位编码核酸序列,(J)ERBB2_Y772_A775dup MHC I类表位编码核酸序列,(K)KRAS_Q61R MHC I类表位编码的核酸序列,(L)CTNNB1_T41A MHC I类表位编码核酸序列,(M)TP53_K132N MHC I类表位编码核酸序列,(N)KRAS_Q61L MHC I类表位编码核酸序列,(O)TP53_R213L MHC I类表位编码核酸序列,(P)BRAF_G466V MHC I类表位编码核酸序列,(Q)KRAS_Q61H MHC I类表位编码核酸序列,(R)CTNNB1_S37F MHC I类表位编码核酸序列,(S)TP53_S127Y MHC I类表位编码核酸序列,(T)TP53_K132E MHC I类表位编码核酸序列,(U)KRAS_G12C MHC I类表位编码核酸序列,并且其中所述肿瘤特异性MHC I类抗原编码核酸序列中的每一个包含:(A)任选地,5'接头序列,和(B)任选地,3'接头序列;(ii)任选地,可操作地连接于所述抗原编码核酸序列的第二启动子核苷酸序列;和(iii)任选地,至少一个MHC II类抗原编码核酸序列;(iv)任选地,至少一个编码GPGPG氨基酸接头序列的核酸序列(SEQ ID NO:56);和(v)任选地,至少一个第二聚(A)序列,其中所述第二聚(A)序列是所述载体主链的原生聚(A)序列或外源聚(A)序列。
在一些方面,所述至少一个抗原编码核酸序列不包含选自SEQ ID NO:19,749和19,865的MHC I类表位。
本文还公开了一种用于递送抗原表达系统的组合物,其包含:所述抗原表达系统,其中所述抗原表达系统包含一个或多个载体,所述一个或多个载体包含:(a)载体主链,其中所述载体主链包含黑猩猩腺病毒载体,任选地其中所述黑猩猩腺病毒载体是ChAdV68载体,或甲病毒载体,任选地其中所述甲病毒载体是委内瑞拉马脑炎病毒载体;和(b)整合在所述26S启动子核苷酸序列与所述聚(A)序列之间的抗原盒,其中所述抗原盒包含:(i)至少一个抗原编码核酸序列,其包含:(I)至少10、11、12、13、14、15、16、17、18、19或20个肿瘤特异性和MHC I类抗原编码核酸序列,它们彼此线性连接并且各自包含:(A)MHC I类表位编码核酸序列,其中所述MHC I类表位编码核酸序列编码长度为7-15个氨基酸的MHC I类表位,并且其中所述MHC I类表位中的至少一个选自SEQ ID NO:57-29,357,(B)5'接头序列,其中所述5'接头序列编码所述MHC I类表位的原生N端氨基酸序列,并且其中所述5'接头序列编码长度为至少3个氨基酸的肽,(C)3'接头序列,其中所述3'接头序列编码所述MHC I类表位的原生C端酸序列,并且其中所述3'接头序列编码长度为至少3个氨基酸的肽,并且其中所述抗原盒可操作地连接于所述26S启动子核苷酸序列,其中所述MHC I类抗原编码核酸序列中的每一个编码长度为13至25个氨基酸的多肽,并且其中每个MHC I类抗原编码核酸序列的每个3'端与下一个MHC I类抗原编码核酸序列的5'端连接,其中例外为所述抗原盒中的最终MHC I类抗原编码核酸序列;和(ii)至少两个MHC II类抗原编码核酸序列,其包含:(I)PADRE MHC II类序列(SEQ ID NO:48),(II)破伤风类毒素MHC II类序列(SEQ ID NO:46),(III)编码连接所述PADRE MHC II类序列和所述破伤风类毒素MHC II类序列的GPGPG氨基酸接头序列的第一核酸序列,(IV)编码连接所述至少两个MHC II类抗原编码核酸序列的5'端与所述肿瘤特异性MHC I类抗原编码核酸序列的GPGPG氨基酸接头序列的第二核酸序列,(V)任选地,编码所述至少两个MHC II类抗原编码核酸序列的3'端处的GPGPG氨基酸接头序列的第三核酸序列。
本文还公开了一种评估患有癌症的受试者的方法,其包括以下步骤:a)确定或已经确定:1)所述受试者是否具有预测或已知递呈基于抗原的疫苗中包含的抗原的HLA等位基因,和以下一项或两项:1)受试者的肿瘤是否表达与所述抗原相关的基因,任选地,其中与正常细胞或组织相比所述基因异常表达,2)所述受试者的肿瘤是否具有与所述抗原相关的突变,b)由(a)的结果确定或已经确定:当所述受试者表达所述HLA等位基因和所述受试者的肿瘤表达所述基因和/或所述受试者的肿瘤具有所述突变时,所述受试者是所述基于抗原的疫苗的治疗的候选者,其中所述抗原包含至少一个选自SEQ ID NO:57-29,357的MHCI类表位序列,以及c)任选地,向所述受试者施用或已经施用所述基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)所述至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列。
本文还公开了一种评估患有癌症的受试者的方法,其包括以下步骤:a)确定或确定所述受试者是否表达:1)A0301 HLA等位基因且所述受试者的肿瘤具有KRAS_G12A突变,2)A0201 HLA等位基因且所述受试者的肿瘤具有KRAS_G12C突变,3)C0802 HLA等位基因或A1101 HLA等位基因且所述受试者的肿瘤具有KRAS_G12D突变,或4)A0301 HLA等位基因或A1101 HLA等位基因或A3101 HLA等位基因或C0102 HLA等位基因或A0302 HLA等位基因且所述受试者的肿瘤具有KRAS_G12V突变,以及b)由(a)的结果确定或已经确定:当所述受试者:1)表达所述A0301等位基因且所述受试者的肿瘤具有所述KRAS_G12A突变,2)表达所述A0201等位基因且所述受试者的肿瘤具有所述KRAS_G12C突变,3)表达所述C0802 HLA等位基因或所述A1101 HLA等位基因且所述受试者的肿瘤具有所述KRAS_G12D突变,或4)表达所述A0301 HLA等位基因或所述A1101 HLA等位基因或所述A3101 HLA等位基因或所述C0102HLA等位基因或所述A0302 HLA等位基因且所述受试者的肿瘤具有KRAS_G12V突变时,所述受试者是所述基于抗原的疫苗的治疗的候选者,以及c)任选地,向所述受试者施用或已经施用所述基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个分别包含所述KRAS_G12A突变、所述KRAS_G12C突变、所述KRAS_G12D突变或所述KRAS_G12V突变的MHC I类表位,或2)MHC I类表位编码核酸序列,其编码至少一个分别包含所述KRAS_G12A突变、所述KRAS_G12C突变、所述KRAS_G12AD突变或所述KRAS_G12V突变的MHC I类表位。
在一些方面,步骤(a)和/或(b)包括从第三方获得数据集,所述第三方已经处理了来自所述受试者的样品。在一些方面,步骤(a)包括从所述受试者获得样品并使用选自以下的方法测定所述样品:外显子组测序、靶向外显子组测序、转录组测序、Sanger测序、基于PCR的基因分型测定、基于质谱的方法、微阵列、Nanostring、ISH和IHC。在一些方面,所述样品包含肿瘤样品、正常组织样品或所述肿瘤样品和所述正常组织样品。在一些方面,所述样品选自组织、体液、血液、肿瘤活检物、脊髓液和针抽吸物。在一些方面,所述基因选自:表34中发现的任何基因。在一些方面,所述基因选自:表32中发现的任何基因。在一些方面,所述癌症选自:肺癌、微卫星稳定结肠癌和胰腺癌。在一些方面,所述HLA等位基因具有至少5%的HLA频率。在一些方面,所述至少一个MHC I类表位由与所述受试者的肿瘤相关的细胞上的HLA等位基因递呈。在一些方面,所述基于抗原的疫苗包含抗原表达系统。在一些方面,所述抗原表达系统包括本文公开的任一种抗原表达系统。在一些方面,所述基于抗原的疫苗包含本文公开的任一种药物组合物。
本文还公开了一种用于治疗患有癌症的受试者的方法,所述方法包括向所述受试者施用基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列,其中所述至少一个MHC I类表位序列选自SEQ ID NO:57-29,357。在一些方面,所述至少一个MHC I类抗原编码核酸序列源自所述患有癌症的受试者的肿瘤。在一些方面,所述至少一个MHC I类抗原编码核酸序列不是源自所述患有癌症的受试者的肿瘤。
本文还公开了一种用于在受试者中诱导免疫应答的方法,所述方法包括所述方法包括向所述受试者施用基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列,其中所述至少一个MHC I类表位序列选自SEQ ID NO:57-29,357。在一些方面,所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因。在一些方面,所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因,并且其中所述至少一个MHC I类表位序列包含选自参考表34中突变的突变。在一些方面,所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因,并且其中所述至少一个MHC I类表位序列包含选自参考表32中突变的突变。
本文还公开了一种用于在受试者中诱导免疫应答的方法,所述方法包括向所述受试者施用基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列,其中所述至少一个MHC I类表位序列选自SEQ ID NO:57-29,357,并且其中所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因。
本文还公开了一种用于在受试者中诱导免疫应答的方法,所述方法包括向所述受试者施用基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列,其中所述至少一个MHC I类表位序列选自SEQ ID NO:57-29,357,并且其中所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因,并且其中所述至少一个MHC I类表位序列包含选自参考表34中突变的突变,并且其中所述受试者表达至少一个表34中所示的HLA等位基因,其与表34中所示的相应突变匹配(例如,KRAS_G13D和C0802)。
本文还公开了一种一种用于在受试者中诱导免疫应答的方法,所述方法包括向所述受试者施用基于抗原的疫苗,其中所述基于抗原的疫苗包含:1)至少一个MHC I类表位,或2)编码所述至少一个MHC I类表位的MHC I类表位编码核酸序列,其中所述至少一个MHCI类表位序列选自SEQ ID NO:57-29,357,并且其中所述受试者表达至少一个预测或已知递呈所述至少一个MHC I类表位序列的HLA等位基因,并且其中所述至少一个MHC I类表位序列包含选自参考表32中突变的突变。在一些方面,所述基于抗原的疫苗包含抗原表达系统。在一些方面,所述抗原表达系统包括本文公开的任一种抗原表达系统。在一些方面,所述基于抗原的疫苗包含本文公开的任一种药物组合物。
在一些方面,新抗原盒的各元件的有序序列以下式描述,其从5'至3'包含:
Pa-(L5b-Nc-L3d)X-(G5e-Uf)Y-G3g
其中P包含第二启动子核苷酸序列,其中a=0或1,N包含MHC I类表位编码核酸序列之一,其中c=1,L5包含5'接头序列,其中b=0或1,L3包含3'接头序列,其中d=0或1,G5包含至少一个编码GPGPG氨基酸接头的核酸序列之一,其中e=0或1,G3包含至少一个编码GPGPG氨基酸接头的核酸序列之一,其中g=0或1,U包含至少一个MHC II类抗原编码核酸序列之一,其中f=1,X=1至400,其中对于每个X,相应的Nc是表位编码核酸序列,并且Y=0、1或2,其中对于每个Y,相应的Uf是抗原编码核酸序列。在一些方面,对于每个X,相应的Nc是不同的MHC I类表位编码核酸序列。在一些方面,对于每个Y,相应的Uf是不同的MHC II类抗原编码核酸序列。
在一些方面,a=0,b=1,d=1,e=1,g=1,h=1,X=20,Y=2,至少一个启动子核苷酸序列是由主链提供的单个26S启动子核苷酸序列,至少一个聚腺苷酸化聚(A)序列是由主链提供的至少100个连续A核苷酸的聚(A)序列,每个N编码长度为7-15个氨基酸的MHC I类表位,L5是编码MHC I表位的原生N端氨基酸序列的原生5'接头序列,并且其中5'接头序列编码长度为至少3个氨基酸的肽,L3是编码MHC I表位的原生核酸末端核酸序列的原生3'接头序列,并且其中3'接头序列编码长度为至少3个氨基酸的肽,U是PADRE II类序列和破伤风类毒素MHC II类序列中的每一者,载体主链包含黑猩猩腺病毒载体,任选地其中所述黑猩猩腺病毒载体是ChAdV68载体,或甲病毒载体,任选地其中所述甲病毒载体是委内瑞拉马脑炎病毒载体,并且并且MHC I类新抗原编码核酸序列中的每一者编码长度为13至25个氨基酸的多肽。
在一些方面,所述新抗原盒整合在所述至少一个启动子核苷酸序列与所述至少一个聚(A)序列之间。在一些方面,其中所述至少一个启动子核苷酸序列可操作地连接于所述新抗原编码核酸序列。
在一些方面,所述一个或多个载体包含一个或多个+-链RNA载体。在一些方面,所述一个或多个+-链RNA载体包含5'7-甲基鸟苷(m7g)帽。在一些方面,通过体外转录产生一个或多个+-链RNA载体。在一些方面,一个或多个载体在哺乳动物细胞内自我复制。
在一些方面,所述主链包含奥拉病毒(Aura virus)、摩根堡病毒(Fort Morganvirus)、委内瑞拉马脑炎病毒(Venezuelan equine encephalitis virus)、罗斯河病毒(Ross River virus)、塞姆利基森林病毒(Semliki Forest virus)、辛德毕斯病毒(Sindbis virus)或马雅鲁病毒(Mayaro virus)的至少一个核苷酸序列。在一些方面,所述载体主链包含委内瑞拉马脑炎病毒的至少一个核苷酸序列。在一些方面,所述主链包含至少用于非结构蛋白介导扩增的序列,26S启动子序列,聚(A)序列,由奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列编码的非结构蛋白1(nsP1)基因、nsP2基因、nsP3基因和nsP4基因。在一些方面,所述主链包含至少用于非结构蛋白介导扩增的序列,26S启动子序列,和由奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列编码的聚(A)序列。在一些方面,所述非结构蛋白介导扩增的序列选自:甲病毒5'UTR、51-nt CSE、24-nt CSE、26S亚基因组启动子序列、19-nt CSE、甲病毒3'UTR或其组合。
在一些方面,所述主链不编码结构病毒粒子蛋白衣壳E2和E1。在一些方面,插入新抗原盒以代替奥拉病毒、摩根堡病毒、委内瑞拉马脑炎病毒、罗斯河病毒、塞姆利基森林病毒、辛德毕斯病毒或马雅鲁病毒的核苷酸序列内的结构病毒粒子蛋白。
在一些方面,委内瑞拉马脑炎病毒(VEE)包含菌株TC-83。在一些方面,委内瑞拉马脑炎病毒包含以SEQ ID NO:3或SEQ ID NO:5所示的序列。在一些方面,委内瑞拉马脑炎病毒包含SEQ ID NO:3或SEQ ID NO:5的序列,其还包含碱基对7544与11175之间的缺失。在一些方面,主链是以SEQ ID NO:6或SEQ ID NO:7所示的序列。在一些方面,插入新抗原盒以代替SEQ ID NO:3或SEQ ID NO:5的序列中所示的碱基对7544与11175之间的缺失。
在一些方面,新抗原盒的插入提供了包含nsP1-4基因和至少一个抗原编码核酸序列的多顺反子RNA的转录,其中所述nsP1-4基因和所述至少一个抗原编码核酸序列在单独的开放阅读框中。
在一些方面,至少一个启动子核苷酸序列是由主链编码的天然26S启动子核苷酸序列。在一些方面,至少一个启动子核苷酸序列是外源RNA启动子。在一些方面,第二启动子核苷酸序列是26S启动子核苷酸序列。在一些方面,第二启动子核苷酸序列包含多个26S启动子核苷酸序列,其中每个26S启动子核苷酸序列提供一个或多个分开的开放阅读框的转录。
在一些方面,腺病毒载体为黑猩猩腺病毒(ChAd)载体,任选地C68载体。在一些方面,腺病毒载体包含以SEQ ID NO:1所示的序列。在一些方面,腺病毒载体包含以SEQ IDNO:1所示的序列,不同之处在于所述序列完全缺失或功能缺失选自以下的至少一个基因:以SEQ ID NO:1所示的序列的黑猩猩腺病毒E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因,任选地其中所述序列完全缺失或功能缺失:以SEQ ID NO:1所示的序列的(1)E1A和E1B;(2)E1A、E1B和E3;或(3)E1A、E1B、E3和E4。在一些方面,腺病毒载体包含获自SEQ ID NO:1序列的基因或调控序列,任选地其中所述基因选自:以SEQ ID NO:1所示的序列的黑猩猩腺病毒反向末端重复序列(ITR)、E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因。
在一些方面,新抗原盒插入腺病毒载体中的E1区、E3区和/或允许并入新抗原盒的任何缺失的AdV区。
在一些方面,腺病毒载体的至少一个启动子序列是诱导型的。在一些方面,腺病毒载体的至少一个启动子序列是非诱导型的。在一些方面,腺病毒载体的至少一个启动子序列是CMV、SV40、EF-1、RSV、PGK或EBV启动子序列。
在一些方面,腺病毒载体的新抗原盒还包含可操作地连接于多个序列中的至少一者的至少一个聚A序列,任选地其中所述聚A序列位于所述多个序列中的至少一个序列的3'。
在一些方面,腺病毒载体由第一代、第二代或辅助依赖性腺病毒载体之一产生。
在一些方面,腺病毒载体包含以SEQ ID NO:1所示序列的碱基对编号577与3407之间的一个或多个缺失,并且任选地其中腺病毒载体还包含碱基对27,141与32,022之间或碱基对27,816与31,332之间的一个或多个缺失。在一些方面,腺病毒载体还包含以SEQ IDNO:1所示序列的碱基对编号3957与10346之间、碱基对编号21787与23370之间以及碱基对编号33486与36193之间的一个或多个缺失。
在一些方面,一个或多个新抗原表达载体的大小各为至少300nt。在一些方面,一个或多个新抗原表达载体的大小各为至少1kb。在一些方面,一个或多个新抗原表达载体的大小各为2kb。在一些方面,一个或多个新抗原表达载体的大小各小于5kb。
在一些方面,至少一个新抗原编码核酸序列中的至少一者编码由MHC I类在肿瘤细胞上递呈的多肽序列或其部分。在一些方面,每个抗原编码核酸序列彼此直接连接。在一些方面,至少一个抗原编码核酸序列中的至少一者连接于具有编码接头的核酸序列的不同抗原编码核酸序列。在一些方面,接头连接两个MHC I类序列或将MHC I类序列连接于MHCII类序列。在一些方面,接头选自:(1)连续甘氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(2)连续丙氨酸残基,长度为至少2、3、4、5、6、7、8、9或10个残基;(3)两个精氨酸残基(RR);(4)丙氨酸、丙氨酸、酪氨酸(AAY);(5)长度为至少2、3、4、5、6、7、8、9或10个氨基酸残基的共有序列,其由哺乳动物蛋白酶体有效加工;和(6)侧接源自同源蛋白质的抗原并且长度为至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或2-20个氨基酸残基的一个或多个原生序列。在一些方面,接头连接两个MHC II类序列或将MHC II类序列连接于MHC I类序列。在一些方面,接头包含序列GPGPG。
在一些方面,至少一个抗原编码核酸序列中的至少一个序列可操作地或直接地连接到单独或连续的序列,所述单独或连续的序列增强至少一个抗原编码核酸序列的表达、稳定性、细胞运输、加工和递呈,和/或免疫原性。在一些方面,单独或连续的序列包含以下中的至少一者:泛素序列、经修饰以增加蛋白酶体靶向的泛素序列(例如,泛素序列在第76位含有Gly至Ala取代)、免疫球蛋白信号序列(例如IgK)、主要组织相容性I类序列、溶酶体相关膜蛋白(LAMP)-1、人树突状细胞溶酶体相关膜蛋白和主要组织相容性II类序列;任选地其中经过修饰以增加蛋白酶体靶向的泛素序列是A76。
在一些方面,至少一个新抗原编码核酸序列中的至少一者编码相对于翻译的相应野生型核酸序列对其相应MHC等位基因的结合亲和力增加的多肽序列或其部分。在一些方面,至少一个新抗原编码核酸序列中的至少一者编码相对于翻译的相应野生型核酸序列对其相应MHC等位基因的结合稳定性增加的多肽序列或其部分。在一些方面,至少一个新抗原编码核酸序列中的至少一者编码相对于翻译的相应野生型核酸序列对其相应MHC等位基因上递呈的可能性增加的多肽序列或其部分。
在一些方面,至少一个突变包括点突变、移码突变、非移码突变、缺失突变、插入突变、剪接变体、基因组重排或蛋白酶体产生的剪接抗原。
在一些方面,肿瘤选自:肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、膀胱癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、成人急性淋巴母细胞性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
在一些方面,至少一个新抗原编码核酸序列包含至少2-10、2、3、4、5、6、7、8、9或10个核酸序列。在一些方面,至少一个新抗原编码核酸序列包含至少11-20、15-20、11-100、11-200、11-300、11-400、11、12、13、14、15、16、17、18、19、20或多至400个核酸序列。
在一些方面,至少一个新抗原编码核酸序列包含至少2-400个核酸序列并且其中所述新抗原编码核酸序列中的至少两者编码肿瘤细胞表面上由MHC I类递呈的多肽序列或其部分。在一些方面,新抗原编码核酸序列中的至少两者编码肿瘤细胞表面上由MHC I类递呈的多肽序列或其部分。在一些方面,当施用于受试者并被翻译时,由至少一个新抗原编码核酸序列编码的至少一个新抗原被递呈在抗原递呈细胞上,产生靶向肿瘤细胞表面上至少一个新抗原的免疫应答。在一些方面,至少一个新抗原编码核酸序列当施用于受试者并被翻译时,在抗原递呈细胞上递呈MHC I类或II类新抗原中的至少一者,产生靶向肿瘤细胞表面上至少一个新抗原的免疫应答,并且任选地其中至少一个新抗原编码核酸序列中的每一者的表达由至少一个启动子核苷酸序列驱动。
在一些方面,每个MHC I类新抗原编码核酸序列编码长度为8至35个氨基酸,任选地长度为9-17、9-25、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34或35个氨基酸的多肽序列。
在一些方面,存在至少一个MHC II类抗原编码核酸序列。在一些方面,存在至少一个MHC II类抗原编码核酸序列并且其包含至少一个MHC II类新抗原编码核酸序列,所述序列包含至少一个突变,使其不同于相应的野生型亲本核酸序列。在一些方面,至少一个MHCII类抗原编码核酸序列的长度为12-20、12、13、14、15、16、17、18、19、20或20-40个氨基酸。在一些方面,存在至少一个MHC II类抗原编码核酸序列并且其包含至少一个通用MHC II类抗原编码核酸序列,任选地其中所述至少一个通用序列包含破伤风类毒素和PADRE中的至少一者。
在一些方面,至少一个启动子核苷酸序列或第二启动子核苷酸序列是诱导型的。在一些方面,至少一个启动子核苷酸序列或第二启动子核苷酸序列是非诱导型的。
在一些方面,至少一个聚(A)序列包含主链原生的聚(A)序列。在一些方面,至少一个聚(A)序列包含主链外源的聚(A)序列。在一些方面,至少一个聚(A)序列可操作地连接于至少一个抗原编码核酸序列中的至少一者。在一些方面,至少一个聚(A)序列为至少20个、至少30个、至少40个、至少50个、至少60个、至少70个、至少80个或至少90个连续A核苷酸。在一些方面,至少一个聚(A)序列为至少100个连续A核苷酸。
在一些方面,新抗原盒还包含以下中的至少一者:内含子序列、土拨鼠肝炎病毒转录后调控元件(WPRE)序列、内部核糖体进入序列(IRES)序列、编码2A自裂解肽序列的核苷酸序列、编码弗林蛋白酶(Furin)裂解位点的核苷酸序列、或已知增强可操作地连接于至少一个抗原编码核酸序列中的至少一者的mRNA的核输出、稳定性或翻译效率的5'或3'非编码区中的序列。
在一些方面,新抗原盒还包含报告基因,其包括但不限于绿色荧光蛋白(GFP)、GFP变体、分泌型碱性磷酸酶、荧光素酶、荧光素酶变体,或可检测的肽或表位。在一些方面,可检测的肽或表位选自HA标签、Flag标签、His标签或V5标签。
在一些方面,一个或多个载体还包含编码至少一种免疫调节物的一个或多个核酸序列。在一些方面,免疫调节物为抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。在一些方面,抗体或其抗原结合片段为Fab片段、Fab'片段、单链Fv(scFv)、呈单特异性或连接在一起的多特异性的单结构域抗体(sdAb)(例如骆驼科抗体结构域)、或全长单链抗体(例如具有通过柔性接头连接的重链和轻链的全长IgG)。在一些方面,抗体的重链和轻链序列是由自裂解序列如2A或IRES分开的连续序列;或抗体的重链和轻链序列由柔性接头如连续甘氨酸残基连接。
在一些方面,免疫调节物是细胞因子。在一些方面,细胞因子是IL-2、IL-7、IL-12、IL-15或IL-21或其各自变体中的至少一者。
在一些方面,至少一个MHC I类新抗原编码核酸序列通过执行以下步骤来选择:(a)获得来自肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表新抗原集合中的每一者的肽序列的数据;(b)将每个新抗原的肽序列输入到递呈模型中,以产生新抗原中的每一者在肿瘤的肿瘤细胞表面上由MHC等位基因中的一者或多者递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据鉴定;以及(c)基于所述数值可能性集合选择所述新抗原集合的子集,以产生经选择的新抗原集合,其用于产生至少一个MHC I类新抗原编码核酸序列。
在一些方面,至少一个MHC I类新抗原编码核酸序列中的每一者通过执行以下步骤来选择:(a)获得来自肿瘤的外显子组、转录组或全基因组肿瘤核苷酸测序数据中的至少一者,其中所述肿瘤核苷酸测序数据用于获得代表新抗原集合中的每一者的肽序列的数据;(b)将每个新抗原的肽序列输入到递呈模型中,以产生新抗原中的每一者在肿瘤的肿瘤细胞表面上由MHC等位基因中的一者或多者递呈的数值可能性集合,所述数值可能性集合已至少基于所接收的质谱数据进行鉴定;以及(c)基于所述数值可能性集合选择所述新抗原集合的子集,以产生经选择的新抗原集合,其用于产生至少一个MHC I类新抗原编码核酸序列。
在一些方面,经选择的新抗原集合的数量为2-20个。
在一些方面,递呈模型表示以下两者之间的依赖性:MHC等位基因中的一对特定等位基因和在肽序列特定位置处特定氨基酸的存在;与在肿瘤细胞表面上由该对MHC等位基因中的特定等位基因递呈在该特定位置处包含特定氨基酸的此类肽序列的可能性。
在一些方面,选择经选择的新抗原集合包括基于递呈模型选择相对于未经选择的新抗原在肿瘤细胞表面上递呈的可能性增加的新抗原。在一些方案,经选择的抗原已被验证为由一种或多种特异性HLA等位基因递呈。在一些方面,选择经选择的新抗原集合包括基于递呈模型选择相对于未经选择的新抗原能够在受试者中诱导肿瘤特异性免疫应答的可能性增加的新抗原。在一些方面,选择经选择的新抗原集合包括基于递呈模型选择相对于未经选择的新抗原能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加的新抗原,任选地其中所述APC是树突状细胞(DC)。在一些方面,选择经选择的新抗原集合包括基于递呈模型选择相对于未经选择的新抗原经由中心或外周耐受性受抑制的可能性降低的新抗原。在一些方面,选择经选择的新抗原集合包括基于递呈模型选择相对于未经选择的新抗原能够在受试者中诱导针对正常组织的自身免疫应答的可能性降低的新抗原。在一些方面,外显子组或转录组核苷酸测序数据通过对肿瘤组织进行测序而获得。在一些方面,测序是下一代测序(NGS)或任何大规模平行测序方法。
在一些方面,新抗原盒包含由新抗原盒中的相邻序列形成的连接表位序列。在一些方面,至少一个或每个连接表位序列对MHC的亲和力大于500nM。在一些方面,每个连接表位序列是非自身的。在一些方面,MHC I类表位中的每一个被预测或证实能够由存在于至少5%的群体中的至少一个HLA等位基因递呈。在一些方面,MHC I类表位中的每一个被预测或证实能够由至少一个HLA等位基因递呈,其中每个抗原/HLA对在群体中具有至少0.01%的抗原/HLA流行度。在一些方面,MHC I类表位中的每一个被预测或证实能够由至少一个HLA等位基因递呈,其中每个抗原/HLA对在群体中具有至少0.1%的抗原/HLA流行度。在一些方面,新抗原盒不编码包含经翻译的野生型核酸序列的非治疗性MHC I类或II类表位核酸序列,其中所述非治疗性表位经预测显示于受试者的MHC等位基因上。在一些方面,经预测的非治疗性MHC I类或II类表位序列是由新抗原盒中的相邻序列形成的连接表位序列。在一些方面,预测是基于通过将非治疗性表位的序列输入到递呈模型中而产生的递呈可能性。在一些方面,新抗原盒中的至少一个抗原编码核酸序列的顺序通过包括以下的一系列步骤来确定:(a)产生对应于所述至少一个抗原编码核酸序列的不同顺序的候选新抗原盒序列集合;(b)对于每个候选新抗原盒序列,基于候选新抗原盒序列中非治疗性表位的递呈来确定递呈评分;以及(c)选择与低于预定阈值的递呈评分相关的候选盒序列作为用于新抗原疫苗的新抗原盒序列。
在一些方面,任何上述组合物还包含纳米颗粒递送媒介物。在一些方面,所述纳米颗粒递送媒介物可以是脂质纳米粒子(LNP)。在一些方面,所述LNP包含可电离的氨基脂质。在一些方面,所述可电离的氨基脂质包含MC3样(二亚油基甲基-4-二甲基氨基丁酸酯)分子。在一些方面,所述纳米颗粒递送媒介物包封新抗原表达系统。
在一些方面,任何上述组合物还包含多个LNP,其中所述LNP包括:新抗原表达系统;阳离子脂质;非阳离子脂质;和抑制LNP聚集的缀合脂质,其中所述多个LNP中至少约95%的LNP:具有非层状形态;或者是电子致密的。
在一些方面,所述非阳离子脂质是(1)磷脂和(2)胆固醇或胆固醇衍生物的混合物。
在一些方面,所述抑制LNP聚集的缀合脂质是聚乙二醇(PEG)-脂质缀合物。在一些方面,所述PEG-脂质缀合物选自:PEG-二酰基甘油(PEG-DAG)缀合物、PEG二烷氧基丙基(PEG-DAA)缀合物、PEG-磷脂缀合物、PEG-神经酰胺(PEG-Cer)缀合物及其混合物。在一些方面,所述PEG-DAA缀合物是选自以下的成员:PEG-二癸氧基丙基(C
在一些方面,新抗原表达系统完全包封在LNP中。
在一些方面,LNP的非层状形态包括反六角形(H
在一些方面,阳离子脂质占LNP中存在的总脂质的约10摩尔%至约50摩尔%。在一些方面,阳离子脂质占LNP中存在的总脂质的约20摩尔%至约50摩尔%。在一些方面,阳离子脂质占LNP中存在的总脂质的约20摩尔%至约40摩尔%。
在一些方面,非阳离子脂质占LNP中存在的总脂质的约10摩尔%至约60摩尔%。在一些方面,非阳离子脂质占LNP中存在的总脂质的约20摩尔%至约55摩尔%。在一些方面,非阳离子脂质占LNP中存在的总脂质的约25摩尔%至约50摩尔%。
在一些方面,缀合脂质占LNP中存在的总脂质的约0.5摩尔%至约20摩尔%。在一些方面,缀合脂质占LNP中存在的总脂质的约2摩尔%至约20摩尔%。在一些方面,缀合脂质占LNP中存在的总脂质的约1.5摩尔%至约18摩尔%。
在一些方面,大于95%的LNP具有非层状形态。在一些方面,大于95%的LNP是电子致密的。
在一些方面,任何上述组合物还包含多个LNP,其中所述LNP包含:阳离子脂质,其占LNP中存在的总脂质的50摩尔%至65摩尔%;抑制LNP聚集的缀合脂质,其占LNP中存在的总脂质的0.5摩尔%至2摩尔%;和非阳离子脂质,其包含:磷脂和胆固醇或其衍生物的混合物,其中磷脂占LNP中存在的总脂质的4摩尔%至10摩尔%,并且胆固醇或其衍生物占LNP中存在的总脂质的30摩尔%至40摩尔%;磷脂和胆固醇或其衍生物的混合物,其中磷脂占LNP中存在的总脂质的3摩尔%至15摩尔%,并且胆固醇或其衍生物占LNP中存在的总脂质的30摩尔%至40摩尔%;或LNP中存在的总脂质的高达49.5摩尔%,并包含磷脂和胆固醇或其衍生物的混合物,其中胆固醇或其衍生物占LNP中存在的总脂质的30摩尔%至40摩尔%。
在一些方面,任何上述组合物还包含多个LNP,其中所述LNP包含:阳离子脂质,其占LNP中存在的总脂质的50摩尔%至85摩尔%;抑制LNP聚集的缀合脂质,其占LNP中存在的总脂质的0.5摩尔%至2摩尔%;和非阳离子脂质,其占LNP中存在的总脂质的13摩尔%至49.5摩尔%。
在一些方面,所述磷脂包含二棕榈酰磷脂酰胆碱(DPPC)、二硬脂酰磷脂酰胆碱(DSPC)或其混合物。
在一些方面,所述缀合脂质包含聚乙二醇(PEG)-脂质缀合物。在一些方面,所述PEG-脂质缀合物包含PEG-二酰基甘油(PEG-DAG)缀合物、PEG-二烷氧基丙基(PEG-DAA)缀合物或其混合物。在一些方面,所述PEG-DAA缀合物包含PEG-二肉豆蔻酰氧基丙基(PEG-DMA)缀合物、PEG-二硬脂酰氧基丙基(PEG-DSA)缀合物或其混合物。在一些方面,所述缀合物的PEG部分具有约2,000道尔顿的平均分子量。
在一些方面,所述缀合脂质占LNP中存在的总脂质的1摩尔%至2摩尔%。
在一些方面,所述LNP包含具有式I结构的化合物:
或其药学上可接受的盐、互变异构体、前药或立体异构体,其中:L
在一些方面,所述LNP包含具有式II结构的化合物:
或其药学上可接受的盐、互变异构体、前药或立体异构体,其中:L
在一些方面,任何上述组合物还包含一种或多种赋形剂,所述赋形剂包含中性脂质、类固醇和聚合物缀合脂质。在一些方面,所述中性脂质包含l,2-二硬脂酰基-sn-甘油-3-磷酸胆碱(DSPC)、l,2-二棕榈酰基-sn-甘油-3-磷酸胆碱(DPPC)、l,2-二肉豆蔻酰基-sn-甘油-3-磷酸胆碱(DMPC)、1-棕榈酰基-2-油酰基-sn-甘油-3-磷酸胆碱(POPC)、1,2-二油酰基-sn-甘油-3-磷酸胆碱(DOPC)和l,2-二油酰基-sn-甘油-3-磷酸乙醇胺(DOPE)中的至少一种。在一些方面,所述中性脂质是DSPC。
在一些方面,所述化合物与中性脂质的摩尔比在约2:1至约8:1的范围内。
在一些方面,所述类固醇是胆固醇。在一些方面,所述化合物与胆固醇的摩尔比在约2:1至1:1的范围内。
在一些方面,所述聚合物缀合脂质是聚乙二醇化脂质。在一些方面,所述化合物与聚乙二醇化脂质的摩尔比在约100:1至约25:1的范围内。在一些方面,所述聚乙二醇化脂质是PEG-DAG、PEG聚乙烯(PEG-PE)、PEG-琥珀酰基-二酰基甘油(PEG-S-DAG)、PEG-cer或PEG二烷氧基丙基氨基甲酸酯。在一些方面,所述聚乙二醇化脂质具有以下结构III:
或其药学上可接受的盐、互变异构体或立体异构体,其中:R
在一些方面,LNP在与聚阴离子核酸混合时自组装成非双层结构。在一些方面,所述非双层结构的直径介于60nm和120nm之间。在一些方面,所述非双层结构的直径为约70nm、约80nm、约90nm或约100nm。在一些方面,其中纳米颗粒递送媒介物具有约100nm的直径。
本文还公开了一种药物组合物,其包含本文公开的任何组合物(例如本文公开的基于甲病毒或基于ChAd的载体)和药学上可接受的载体。在一些方面,所述药物组合物还包含佐剂。在一些方面,所述药物组合物还包含免疫调节物。在一些方面,所述免疫调节物是抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。
本文还公开了分离的核苷酸序列或分离的核苷酸序列集合,其包含任何上述组合物权利要求的新抗原盒和从SEQ ID NO:3或SEQ ID NO:5的序列获得的一种或多种元件,任选地其中所述一种或多种元件选自非结构蛋白介导扩增所必需的序列,26S启动子核苷酸序列,聚(A)序列和以SEQ ID NO:3或SEQ ID NO:5所示序列的nsP1-4基因,并且任选地其中所述核苷酸序列是cDNA。在一些方面,所述序列或分离的核苷酸序列集合包含插入在以SEQID NO:6或SEQ ID NO:7所示序列的第7544位的本文公开的新抗原盒。在一些方面,分离的核苷酸序列还包含从SEQ ID NO:3或SEQ ID NO:5的序列获得的一种或多种元件5'的T7或SP6 RNA聚合酶启动子核苷酸序列,和任选地聚(A)序列3'的一个或多个限制性位点。在一些方面,本文公开的新抗原盒插入在SEQ ID NO:8或SEQ ID NO:9的第7563位。在另一方面,以SEQ ID NO:8或SEQ ID NO:9所示的序列还包含在第17位插入的另一个腺嘌呤核苷酸。
本文还公开了一种分离的核苷酸序列,其包含本文公开的新抗原盒和本文公开的至少一种启动子。在一些方面,分离的核苷酸序列还包含基于ChAd的基因。在一些方面,基于ChAd的基因从SEQ ID NO:1的序列获得,任选地其中所述基因选自以SEQ ID NO:1所示序列的黑猩猩腺病毒ITR、E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因,并且任选地其中核苷酸序列是cDNA。
本文还公开了一种分离的细胞,其包含本文公开的分离的核苷酸序列,任选地其中所述细胞是BHK-21、CHO、HEK293或其变体、911、HeLa、A549、LP-293、PER.C6或AE1-2a细胞。
本文还公开了一种载体,其包含本文公开的分离的核苷酸序列。
本文还公开了一种试剂盒,其包含本文公开的载体或组合物和使用说明书。
本文还公开了一种用于治疗患有癌症的受试者的方法,所述方法包括向所述受试者施用本文公开的载体或本文公开的药物组合物。在一些方面,至少一个MHC I类新抗原编码核酸序列来源于患有癌症的受试者的肿瘤。在一些方面,至少一个MHC I类新抗原编码核酸序列不是源自患有癌症的受试者的肿瘤。
本文还公开了一种用于在受试者中诱导免疫应答的方法,所述方法包括向所述受试者施用本文所述的任何组合物、载体或药物组合物。在一些方面,受试者表达至少一个预测或已知递呈MHC I类表位的HLA等位基因。在一些方面,受试者表达至少一个预测或已知递呈所述MHC I类表位的HLA等位基因,并且其中所述MHC I类表位包含选自参考表34中突变的突变。在一些方面,受试者表达至少一个预测或已知递呈所述MHC I类表位的HLA等位基因,并且其中所述MHC I类表位包含选自参考表32中突变的突变。
在一些方面,所述载体或组合物经肌肉内(IM)、皮内(ID)、或皮下(SC)、或静脉内(IV)施用。
在一些方面,本文所述的方法还包括施用一种或多种免疫调节物,任选地其中所述免疫调节物在施用组合物或药物组合物之前、同时或之后施用。在一些方面,所述一种或多种免疫调节物选自:抗CTLA4抗体或其抗原结合片段、抗PD-1抗体或其抗原结合片段、抗PD-L1抗体或其抗原结合片段、抗4-1BB抗体或其抗原结合片段、或抗OX-40抗体或其抗原结合片段。在一些方面,所述免疫调节物经静脉内(IV)、肌肉内(IM)、皮内(ID)或皮下(SC)施用。在一些方面,皮下施用是在组合物或药物组合物施用部位附近或者靠近一个或多个载体或组合物引流淋巴结。
在一些方面,本文所述的方法还包括向受试者施用第二疫苗组合物。在一些方面,所述第二疫苗组合物在施用上述组合物或药物组合物之前施用。在一些方面,所述第二疫苗组合物在施用上述组合物或药物组合物之后施用。在一些方面,所述第二疫苗组合物与上述组合物或药物组合物相同。在一些方面,所述第二疫苗组合物不同于上述组合物或药物组合物。在一些方面,所述第二疫苗组合物包含编码至少一个抗原编码核酸序列的黑猩猩腺病毒载体。在一些方面,由黑猩猩腺病毒载体编码的至少一个抗原编码核酸序列与任何上述组合物或载体的至少一个抗原编码核酸序列相同。
本文还公开了一种制造任何上述组合物的一个或多个载体的方法,所述方法包括:获得包含主链和新抗原盒的线性化DNA序列;通过将所述线性化DNA序列添加到体外转录反应中来体外转录线性化DNA序列,所述体外转录反应包含将线性化DNA序列转录成RNA的所有必需组分,任选地还包括将m7g帽体外添加到所得RNA中;以及从体外转录反应中分离出所述一个或多个载体。在一些方面,线性化DNA序列通过线性化DNA质粒序列或通过使用PCR扩增来产生。在一些方面,使用细菌重组或全基因组DNA合成或在细菌细胞中使用合成DNA扩增的全基因组DNA合成中的一种产生DNA质粒序列。在一些方面,从体外转录反应中分离一个或多个载体涉及苯酚氯仿提取、基于二氧化硅柱的纯化或类似RNA纯化方法中的一种或多种。
本文还公开了一种制造本文公开的任何组合物的方法,所述方法包括:提供纳米颗粒递送媒介物的组分;提供新抗原表达系统;以及提供足以使所述纳米颗粒递送媒介物和所述新抗原表达系统产生用于递送新抗原表达系统的组合物的条件。在一些方面,所述条件通过微流体混合来提供。
本文还公开了一种制造本文公开的腺病毒载体的方法,所述方法包括:获得包含至少一个启动子序列和新抗原盒的质粒序列;将所述质粒序列转染到一个或多个宿主细胞中;以及从所述一个或多个宿主细胞中分离腺病毒载体。
在一些方面,分离包括:裂解宿主细胞以获得包含腺病毒载体的细胞裂解物;以及从所述细胞裂解物中纯化腺病毒载体。
在一些方面,使用细菌重组或全基因组DNA合成或在细菌细胞中使用合成DNA扩增的全基因组DNA合成中的一种产生质粒序列。在一些方面,一个或多个宿主细胞是CHO、HEK293或其变体、911、HeLa、A549、LP-293、PER.C6和AE1-2a细胞中的至少一者。在一些方面,从细胞裂解物中纯化腺病毒载体涉及色谱分离、离心、病毒沉淀和过滤中的一种或多种。
附图说明
参照以下描述和附图将更好地理解本发明的这些和其它特征、方面和优势,在附图中:
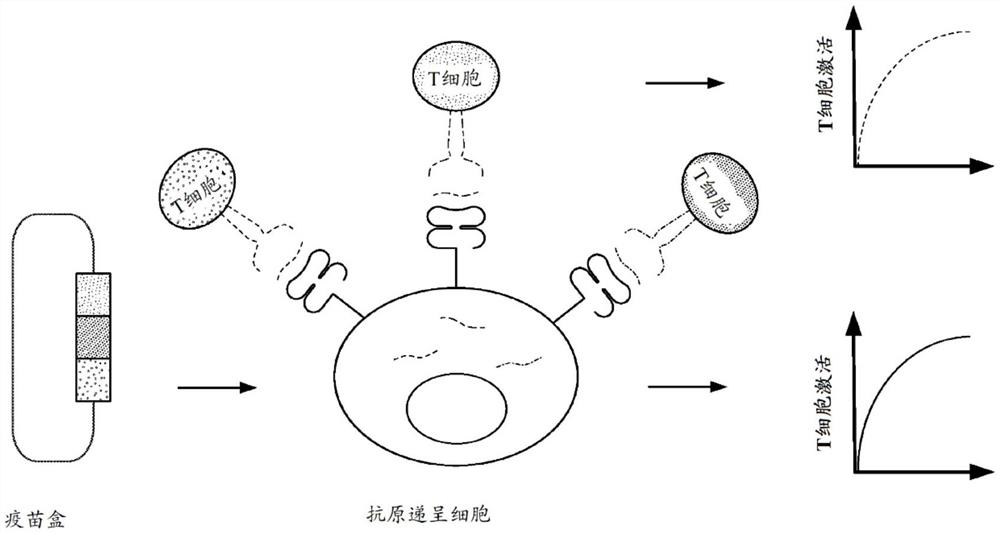
图1示出体外T细胞活化分析的开发。示意性展示该分析,其中将疫苗盒递送到抗原递呈细胞引起独特肽抗原的表达、加工和MHC限制性递呈。经工程改造成具有匹配特定肽-MHC组合的T细胞受体的报告T细胞经活化,引起荧光素酶表达。
图2A示出对短盒中接头序列的评估并且显示在相对于彼此相同的位置中串接的五个I类MHC限制性表位(表位1至5),接着是两个通用II类MHC表位(MHC-II)。使用不同接头产生各种迭代。在一些情况下,T细胞表位彼此直接连接。在其它情况下,T细胞表位侧接于其天然序列的一侧或两侧上。在其它迭代中,T细胞表位由非天然序列AAY、RR及DPP连接。
图2B示出对短盒中接头序列的评估并且显示关于嵌入所述短盒中的T细胞表位的序列信息。
图3示出对添加到模型疫苗盒中的细胞靶向序列的评估。所述靶向盒用泛素(Ub)、信号肽(SP)和/或跨膜(TM)结构域延伸该短盒设计,特征在于紧邻五个标志物人T细胞表位(表位1至5)以及两个小鼠T细胞表位SIINFEKL(SII)和SPSYAYHQF(A5),并且使用非天然接头AAY-或天然接头侧接两侧上的T细胞表位(25聚体)。
图4示出对短盒中接头序列的体内评估。A)使用HLA-A2转基因小鼠进行疫苗盒的体内评估的实验设计。
图5A示出对21聚体长盒中表位位置的影响的体内评估并且显示长盒的设计需要用25聚体天然序列中所包含的另外的熟知T细胞I类表位(表位6至21)隔开的包含在25聚体天然序列中的五个标志物I类表位(表位1至5)(接头=天然侧接序列),以及两个通用II类表位(MHC-II0,其中仅I类表位的相对位置改变。
图5B示出对21聚体长盒中表位位置的影响的体内评估并且显示关于所用T细胞表位的序列信息。
图6A示出临床前IND授权研究(IND-enabling study)的最终盒设计并且显示最终盒的设计包含在25聚体天然序列中所包含的20个MHC I表位(接头=天然侧接序列)以及2个通用MHC II类表位,所述20个MHC I表位由6个非人灵长类动物(NHP)表位、5个人表位、9个鼠类表位构成。
图6B示出临床前IND授权研究的最终盒设计并且显示递呈在非人灵长类动物、小鼠和人来源的I类MHC上的所用T细胞表位的序列信息,以及2个通用MHC II类表位PADRE和破伤风类毒素的序列。
图7A示出在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制,并且使用光学显微镜(40×放大倍率)观测到ChAdV68.4WTnt.GFP病毒斑块。
图7B示出在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制,并且使用荧光显微镜在40×放大倍率下观测到ChAdV68.4WTnt.GFP病毒斑块。
图7C示出在转染之后产生ChAdV68.4WTnt.GFP病毒。使用磷酸钙方案,用ChAdV68.4WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制,并且使用荧光显微镜在100×放大倍率下观测到ChAdV68.4WTnt.GFP病毒斑块。
图8A示出转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺(lipofectamine)方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用光学显微镜(40×放大倍率)观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图8B示出转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用荧光显微镜在40×放大倍率下观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图8C示出转染之后产生ChAdV68.5WTnt.GFP病毒。使用脂染胺方案,用ChAdV68.5WTnt.GFP DNA转染HEK293A细胞。在转染之后10天观察到病毒复制(斑块)。制备溶解产物并用于再感染T25烧瓶中的293A细胞。3天后,使用荧光显微镜在100×放大倍率下观测到ChAdV68.5WTnt.GFP病毒斑块并拍照。
图9示出病毒粒子制造方案。
图10示出甲病毒源性VEE自我复制型RNA(srRNA)载体。
图11示出在用VEE-荧光素酶srRNA接种C57BL/6J小鼠之后的体内报告基因表达。显示出在各种时间点用VEE-荧光素酶srRNA免疫接种C57BL/6J小鼠(每只小鼠10ug,两侧肌肉内注射,MC3包封)之后的代表性荧光素酶信号图像。
图12A示出在带有B16-OVA肿瘤的小鼠中免疫接种用MC3 LNP配制的VEE srRNA之后14天测量的T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射10ug VEE-荧光素酶srRNA(对照)、VEE-UbAAY srRNA(Vax)、VEE-荧光素酶srRNA和抗CTLA-4(aCTLA-4)或VEE-UbAAY srRNA以及抗CTLA-4(Vax+aCTLA-4)。此外,在第7天开始,用抗PD1 mAb治疗所有小鼠。每组由8只小鼠组成。在免疫接种之后14天,处死小鼠并收集脾和淋巴结。通过IFN-γELISPOT评定SIINFEKL特异性T细胞反应并且以每106个脾细胞的斑点形成细胞(SFC)数报道。线表示中值。
图12B示出在带有B16-OVA肿瘤的小鼠中免疫接种用MC3 LNP配制的VEE srRNA之后14天测量的T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射10ug VEE-荧光素酶srRNA(对照)、VEE-UbAAY srRNA(Vax)、VEE-荧光素酶srRNA和抗CTLA-4(aCTLA-4)或VEE-UbAAY srRNA以及抗CTLA-4(Vax+aCTLA-4)。此外,在第7天开始,用抗PD1 mAb治疗所有小鼠。每组由8只小鼠组成。在免疫接种之后14天,处死小鼠并收集脾和淋巴结。通过MHCI-五聚体染色评定SIINFEKL特异性T细胞反应,以五聚体阳性细胞占CD8阳性细胞的百分比报道。线表示中值。
图13A示出在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过IFN-γELISPOT测量T细胞反应。在用腺病毒免疫接种后14天,处死小鼠并收集脾和淋巴结。
图13B示出在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过IFN-γELISPOT测量T细胞反应。在用腺病毒免疫接种后14天以及在用srRNA加强免疫后14天(初免之后第28天),处死小鼠并收集脾和淋巴结。
图13C示出在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过MHC I类五聚体染色测量T细胞反应。在用腺病毒免疫接种后14天,处死小鼠并收集脾和淋巴结。
图13D示出在带有B16-OVA肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。向带有B16-OVA肿瘤的C57BL/6J小鼠注射表达GFP的腺病毒(Ad5-GFP)并用经MC3LNP配制的VEE-荧光素酶srRNA(对照)加强免疫或者注射Ad5-UbAAY并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。第三组用Ad5-GFP初免/VEE-荧光素酶srRNA加强免疫与抗CTLA-4的组合(aCTLA-4)治疗,而第四组用Ad5-UbAAY初免/VEE-UbAAY加强免疫与抗CTLA-4的组合(Vax+aCTLA-4)治疗。此外,在第21天开始,用抗PD-1mAb治疗所有小鼠。通过MHC I类五聚体染色测量T细胞反应。在用腺病毒免疫接种后14天以及在用srRNA加强免疫后14天(初免之后第28天),处死小鼠并收集脾和淋巴结。
图14A示出在带有CT26(Balb/c)肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。对小鼠免疫接种Ad5-GFP并且在腺病毒初免之后15天,用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫,或者用Ad5-UbAAY进行初免并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。向另一个组施用Ad5-GFP/VEE-荧光素酶srRNA初免/加强免疫与抗PD-1的组合(aPD1),而第四组接受Ad5-UbAAY/VEE-UbAAY srRNA初免/加强免疫与抗PD-1mAb的组合(Vax+aPD1)。使用IFN-γELISPOT测量T细胞对AH1肽的反应。在用腺病毒免疫接种后12天,处死小鼠并收集脾和淋巴结。
图14B示出在带有CT26(Balb/c)肿瘤的小鼠中进行异源初免/加强免疫之后的抗原特异性T细胞反应。对小鼠免疫接种Ad5-GFP并且在腺病毒初免之后15天,用经MC3 LNP配制的VEE-荧光素酶srRNA(对照)加强免疫,或者用Ad5-UbAAY进行初免并用VEE-UbAAYsrRNA(Vax)加强免疫。还用IgG对照mAb治疗对照组和Vax组。向另一个组施用Ad5-GFP/VEE-荧光素酶srRNA初免/加强免疫与抗PD-1的组合(aPD1),而第四组接受Ad5-UbAAY/VEE-UbAAY srRNA初免/加强免疫与抗PD-1mAb的组合(Vax+aPD1)。使用IFN-γELISPOT测量T细胞对AH1肽的反应。在用腺病毒免疫接种后12天以及在用srRNA加强免疫后6天(初免之后第21天),处死小鼠并收集脾和淋巴结。
图15示出ChAdV68引起针对小鼠中小鼠肿瘤抗原的T细胞反应。对小鼠免疫接种ChAdV68.5WTnt.MAG25mer,并且在C57BL/6J雌性小鼠中测量针对MHC I类表位SIINFEKL(OVA)的T细胞反应并在Balb/c小鼠中测量针对MHC I类表位AH1-A5的T细胞反应。递呈在ELISpot分析中测量的每10
图16示出在CT26肿瘤模型中单次免疫接种ChAdV6、ChAdV+抗PD-1、srRNA、srRNA+抗PD-1或单独抗PD-1之后的细胞免疫应答。使用ELISpot测量来自每组的6只小鼠的脾细胞中抗原特异性IFN-γ的产生。结果呈现为每10
图17示出在CT26肿瘤模型中单次免疫接种ChAdV6、ChAdV+抗PD-1、srRNA、srRNA+抗PD-1或单独抗PD-1之后的CD8 T细胞反应。使用ICS测量CD8 T细胞中抗原特异性IFN-γ的产生并且结果呈现为抗原特异性CD8 T细胞占总CD8 T细胞的百分比。每个组的中值以水平线指示。P值使用Dunnett多重比较检验确定;***P<0.0001,**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图18示出在CT26肿瘤模型中用ChAdV/srRNA异源初免/加强免疫、srRNA/ChAdV异源初免/加强免疫或srRNA/srRNA同源初免/加强免疫进行免疫接种之后的肿瘤生长情况。还示出与在初免和加强免疫期间施用或不施用抗PD1的初免/加强免疫的比较。每周两次测量肿瘤体积并且呈现研究的前21天的平均肿瘤体积。研究起始时每组22-28只小鼠。误差条表示平均值的标准误差(SEM)。P值使用Dunnett检验确定;***P<0.0001,**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mer srRNA。
图19示出在CT26肿瘤模型中用ChAdV/srRNA异源初免/加强免疫、srRNA/ChAdV异源初免/加强免疫或srRNA/srRNA同源初免/加强免疫进行免疫接种之后的存活情况。还示出与在初免和加强免疫期间施用或不施用抗PD1的初免/加强免疫的比较。P值使用对数秩检验确定;***P<0.0001,**P<0.001,*P<0.01。ChAdV=ChAdV68.5WTnt.MAG25mer;srRNA=VEE-MAG25mersrRNA。
图20示出使用ELISpot测量的抗原特异性细胞免疫应答。在第一次加强免疫之后1、2、3、4、5、6、8、9或10周,使用ELISpot测量VEE-MAG25mer srRNA-LNP1(30μg)(图20A)、VEE-MAG25mer srRNA-LNP1(100μg)(图20B)、或VEE-MAG25mer srRNA-LNP2(100μg)(图20C)同源初免/加强免疫或ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫组(图20D)的PBMC中针对六个不同mamu A01限制性表位的抗原特异性IFN-γ产生情况(每组6只恒河猴)。结果以堆叠条形图格式对于各表位以每10
图21显示使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周,使用ELISpot测量在用ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamu A01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每10
图22显示使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后4、5、6、7、8、10、11、12、13、14或15周,使用ELISpot测量在用VEE-MAG25mer srRNA LNP2同源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamuA01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每10
图23显示使用ELISpot测量的抗原特异性细胞免疫应答。在免疫接种之前以及在初始免疫接种之后4、5、6、7、8、10、11、12、13、14或15周,使用ELISpot测量在用VEE-MAG25mer srRNA LNP1同源初免/加强免疫方案免疫接种后的PBMC中针对六个不同mamuA01限制性表位的抗原特异性IFN-γ产生情况。结果以堆叠条形图格式对于每个表位以每10
图24A和图24B显示从Promega的动态范围标准生成的示例肽谱。
图25显示EDGE得分与通过靶向MS检测到候选共有新抗原肽的可能性之间的相关性。
图26显示用突变的肽HLA-A*11:01四聚体染色的来自患者的扩增的TIL。显示了CD8+细胞的流式细胞术门控策略(左图)和KRAS-G12V/HLA-A*11:01四聚体对CD8+细胞的染色(右图)。
图27示出一般TCR测序策略和工作流程。
图28显示使用KRAS-G12V/HLA-A*11:01四聚体的代表性实例的TCR测序策略。
图29示出具有30个(L)、40个(XL)或50个(XXL)表位的大抗原盒的来自多种物种的模型表位的一般组织。
图30显示ChAd载体表达长的盒,如通过使用识别所有盒共有的序列的抗II类(PADRE)抗体的如上述蛋白质印迹指示的。用表达可变大小的大盒(chAd68-50XXL、chAd68-40XL和chAd68-30L)的chAd68载体感染HEK293细胞。感染设定为MOI为0.2。感染后二十四小时,将蛋白酶体抑制剂MG132添加到一组感染孔中(用加号表示)。另一组用病毒处理的孔未用MG132处理(以减号表示)。未感染的HEK293细胞(293F)用作阴性对照。感染后四十八小时,收集细胞沉淀并通过SDS/PAGE电泳和使用兔抗II类PADRE抗体的免疫印迹进行分析。使用HRP抗兔抗体和ECL化学发光底物进行检测。
图31显示在chAd68大盒免疫的小鼠中,通过ICS针对AH1(上)和SIINFEKL(下)检测到的CD8+免疫应答。数据表示为针对模型表位的IFNg+细胞占总CD8细胞的百分比。
图32显示chAd68大盒疫苗接种后对LD-AH1+(上)和Kb-SIINFEKL+(下)四聚体的CD8+应答。数据表示为对模型四聚体肽复合物有反应性的总CD8细胞的百分比。*p<0.05,**p<0.01,通过Tukey检验的ANOVA。所有p值与MAG 20抗原盒比较。
图33显示在甲病毒大盒处理的小鼠中,通过ICS检测到的针对AH1(上)和SIINFEKL(下)的CD8+免疫应答。数据表示为针对模型表位的IFNg+细胞占总CD8细胞的百分比。*p<0.05,**p<0.01,***p<0.001,通过Tukey检验的ANOVA。所有p值与MAG 20抗原盒比较。
图34示出用于评估恒河猴中含抗原盒的载体的免疫原性的疫苗接种策略。三角形表示在第0和32周时chAd68疫苗接种(1e12 vp/动物)。圆圈表示在第0、4、12、20、28和32周时甲病毒疫苗接种。正方形表示施用抗CTLA4抗体。
图35显示仅给予chAd-MAG(第4组)的恒河猴中CD8+抗表位应答的时程。显示了平均SFC/1e6脾细胞。
图36显示给予IV递送的chAd-MAG加抗CTLA4抗体(伊匹单抗)的恒河猴中CD8+抗表位应答的时程(第5组)。显示了平均SFC/1e6脾细胞。
图37显示给予SC递送的chAd-MAG加抗CTLA4抗体(伊匹单抗)的恒河猴中CD8+抗表位应答的时程(第6组)。显示了平均SFC/1e6脾细胞。
图38显示通过ELISpot测量的通过ChAdV68/samRNA疫苗方案产生的抗原特异性记忆应答。结果以单独的点图表示,每个点代表一只动物。显示了免疫前的基线(左图)和初免后18个月的记忆应答(右图)。
图39显示使用组合的四聚体染色和CD45RA/CCR7共染色的流式细胞术对抗原特异性CD8+T细胞的记忆细胞表型分析。
图40显示在研究第18个月时,四个Mamu-A*01四聚体+CD8+T细胞群体的总和中的记忆细胞类型的分布。记忆细胞的表征如下:CD45RA+CCR7+=初始,CD45RA+CCR7-=效应子(Teff),CD45RA-CCR7+=中央记忆(Tcm),CD45RA-CCR7-=效应记忆(Tem)。
图41显示在具有CT26肿瘤的小鼠中识别CT26肿瘤抗原AH1的CD8+T细胞的频率。使用单因素ANOVA和Tukey多重比较检验确定P值。**P<0.001,*P<0.05。ChAdV=ChAdV68.5WTnt.MAG25mer;aCTLA4=抗CTLA4抗体,克隆9D9。
具体实施方式
I.定义
一般说来,权利要求书和说明书中使用的术语意图解释为具有与本领域普通技术人员所理解的普通含义。为清楚起见,以下定义某些术语。如果普通含义与所提供的定义之间存在矛盾,应使用所提供的定义。
如本文所用,术语“抗原”是诱导免疫应答的物质。抗原可以是新抗原。抗原可以是“共有抗原”,其为特定群体(例如特定癌症患者群体)中发现的抗原。
如本文所用,术语“新抗原”是具有至少一个使其不同于相应野生型抗原的改变的抗原,例如经由肿瘤细胞中的突变或特异性针对肿瘤细胞的翻译后修饰。新抗原可以包括多肽序列或核苷酸序列。突变可以包括移码或非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合,或产生neoORF的任何基因组或表达改变。突变还可以包括剪接变体。特异性针对肿瘤细胞的翻译后修饰可以包括异常磷酸化。特异性针对肿瘤细胞的翻译后修饰还可以包括蛋白酶体产生的剪接抗原。参见Liepe等人,A large fraction ofHLA class I ligands are proteasome-generated spliced peptides;Science.2016年10月21日;354(6310):354-358。表A和AACR GENIE结果(SEQ ID NO:10,755-29,357)显示了示例性共有新抗原;还显示了每种抗原的相应HLA等位基因。这样的共有新抗原可用于通过施用在受试者中诱导免疫应答。可通过使用多种诊断方法(例如下文进一步描述的患者选择方法)来鉴定受试者用于施用。
如本文所用,术语“肿瘤抗原”是存在于受试者的肿瘤细胞或组织中但不存在于受试者的相应正常细胞或组织中或源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的多肽的抗原。
如本文所用,术语“基于抗原的疫苗”是基于一个或多个抗原(例如多个抗原)的疫苗组合物。疫苗可以是基于核苷酸的(例如,基于病毒的、基于RNA的或基于DNA的)、基于蛋白质的(例如,基于肽的)或其组合。
如本文所用,术语“候选抗原”是产生可代表抗原的序列的突变或其它畸变。
如本文所用,术语“编码区”是编码蛋白质的基因的部分。
如本文所用,术语“编码突变”是在编码区中出现的突变。
如本文所用,术语“ORF”意指开放阅读框。
如本文所用,术语“NEO-ORF”是由突变或其它畸变如剪接而产生的肿瘤特异性ORF。
如本文所用,术语“错义突变”是引起一个氨基酸被另一个氨基酸取代的突变。
如本文所用,术语“无义突变”是引起氨基酸被终止密码子取代或引起典型起始密码子移除的突变。
如本文所用,术语“移码突变”是引起蛋白质框架改变的突变。
如本文所用,术语“插入缺失”是一个或多个核酸的插入或缺失。
如本文所用,在两个或更多个核酸或多肽序列的上下文中,术语“同一性”百分比是指当出于最大对应性比较和比对时,两个或更多个序列或子序列具有指定百分比的核苷酸或氨基酸残基是相同的,如使用下文所述的序列比较算法(例如BLASTP和BLASTN,或技术人员可用的其它算法)之一测量或通过目测检查所测量。取决于应用,“同一性”百分比可以存在于所比较的序列区域上,例如在功能结构域上,或者存在于待比较的两个序列的全长上。
关于序列比较,通常一个序列充当与测试序列进行比较的参考序列。当使用序列比较算法时,将测试序列和参考序列输入计算机中,必要时指定子序列坐标,并且指定序列算法程序参数。然后,序列比较算法基于指定程序参数计算测试序列相对于参考序列的序列同一性百分比。或者,可以通过组合在所选序列位置(例如序列基序)处特定核苷酸,或对于翻译的序列来说特定氨基酸的存在或不存在来确定序列相似性或不相似性。
用于比较的最佳序列比对可以例如通过Smith和Waterman,Adv.Appl.Math.2:482(1981)的局部同源性算法;Needleman和Wunsch,J.Mol.Biol.48:443(1970)的同源性比对算法;Pearson和Lipman,Proc.Nat'l.Acad.Sci.USA 85:2444(1988)的相似性搜索方法;这些算法的计算机化实施(Wisconsin Genetics软件包中的GAP、BESTFIT、FASTA和TFASTA;Genetics Computer Group,575Science Dr.,Madison,Wis.);或通过目测检查(一般参见Ausubel等人,见下文)来进行。
适于测定序列同一性和序列相似性百分比的算法的一个实例是Altschul等人,J.Mol.Biol.215:403-410(1990)中描述的BLAST算法。执行BLAST分析的软件可通过国家生物技术信息中心(National Center for Biotechnology Information)公开获得。
如本文所用,术语“无终止或通读”是引起天然终止密码子移除的突变。
如本文所用,术语“表位”是抗原中通常由抗体或T细胞受体结合的特异性部分。
如本文所用,术语“免疫原性”是例如通过T细胞、B细胞或两者引发免疫应答的能力。
如本文所用,术语“HLA结合亲和力”、“MHC结合亲和力”意指特异性抗原与特异性MHC等位基因之间的结合亲和力。
如本文所用,术语“诱饵”是用于从样品富集DNA或RNA的特定序列的核酸探针。
如本文所用,术语“变体”是受试者的核酸与用作对照的参考人基因组之间的差异。
如本文所用,术语“变体识别(variant call)”是对通常由测序确定的变体存在的算法确定。
如本文所用,术语“多态性”是生殖系变体,即在个体的所有携带DNA的细胞中发现的变体。
如本文所用,术语“体细胞变体”是在个体的非生殖系细胞中产生的变体。
如本文所用,术语“等位基因”是基因的一种形式,或基因序列的一种形式,或蛋白质的一种形式。
如本文所用,术语“HLA型”是HLA基因等位基因的补体。
如本文所用,术语“无义介导的衰变”或“NMD”是由过早终止密码子所致的细胞对mRNA的降解。
如本文所用,术语“躯干突变”是起源于肿瘤发展早期且存在于大多数肿瘤细胞中的突变。
如本文所用,术语“亚克隆突变”是起源于肿瘤发展后期且仅存在于肿瘤细胞子集中的突变。
如本文所用,术语“外显子组”是编码蛋白质的基因组的子集。外显子组可以是基因组的集合外显子。
如本文所用,术语“逻辑回归”是来自统计的二进制数据的回归模型,其中因变量等于1的概率的逻辑被建模为因变量的线性函数。
如本文所用,术语“神经网络”是用于分类或回归的机器学习模型,其由多层线性变换,接着是通常经由随机梯度下降和反向传播训练的逐元素非线性组成。
如本文所用,术语“蛋白质组”是由细胞、细胞群或个体表达和/或翻译的全部蛋白质的集合。
如本文所用,术语“肽组”是由MHC-I或MHC-II在细胞表面上递呈的所有肽的集合。肽组可以指细胞或细胞集合(例如肿瘤肽组,意味着构成肿瘤的所有细胞的肽组的联合)的特性。
如本文所用,术语“ELISPOT”意指酶联免疫吸附斑点分析,这是一种用于监测人和动物的免疫应答的常用方法。
如本文所用,术语“葡聚糖肽多聚体”是在流式细胞测量术中用于抗原特异性T细胞染色的基于葡聚糖的肽-MHC多聚体。
如本文所用,术语“耐受性或免疫耐受性”是对一种或多种抗原(例如自身抗原)免疫无反应性的状态。
如本文所用,术语“中心耐受性”是通过缺失自身反应性T细胞克隆或通过促进自身反应性T细胞克隆分化成免疫抑制性调节性T细胞(Treg)而在胸腺中遭受的耐受性。
如本文所用,术语“外周耐受性”是通过下调或不激活经受中心耐受性的自身反应性T细胞或促进这些T细胞分化成Treg而在外周遭受的耐受性。
术语“样品”可以包括借助于包括静脉穿刺、排泄、射精、按摩、活组织检查、针抽吸、灌洗样品、刮取、手术切口或干预在内的手段,或本领域中已知的其它手段从受试者获取单个细胞或多个细胞或细胞碎片或体液等分试样。
术语“受试者”涵盖人或非人,无论体内、离体或体外,雄性或雌性的细胞、组织或生物体。术语受试者包括含人在内的哺乳动物。
术语“哺乳动物”涵盖人和非人,并且包括但不限于人、非人灵长类动物、犬科动物、猫科动物、鼠科动物、牛科动物、马科动物和猪科动物。
术语“临床因素”是指受试者状况的量度,例如疾病活动性或严重程度。“临床因素”涵盖受试者健康状况的所有标志物,包括非样品标志物,和/或受试者的其它特征,例如但不限于年龄和性别。临床因素可以是可在确定条件下评估来自受试者的样品(或样品群体)或受试者而获得的评分、值或值集合。临床因素也可以由标志物和/或其它参数(例如基因表达替代物)进行预测。临床因素可以包括肿瘤类型、肿瘤亚型和吸烟史。
术语“源于肿瘤的抗原编码核酸序列”是指例如经由RT-PCR直接从肿瘤提取的核酸序列;或通过肿瘤测序获得的序列数据,然后使用测序数据例如经由本领域中已知的各种合成或基于PCR的方法合成核酸序列。
术语“甲病毒”是指披膜病毒科(Togaviridae)的成员,并且是正义单链RNA病毒。甲病毒通常分类为旧世界,例如辛德毕斯、罗斯河、马雅鲁、基孔肯尼亚(Chikungunya)和塞姆利基森林病毒,或新世界,例如东部马脑炎、奥拉、摩根堡或委内瑞拉马脑炎及其衍生病毒株TC-83。甲病毒通常是自我复制的RNA病毒。
术语“甲病毒主链”是指允许病毒基因组自我复制的甲病毒的最小序列。最小序列可包括用于非结构蛋白质介导的扩增的保守序列、非结构蛋白质1(nsP1)基因、nsP2基因、nsP3基因、nsP4基因和聚A序列,以及用于亚基因组病毒RNA表达的序列,包括26S启动子元件。
术语“用于非结构蛋白质介导的扩增的序列”包括本领域技术人员熟知的甲病毒保守序列元件(CSE)。CSE包括(但不限于)甲病毒5'UTR、51-nt CSE、24-nt CSE或其它26S亚基因组启动子序列、19-nt CSE和甲病毒3'UTR。
术语“RNA聚合酶”包括催化由DNA模板产生RNA多核苷酸的聚合酶。RNA聚合酶包括(但不限于)源自噬菌体的聚合酶,包括T3、T7和SP6。
术语“脂质”包括疏水性和/或两亲性分子。脂质可以是阳离子、阴离子或中性的。脂质可以是合成或天然来源的,并且在一些情况下是可生物降解的。脂质可包括胆固醇、磷脂、脂质缀合物包括(但不限于)聚乙二醇(PEG)缀合物(聚乙二醇化脂质)、蜡、油、甘油酯、脂肪和脂溶性维生素。脂质也可包括二亚油基甲基-4-二甲基胺基丁酸酯(MC3)和MC3样分子。
术语“脂质纳米粒子”或“LNP”包括使用含脂质膜围绕水性内部形成的小泡样结构,也称为脂质体。脂质纳米粒子包括具有通过表面活性剂稳定的固体脂质核心的基于脂质的组合物。核心脂质可以是脂肪酸、酰基甘油、蜡和这些表面活性剂的混合物。生物膜脂质,例如磷脂、鞘磷脂、胆汁盐(牛磺胆酸钠)和固醇(胆固醇),可用作稳定剂。脂质纳米粒子可使用限定比率的不同脂质分子形成,包括(但不限于)限定比率的一种或多种阳离子脂质、阴离子脂质或中性脂质。脂质纳米粒子可将分子包封在外膜壳内,并且随后可与靶细胞接触以将包封的分子递送到宿主细胞胞溶质。脂质纳米粒子可用非脂质分子修饰或官能化,包括在其表面上。脂质纳米粒子可为单层的(单层)或多层的(多层)。脂质纳米粒子可与核酸复合。单层脂质纳米粒子可与核酸复合,其中核酸在水性内部。多层脂质纳米粒子可与核酸复合,其中核酸在水性内部,或者形成或包夹在其间。
缩写:MHC:主要组织相容性复合物;HLA:人白细胞抗原或人MHC基因座;NGS:下一代测序;PPV:阳性预测值;TSNA:肿瘤特异性新抗原;FFPE:福尔马林固定、石蜡包埋;NMD:无义介导的衰变;NSCLC:非小细胞肺癌;DC:树突状细胞。
除非上下文另外清楚地规定,否则如本说明书和所附权利要求中所用,单数形式“一个(种)”和“所述”包括多个指示物。
除非特别说明或从上下文中可以明显看出,否则如本文所用,术语“约”应理解为在本领域的正常公差范围内,例如在平均值的2个标准偏差内。约可以理解为所述值的10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%、0.05%或0.01%内。除非上下文另有明确说明,否则本文提供的所有数值均由术语约修饰。
本文中未直接定义的任何术语应理解为具有与本发明领域内所理解的通常与之相关的含义。本文论述的某些术语是为了向从业人员描述本发明各方面的组合物、装置、方法等以及其制备或使用提供额外的指导。应了解,相同的事物可以按超过一种方式表示。因此,替代性措辞和同义词可以用于本文所论述的任何一个或多个术语。无论本文中是否阐述或论述术语都无关紧要。提供了一些同义词或可取代的方法、材料等。除非明确陈述,否则对一个或数个同义词或等效物的叙述不排除其它同义词或等效物的使用。实例,包括术语实例的使用只是出于说明的目的,且并非在本文中限制本发明各方面的范围和含义。
说明书正文内引用的所有参考文献、颁布的专利和专利申请都是以引用的方式整体并入本文中用于所有目的。
II.鉴定抗原的方法
用于鉴定共有抗原(例如新抗原)的方法包括鉴定来自受试者肿瘤的抗原的方法,这些抗原可能被递呈于肿瘤或免疫细胞(包括专职抗原递呈细胞如树突状细胞)的细胞表面上,和/或可能是免疫原性的。举例来说,一种此类方法可以包括以下步骤:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中所述肿瘤核苷酸测序和/或表达数据被用于获得代表抗原集合中的每一者的肽序列的数据(例如,在新抗原的情况下,其中每个新抗原的肽序列包含使其不同于相应野生型肽序列的至少一个改变,或在没有突变的共有抗原的情况下,其中肽源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中以产生所述抗原各自被一个或多个MHC等位基因递呈于受试者肿瘤细胞的肿瘤细胞表面或肿瘤中存在的细胞上的数值可能性集合,所述数值可能性集合已被至少基于接收到的质谱数据进行鉴定;以及基于所述数值可能性集合选择所述抗原集合的子集,以产生经选择的抗原的集合。
递呈模型可以包括针对包含相应标记集合的参考数据集合(也称为训练数据集)训练的统计回归或机器学习(例如深度学习)模型,其中所述参考数据集合是从多个不同受试者中的每一者获得,其中任选地,一些受试者可以患有肿瘤,并且其中所述参考数据集合包含以下中的至少一者:代表来自肿瘤组织的外显子组核苷酸序列的数据、代表来自正常组织的外显子组核苷酸序列的数据、代表来自肿瘤组织的转录组核苷酸序列的数据、代表来自肿瘤组织的蛋白质组序列的数据和代表来自肿瘤组织的MHC肽组序列的数据,以及代表来自正常组织的MHC肽组序列的数据。参考数据可以另外包括工程改造成表达预定MHC等位基因且随后暴露于合成蛋白质的单等位基因细胞系、正常和肿瘤人细胞系,以及新鲜和冷冻原始样品的质谱数据、测序数据、RNA测序数据、表达分析数据和蛋白质组数据,以及T细胞分析(例如ELISPOT)。在某些方面,参考数据集合包括每种形式的参考数据。
递呈模型可以包含至少部分从参考数据集合得到的特征集合,并且其中所述特征集合包含等位基因依赖性特征和等位基因非依赖性特征中的至少一种。在某些方面,包括每一特征。
用于鉴定共有抗原的方法还包括通过从受试者的一个或多个肿瘤细胞中鉴定一个或多个可能递呈于肿瘤细胞表面上的抗原来产生用于构建个性化癌症疫苗的输出的方法。举例来说,一种这样的方法可以包括以下步骤:从受试者的肿瘤细胞和正常细胞获得外显子组、转录组或全基因组核苷酸测序和/或表达数据中的至少一者,其中所述核苷酸测序和/或表达数据用于获得代表通过比较来自肿瘤细胞的核苷酸测序和/或表达数据与来自正常细胞的核苷酸测序和/或表达数据而鉴定的抗原集合中的每一者的肽序列的数据(例如,在新抗原的情况下,其中每个新抗原的肽序列包含使其不同于相应野生型肽序列的至少一个改变,或在没有突变的共有抗原的情况下,其中肽源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽);将每个抗原的肽序列编码成相应的数值向量,每个数值向量包括有关构成肽序列的多个氨基酸和肽序列中的氨基酸位置集合的信息;使用计算机处理器将所述数值向量输入到深度学习递呈模型中,以生成所述抗原集合的递呈可能性集合,所述集合中的每个递呈可能性表示一个或多个II类MHC等位基因在受试者的肿瘤细胞表面上递呈相应抗原的可能性,即深度学习递呈模型;基于所述递呈可能性集合,选择所述抗原集合的子集,以生成经选择的抗原集合;以及基于所述经选择的抗原集合,生成用于构建个性化癌症疫苗的输出。
鉴定抗原(包括新抗原)的具体方法是本领域技术人员已知的,例如在国际专利申请出版物WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细描述的方法,其出于所有目的以全文引用的方式并入本文中。
本文公开了治疗患有肿瘤的受试者的方法,包括进行本文所述的任何抗原鉴定方法的步骤,并且还包括获得包含经选择的抗原集合的肿瘤疫苗,以及将所述肿瘤疫苗施用于所述受试者。
本文公开的方法还可以包括鉴定一个或多个T细胞,所述T细胞针对子集中的至少一个抗原具有抗原特异性。在一些实施方案中,鉴定包括在使一个或多个抗原特异性T细胞扩增的条件下,将一个或多个T细胞与子集中的一个或多个抗原共培养。在其它实施方案中,鉴定包括在允许T细胞和四聚体之间结合的条件下,使一个或多个T细胞与包含子集中一个或多个抗原的四聚体接触。在其它实施方案中,本文公开的方法还可以包括鉴定一个或多个已鉴定T细胞的一个或多个T细胞受体(TCR)。在某些实施方案中,鉴定一个或多个T细胞受体包括对一个或多个已鉴定T细胞的T细胞受体序列进行测序。本文公开的方法可以进一步包括对多个T细胞进行基因工程改造以表达一个或多个已鉴定T细胞受体中的至少一者;在使多个T细胞扩增的条件下培养所述多个T细胞;以及将扩增的T细胞注入受试者体内。在一些实施方案中,对多个T细胞进行基因工程改造以表达一个或多个已鉴定T细胞受体中的至少一者包括将一个或多个已鉴定T细胞的T细胞受体序列克隆到表达载体中;以及用所述表达载体转染多个T细胞中的每一者。在一些实施方案中,本文公开的方法还包括在扩增一个或多个已鉴定T细胞的条件下培养所述一个或多个已鉴定T细胞;以及将扩增的T细胞注入受试者体内。
本文还公开了一种分离T细胞,其针对子集中的至少一个经选择的抗原具有抗原特异性。
本文还公开了一种用于制造肿瘤疫苗的方法,其包括以下步骤:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中该肿瘤核苷酸测序和/或表达数据用于获得代表抗原集合中的每一者的肽序列的数据(例如,在新抗原的情况下,其中每个新抗原的肽序列包含使其不同于相应野生型肽序列的至少一个改变,或在没有突变的共有抗原的情况下,其中肽源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中,以产生抗原中的每一者在受试者肿瘤细胞的肿瘤细胞表面上由一个或多个MHC等位基因递呈的数值可能性集合,该数值可能性集合已至少基于所接收的质谱数据进行鉴定;以及基于该数值可能性集合选择该抗原集合的子集,以产生经选择的抗原集合;以及产生或已产生包含该经选择的抗原集合的肿瘤疫苗。
本文还公开了一种包括经选择的抗原集合的肿瘤疫苗,该经选择的抗原集合通过执行包括以下步骤的方法来选择:从受试者的肿瘤细胞获得外显子组、转录组或全基因组肿瘤核苷酸测序和/或表达数据中的至少一者,其中该肿瘤核苷酸测序和/或表达数据用于获得代表抗原集合中的每一者的肽序列的数据(例如,在新抗原的情况下,其中每个新抗原的肽序列包含使其不同于相应野生型肽序列的至少一个改变,或在没有突变的共有抗原的情况下,其中肽源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽);将每个抗原的肽序列输入到一个或多个递呈模型中,以产生抗原中的每一者在受试者肿瘤细胞的肿瘤细胞表面上被一个或多个MHC等位基因递呈的数值可能性集合,该数值可能性集合已至少基于所接收的质谱数据进行鉴定;以及基于该数值可能性集合选择该抗原集合的子集,以产生经选择的抗原集合;以及产生或已产生包含该经选择的抗原集合的肿瘤疫苗。
肿瘤疫苗可包括核苷酸序列、多肽序列、RNA、DNA、细胞、质粒或载体中的一者或多者。
肿瘤疫苗可包括在肿瘤细胞表面上递呈的一个或多个抗原。
肿瘤疫苗可包括在受试者中具有免疫原性的一个或多个抗原。
肿瘤疫苗可能不包括在受试者中诱导针对正常组织的自身免疫应答的一个或多个抗原。
肿瘤疫苗可包括佐剂。
肿瘤疫苗可包括赋形剂。
本文公开的方法还可包括基于递呈模型选择相对于未经选择的抗原在肿瘤细胞表面上递呈的可能性增加的抗原。
本文公开的方法还可包括基于递呈模型选择相对于未经选择的抗原能够在受试者中诱导肿瘤特异性免疫应答的可能性增加的抗原。
本文公开的方法还可包括基于递呈模型选择相对于未经选择的抗原能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加的抗原,任选地其中该APC是树突状细胞(DC)。
本文公开的方法还可包括基于递呈模型选择相对于未经选择的抗原经由中心或外周耐受性受抑制的可能性降低的抗原。
本文公开的方法还可包括基于递呈模型选择相对于未经选择的抗原能够在受试者中诱导针对正常组织的自身免疫应答的可能性降低的抗原。
外显子组或转录组核苷酸测序和/或表达数据可通过对肿瘤组织进行测序而获得。
测序可是下一代测序(NGS)或任何大规模平行测序方法。
数值可能性集合可通过至少MHC-等位基因相互作用特征来进一步鉴定,所述特征包含以下中的至少一者:经预测的MHC等位基因与抗原编码肽结合的亲和力;经预测的抗原编码肽-MHC复合物的稳定性;抗原编码肽的序列和长度;如通过质谱蛋白质组学或其它手段所评定,在来自表达特定MHC等位基因的其它个体的细胞中递呈具有类似序列的抗原编码肽的概率;所讨论的受试者中特定MHC等位基因的表达水平(例如,如通过RNA-seq或质谱法所测量);在表达特定MHC等位基因的其他不同受试者中由特定MHC等位基因递呈的总体新抗原编码肽序列独立性概率;在其他不同受试者中由同一家族分子(例如HLA-A、HLA-B、HLA-C、HLA-DQ、HLA-DR、HLA-DP)中的MHC等位基因递呈的总体新抗原编码肽序列独立性概率。
数值可能性集合通过至少MHC-等位基因非相互作用特征来进一步鉴定,所述特征包含以下中的至少一者:在其源蛋白序列内侧接新抗原编码肽的C端和N端序列;新抗原编码肽中蛋白酶裂解基序的存在,任选地根据相应蛋白酶在肿瘤细胞中的表达加权(如通过RNA-seq或质谱法所测量);如在适当细胞类型中所测量的源蛋白的周转率;源蛋白的长度,任选地考虑在肿瘤细胞中最高度表达的特异性剪接变体(“同种型”),如通过RNA-seq或蛋白质组质谱法所测量,或如DNA或RNA序列数据中所检测的生殖系或体细胞剪接突变的批注所预测;蛋白酶体、免疫蛋白酶体、胸腺蛋白酶体或其它蛋白酶在肿瘤细胞中的表达水平(其可通过RNA-seq、蛋白质组质谱法或免疫组织化学测量);新抗原编码肽的源基因的表达(例如,如通过RNA-seq或质谱法所测量);新抗原编码肽的源基因在细胞周期的各种阶段期间的典型组织特异性表达;源蛋白和/或其结构域的综合特征目录,如例如uniProt或PDBhttp://www.rcsb.org/pdb/home/home.do中可见;描述含有该肽的源蛋白的结构域特性的特征,例如:二级或三级结构(例如α螺旋对β折叠);替代性剪接;在其他不同受试者中由所讨论的新抗原编码肽的源蛋白递呈肽的概率;由于技术偏差,肽将不会由质谱法检测到或过量表示的概率;通过RNASeq(其无需含有肽的源蛋白)所测量的提供关于肿瘤细胞、基质或肿瘤浸润淋巴细胞(TIL)状态的信息的各种基因模块/通路的表达;新抗原编码肽的源基因在肿瘤细胞中的拷贝数;肽结合于TAP的概率或经测量或经预测的肽对TAP的结合亲和力;TAP在肿瘤细胞中的表达水平(其可通过RNA-seq、蛋白质组质谱法、免疫组织化学测量);存在或不存在肿瘤突变,包括(但不限于):已知癌症驱动基因(例如EGFR、KRAS、ALK、RET、ROS1、TP53、CDKN2A、CDKN2B、NTRK1、NTRK2、NTRK3)及编码抗原递呈机制中所涉及的蛋白质的基因(例如B2M、HLA-A、HLA-B、HLA-C、TAP-1、TAP-2、TAPBP、CALR、CNX、ERP57、HLA-DM、HLA-DMA、HLA-DMB、HLA-DO、HLA-DOA、HLA-DOB、HLA-DP、HLA-DPA1、HLA-DPB1、HLA-DQ、HLA-DQA1、HLA-DQA2、HLA-DQB1、HLA-DQB2、HLA-DR、HLA-DRA、HLA-DRB1、HLA-DRB3、HLA-DRB4、HLA-DRB5或编码蛋白酶体或免疫蛋白酶体组分的基因中的任一者)中的驱动突变。递呈依赖于肿瘤中经受功能丧失性突变的抗原递呈机制的组分的肽具有降低的递呈概率;存在或不存在功能性生殖系多态性,包括(但不限于):在编码抗原递呈机制中所涉及的蛋白质的基因(例如B2M、HLA-A、HLA-B、HLA-C、TAP-1、TAP-2、TAPBP、CALR、CNX、ERP57、HLA-DM、HLA-DMA、HLA-DMB、HLA-DO、HLA-DOA、HLA-DOB、HLA-DP、HLA-DPA1、HLA-DPB1、HLA-DQ、HLA-DQA1、HLA-DQA2、HLA-DQB1、HLA-DQB2、HLA-DR、HLA-DRA、HLA-DRB1、HLA-DRB3、HLA-DRB4、HLA-DRB5或编码蛋白酶体或免疫蛋白酶体组分的基因中的任一者)中;肿瘤类型(例如NSCLC、黑素瘤);临床肿瘤亚型(例如鳞状肺癌对非鳞状肺癌);吸烟史;该肽的源基因在相关肿瘤类型或临床亚型中的典型表达,任选地通过驱动突变分层。
至少一个改变可为移码或非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合、或产生neoORF的任何基因组或表达改变。
肿瘤细胞可选自:肺癌、黑素瘤、乳癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌。
本文公开的方法还可包括获得包含经选择的新抗原集合或其子集的肿瘤疫苗,任选地另外包括向受试者施用该肿瘤疫苗。
当呈多肽形式时,经选择的新抗原集合中的至少一个新抗原可包括以下中的至少一者:对于长度为8-15、8、9、10、11、12、13、14或15个氨基酸的MHC I类多肽,对于长度为6-30、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC II类多肽,与MHC的结合亲和力为IC50值小于1000nM,在亲本蛋白质序列中在该多肽内部或附近存在促进蛋白酶体裂解的序列基序,以及存在促进TAP转运的序列基序。对于MHC II类,在肽内部或附近存在促进通过细胞外或溶酶体蛋白酶(例如组织蛋白酶)裂解或HLA-DM催化的HLA结合的序列基序。
本文公开了用于鉴定可能在肿瘤细胞的肿瘤细胞表面上递呈的一个或多个新抗原的方法,其包括执行以下步骤:接收包含与从源自多个新鲜或冷冻肿瘤样品的主要组织相容性复合体(MHC)洗脱的多个分离肽相关的数据的质谱数据;通过至少鉴定肿瘤样品中存在且递呈在与各训练肽序列相关的一个或多个MHC等位基因上的训练肽序列集合来获得训练数据集;基于训练肽序列获得训练蛋白质序列集合;以及使用训练蛋白质序列和训练肽序列训练递呈模型的数值参数集合,所述递呈模型提供来自肿瘤细胞的肽序列在肿瘤细胞表面上由一个或多个MHC等位基因递呈的多个数值可能性。
递呈模型可表示以下两者之间的依赖性:MHC等位基因中的一对特定等位基因和在肽序列特定位置处特定氨基酸的存在;与在肿瘤细胞表面上由该对MHC等位基因中的特定等位基因递呈在该特定位置处包含特定氨基酸的此类肽序列的可能性。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自在肿瘤细胞表面上递呈的可能性增加而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够在受试者中诱导肿瘤特异性免疫应答的可能性增加而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够由专职抗原递呈细胞(APC)递呈于初始T细胞的可能性增加而被选择,任选地其中该APC是树突状细胞(DC)。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自经由中心或外周耐受性受抑制的可能性降低而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于一个或多个不同肿瘤新抗原各自能够在受试者中诱导针对正常组织的自身免疫应答的可能性降低而被选择。
本文公开的方法还可包括选择新抗原的子集,其中新抗原的子集是因为相对于APC各自将在肿瘤细胞中经差异性翻译后修饰的可能性降低而被选择,任选地其中该APC是树突状细胞(DC)。
除非另外指明,否则本文方法的实践将采用本领域技能范围内的蛋白质化学、生物化学、重组DNA技术和药理学的常规方法。这些技术在文献中充分解释。参见例如T.E.Creighton,Proteins:Structures and Molecular Properties(W.H.Freeman andCompany,1993);A.L.Lehninger,Biochemistry(Worth Publishers,Inc.,现行版);Sambrook等人,Molecular Cloning:A Laboratory Manual(第2版,1989);Methods InEnzymology(S.Colowick和N.Kaplan编,Academic Press,Inc.);Remington′sPharmaceutical Sciences,第18版(Easton,Pennsylvania:Mack Publishing Company,1990);Carey和Sundberg Advanced Organic Chemistry第3版.(Plenum Press)A卷和B卷(1992)。
III.鉴定新抗原中的肿瘤特异性突变
本文还公开用于鉴定某些突变(例如癌细胞中存在的变体或等位基因)的方法。具体来说,这些突变可存在于患有癌症的受试者的癌细胞的基因组、转录组、蛋白质组或外显子组中,而非受试者的正常组织中。鉴定肿瘤特异型的新抗原(包括共有新抗原)的聚体方法是本领域技术人员已知的,例如在国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细描述的,其出于所有目的以全文引用的方式并入本文中。
如果肿瘤中的基因突变引起肿瘤中特有的蛋白质的氨基酸序列变化,则认为其可用于免疫靶向肿瘤。有用的突变包括:(1)非同义突变,导致蛋白质中的氨基酸不同;(2)通读突变,其中终止密码子被修饰或缺失,导致翻译在C端具有新颖肿瘤特异性序列的较长蛋白质;(3)剪接位点突变,导致在成熟mRNA中包含内含子且因此导致特有的肿瘤特异性蛋白质序列;(4)染色体重排,在2种蛋白质的接合处产生具有肿瘤特异性序列的嵌合蛋白质(即基因融合);(5)框移突变或缺失,导致具有新颖肿瘤特异性蛋白质序列的新的开放阅读框。突变还可包括非移码插入缺失、错义或无义取代、剪接位点改变、基因组重排或基因融合、或产生neoORF的任何基因组或表达改变中的一个或多个。
由例如肿瘤细胞中的剪接位点、移码、通读或基因融合突变产生的具有突变的肽或突变多肽可通过对肿瘤与正常细胞中的DNA、RNA或蛋白质进行测序来鉴定。
突变还可包括先前鉴定的肿瘤特异性突变。已知肿瘤突变可见于癌症体细胞突变目录(COSMIC)数据库。
多种方法可用于检测个体的DNA或RNA中特定突变或等位基因的存在。本领域中的进步已提供精确、容易且便宜的大规模SNP基因分型。举例来说,已描述数种技术,包括动态等位基因特异性杂交(DASH)、微孔板阵列对角线凝胶电泳(MADGE)、焦磷酸测序、寡核苷酸特异性连接、TaqMan系统以及各种DNA“芯片”技术,例如Affymetrix SNP芯片。这些方法利用通常通过PCR扩增靶基因区。仍有其它方法,基于通过侵入性裂解产生小信号分子,接着进行质谱法或固定化挂锁探针和滚环扩增。下文汇总本领域中已知用于检测特异性突变的数种方法。
基于PCR的检测手段可包括同时多重扩增多个标志物。举例来说,选择PCR引物以产生大小不重叠且可同时分析的PCR产物是本领域中所熟知的。或者,可用经差异性标记且因此可各自经差异性检测的引物扩增不同的标志物。当然,基于杂交的检测手段允许样品中多个PCR产物的差异检测。本领域中已知其它技术以允许多个标志物的多重分析。
已开发数种方法以便于基因组DNA或细胞RNA中单核苷酸多态性的分析。举例来说,单碱基多态性可通过使用特殊化核酸外切酶抗性核苷酸来检测,如例如Mundy,C.R.(美国专利第4,656,127号)中所公开。根据该方法,允许与紧靠着多形位点3'的等位基因序列互补的引物与获自特定动物或人的靶分子杂交。如果靶分子上的多形位点含有与所存在的特定核酸外切酶抗性核苷酸衍生物互补的核苷酸,则该衍生物将并入到杂交引物的末端上。这种并入使得引物对核酸外切酶具有抗性,从而允许其检测。由于样品的核酸外切酶抗性衍生物的身份是已知的,故引物已对核酸外切酶具有抗性的发现揭露靶分子的多形位点中存在的核苷酸与反应中所用的核苷酸衍生物互补。这种方法的优势在于其不需要确定大量无关序列数据。
可使用基于溶液的方法确定多形位点的核苷酸的身份。Cohen,D.等人(法国专利2,650,840;PCT申请第WO91/02087号)。如在美国专利第4,656,127号的芒迪方法(Mundymethod)中,采用与紧靠着多形位点3'的等位基因序列互补的引物。该方法使用经标记的双脱氧核苷酸衍生物确定该位点的核苷酸的身份,如果该核苷酸与多形位点的核苷酸互补,则将并入到引物的末端上。
称为遗传位分析或GBA的替代方法由Goelet,P.等人(PCT申请第92/15712号)描述。Goelet,P.等人的方法使用经标记的终止子和与多形位点3'序列互补的引物的混合物。所并入的经标记的终止子因此通过所评估的靶分子的多形位点中存在的核苷酸确定且与其互补。与Cohen等人(法国专利2,650,840;PCT申请第WO91/02087号)的方法相比,Goelet,P.等人的方法可为非均相分析,其中引物或靶分子固定于固相。
已描述数种用于分析DNA中多形位点的引物引导的核苷酸并入程序(Komher,J.S.等人,Nucl.Acids.Res.17:7779-7784(1989);Sokolov,B.P.,Nucl.Acids Res.18:3671(1990);Syvanen,A.-C.等人,Genomics 8:684-692(1990);Kuppuswamy,M.N.等人,Proc.Natl.Acad.Sci.(U.S.A.)88:1143-1147(1991);Prezant,T.R.等人,Hum.Mutat.1:159-164(1992);Ugozzoli,L.等人,GATA 9:107-112(1992);Nyren,P.等人,Anal.Biochem.208:171-175(1993))。这些方法与GBA的不同之处在于其利用并入经标记的脱氧核苷酸来区分多形位点处的碱基。在这种格式中,由于信号与并入的脱氧核苷酸的数量成比例,故在同一核苷酸的操作中发生的多态性可产生与操作的长度成比例的信号(Syvanen,A.-C.等人,Amer.J.Hum.Genet.52:46-59(1993))。
许多方案直接从数百万个单独的DNA或RNA分子中并行获取序列信息。实时单分子合成测序技术依赖于荧光核苷酸的检测,因为其并入到与正测序的模板互补的DNA的新生链中。在一种方法中,将长度为30-50个碱基的寡核苷酸在5'端共价锚定于玻璃盖玻片上。这些锚定链执行两种功能。首先,如果模板被配置成具有与表面结合的寡核苷酸互补的捕捉尾部,则其充当靶模板链的捕捉位点。它们还充当模板引导的引物延伸的引物,形成序列阅读的基础。捕捉引物充当固定位点以便使用多个合成、检测及化学裂解染料接头来移除染料的循环进行序列测定。各循环由以下组成:添加聚合酶/经标记的核苷酸混合物,冲洗,成像和染料的裂解。在一个替代方法中,聚合酶被荧光供体分子修饰并固定在载玻片上,而各核苷酸用连接至γ-磷酸的受体荧光部分进行颜色编码。系统检测经荧光标记的聚合酶与经荧光修饰的核苷酸之间的相互作用,因为核苷酸并入到从头链中。也存在其它合成测序技术。
可使用任何适合的合成测序平台鉴定突变。如上所述,目前可用四种主要合成测序平台:来自Roche/454Life Sciences的基因组测序仪、来自Illumina/Solexa的1G分析仪、来自Applied BioSystems的SOLiD系统和来自Helicos Biosciences的Heliscope系统。合成测序平台也已由Pacific BioSciences和VisiGen Biotechnologies描述。在一些实施方案中,将要测序的多个核酸分子结合于支撑物(例如固体支撑物)。为将核酸固定于支撑物上,可在模板的3'和/或5'端添加捕捉序列/通用引发位点。核酸可通过将捕捉序列与共价连接于支撑物的互补序列杂交而结合于支撑物。捕捉序列(也称为通用捕捉序列)是与连接至支撑物的序列互补的核酸序列,其可双重充当通用引物。
作为捕捉序列的替代方案,偶联对(例如抗体/抗原、受体/配体或如例如美国专利申请第2006/0252077号中所述的抗生物素蛋白-生物素对)的一个成员可连接于各片段,而被捕捉在用该偶联对的相应第二成员涂布的表面上。
在捕捉后,可例如通过单分子检测/测序来分析序列,例如,如实施例和美国专利第7,283,337号中所述,包括模板依赖性合成测序。在合成测序中,表面结合的分子在聚合酶存在下暴露于多个经标记的三磷酸核苷酸。模板的序列通过并入到生长链的3'端的经标记的核苷酸的顺序来确定。这可实时进行或可以分步重复模式进行。对于实时分析,可将不同的光学标记并入各核苷酸并且可利用多个激光刺激并入的核苷酸。
测序还可包括其它大规模平行测序或下一代测序(NGS)技术和平台。大规模平行测序技术和平台的额外实例是Illumina HiSeq或MiSeq、Thermo PGM或Proton、Pac Bio RSII或Sequel、Qiagen的Gene Reader和Oxford Nanopore MinION。可使用其它类似的当前大规模平行测序技术,以及这些技术的后代。
可利用任何细胞类型或组织来获得用于本文所述的方法的核酸样品。举例来说,DNA或RNA样品可获自肿瘤或体液,例如通过已知技术(例如静脉穿刺)获得的血液或唾液。或者,可对干燥样品(例如头发或皮肤)执行核酸测试。另外,可从肿瘤获得样品用于测序并且可从正常组织获得另一样品用于测序,其中正常组织具有与肿瘤相同的组织类型。可从肿瘤获得样品用于测序并且可从正常组织获得另一样品用于测序,其中正常组织相对于肿瘤具有不同的组织类型。
肿瘤可包括肺癌、黑素瘤、乳腺癌、卵巢癌、前列腺癌、肾癌、胃癌、结肠癌、睾丸癌、头颈癌、胰腺癌、脑癌、B细胞淋巴瘤、急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病、非小细胞肺癌和小细胞肺癌中的一个或多个。
或者,可使用蛋白质质谱法鉴定或验证与肿瘤细胞上的MHC蛋白质结合的突变肽的存在。肽可从肿瘤细胞或从肿瘤免疫沉淀的HLA分子酸洗脱,然后使用质谱法鉴定。
IV.抗原
抗原可包括核苷酸或多肽。举例来说,抗原可为编码多肽序列的RNA序列。可用于疫苗中的抗原可因此包括核苷酸序列或多肽序列。共有新抗原示于表A(参见SEQ ID NO:10,755-21,015)和AACR GENIE结果(参见SEQ ID NO:21,016-29,357)中。共有抗原示于表1.2(参见SEQ ID NO:57-10,754)中。
本文公开了包含通过本文公开的方法鉴定的肿瘤特异性突变的分离肽、包含已知肿瘤特异性突变的肽和通过本文公开的方法鉴定的突变多肽或其片段。新抗原肽可描述于其编码序列的上下文中,其中新抗原包括编码相关多肽序列的核苷酸序列(例如DNA或RNA)。
本文还公开了这样的肽,其源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽,例如已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中异常表达的任何多肽。可例如在COSMIC数据库中发现可获得抗原肽的适合的多肽。COSMIC策划关于人癌症体细胞突变的综合信息。该肽含有肿瘤特异性突变。
由抗原核苷酸序列编码的一个或多个多肽可包含以下中的至少一者:对于长度为8-15、8、9、10、11、12、13、14或15个氨基酸的MHC I类肽,与MHC的结合亲和力为IC50值小于1000nM,在肽内部或附近存在促进蛋白酶体裂解的序列基序,以及存在促进TAP转运的序列基序。对于长度为6-30、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC II类肽,在肽内部或附近存在促进通过细胞外或溶酶体蛋白酶(例如组织蛋白酶)裂解或HLA-DM催化的HLA结合的序列基序。
一个或多个抗原可递呈在肿瘤的表面上。
一个或多个抗原在具有肿瘤的受试者中可为免疫原性的,例如能够在受试者中引发T细胞反应或B细胞反应。
在疫苗生产背景下,对于具有肿瘤的受试者,可不考虑在受试者中诱导自身免疫应答的一个或多个抗原。
至少一个抗原肽分子的大小可包含(但不限于)约5个、约6个、约7个、约8个、约9个、约10个、约11个、约12个、约13个、约14个、约15个、约16个、约17个、约18个、约19个、约20个、约21个、约22个、约23个、约24个、约25个、约26个、约27个、约28个、约29个、约30个、约31个、约32个、约33个、约34个、约35个、约36个、约37个、约38个、约39个、约40个、约41个、约42个、约43个、约44个、约45个、约46个、约47个、约48个、约49个、约50个、约60个、约70个、约80个、约90个、约100个、约110个、约120个或更多个氨基分子残基,以及其中可导出的任何范围。在具体实施方案中,抗原肽分子等于或小于50个氨基酸。
抗原肽和多肽可为:对于MHC I类,15个残基或更小的长度且通常由约8至约11个残基、特别是9或10个残基组成;对于MHC II类,6-30个残基(包括端点)。
如果需要,可以数种方式设计较长的肽。在一种情况下,当HLA等位基因上肽的递呈可能性经预测或已知时,较长的肽可由以下任一者组成:(1)个别递呈的具有朝向各相应基因产物的N端及C端延伸2-5个氨基酸的肽;(2)所递呈的肽中的一些或全部与各自的延伸序列的串接。在另一种情况下,当测序揭露肿瘤中所存在的长(>10个残基)新表位序列(例如归因于产生新颖肽序列的移码、通读或内含子包含)时,较长的肽将由以下组成:(3)新颖肿瘤特异性氨基酸的整个延伸段,因此绕过对基于计算或活体外测试选择最强HLA递呈的较短肽的需要。在两种情况下,使用较长的肽允许患者细胞进行内源性加工,并且可引起更有效的抗原递呈和诱导T细胞反应。
抗原肽和多肽可递呈在HLA蛋白质上。在一些方面,抗原肽和多肽以比野生型肽更大的亲和力递呈在HLA蛋白质上。在一些方面,抗原肽或多肽的IC50可至少小于5000nM、至少小于1000nM、至少小于500nM、至少小于250nM、至少小于200nM、至少小于150nM、至少小于100nM、至少小于50nM或更小。
在一些方面,抗原肽和多肽在施用于受试者时不诱导自身免疫应答和/或引起免疫耐受性。
还提供了包含至少两种或更多种抗原肽的组合物。在一些实施方案中,组合物含有至少两种不同的肽。至少两种不同的肽可源自相同的多肽。不同的多肽意指肽根据长度、氨基酸序列或两者而变化。所述肽源自已知或已发现含有肿瘤特异性突变的任何多肽,或肽源自已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的任何多肽,例如已知或已发现与正常细胞或组织相比在肿瘤细胞或癌组织中异常表达的任何多肽。可例如在COSMIC数据库或AACR Genomics Evidence Neoplasia InformationExchange(GENIE)数据库中发现可获得抗原肽的适合的多肽。COSMIC策划关于人癌症体细胞突变的综合信息。AACR GENIE汇总并关联临床级别的癌症基因组数据与成千上万癌症患者的临床结果。该肽含有肿瘤特异性突变。在一些方面,肿瘤特异性突变为特定癌症类型的驱动突变。
具有所需活性或特性的抗原肽和多肽可经修饰以提供某些所需属性,例如改进的药理学特征,同时增加或至少保留未修饰肽的实质上所有生物活性以结合所需MHC分子并活化适当T细胞。举例来说,抗原肽和多肽可进行各种变化,例如保守或非保守取代,其中这些变化可在其使用中提供某些优势,例如改进的MHC结合、稳定性或递呈。保守取代意指氨基酸残基用生物学和/或化学上类似的另一个氨基酸残基置换,例如一个疏水性残基置换另一个,或一个极性残基置换另一个。取代包括以下组合,例如Gly、Ala;Val、Ile、Leu、Met;Asp、Glu;Asn、Gln;Ser、Thr;Lys、Arg;以及Phe、Tyr。单氨基酸取代的效应也可使用D-氨基酸探测。这类修饰可使用熟知的肽合成程序进行,如例如Merrifield,Science 232:341-347(1986),Barany和Merrifield,The Peptides,Gross和Meienhofer编(N.Y.,AcademicPress),第1-284页(1979);以及Stewart和Young,Solid Phase Peptide Synthesis,(Rockford,Ill.,Pierce),第2版(1984)中所述。
肽和多肽用各种氨基酸模拟物或非天然氨基酸修饰可在提高肽和多肽的体内稳定性方面特别有用。稳定性可以多种方式加以分析。举例来说,肽酶和各种生物介质(例如人血浆和血清)已用于测试稳定性。参见例如Verhoef等人,Eur.J.Drug MetabPharmacokin.11:291-302(1986)。肽的半衰期可使用25%人血清(v/v)分析方便地确定。方案一般如下。汇集的人血清(AB型,非加热不活化)在使用之前通过离心去脂。血清然后用RPMI组织培养基稀释至25%并用于测试肽稳定性。在预定时间间隔下,移出少量反应溶液并添加至6%三氯乙酸或乙醇水溶液中。将混浊的反应样品冷却(4℃)15分钟,然后旋转集结沉淀的血清蛋白质。然后使用稳定性特异性色谱条件通过逆相HPLC确定肽的存在。
肽和多肽可经修饰以提供除改进的血清半衰期以外的所需属性。举例来说,肽诱导CTL活性的能力可通过与含有至少一个能够诱导T辅助细胞反应的表位的序列连接来增强。免疫原性肽/T辅助细胞缀合物可通过间隔分子连接。间隔子通常由相对较小的中性分子如氨基酸或氨基酸模拟物构成,其在生理条件下实质上不带电。间隔子通常选自例如Ala、Gly或非极性氨基酸或中性极性氨基酸的其它中性间隔子。应理解,任选地存在的间隔子无需由相同残基组成,且因此可以是杂寡聚物或均寡聚物。当存在时,间隔子将通常为至少一个或两个残基,更通常三至六个残基。或者,肽可在无间隔子的情况下连接到T辅助肽。
抗原肽可直接或经由在肽的氨基或羧基端处的间隔子连接到T辅助肽。抗原肽或T辅助肽的氨基端可经酰化。示例性T辅助肽包括破伤风类毒素830-843、流感307-319、疟疾环子孢子382-398和378-389。
蛋白质或肽可通过本领域技术人员已知的任何技术制造,包括通过标准分子生物学技术表达蛋白质、多肽或肽;从天然来源分离蛋白质或肽;或化学合成蛋白质或肽。先前已公开对应于各种基因的核苷酸和蛋白质、多肽和肽序列,并且可见于本领域一般技术人员已知的计算机化数据库中。一个此类数据库是位于美国国家卫生研究院(NationalInstitutes of Health)网站的国家生物技术信息中心的Genbank和GenPept数据库。已知基因的编码区可使用本文所公开或本领域一般技术人员应知晓的技术扩增和/或表达。或者,蛋白质、多肽和肽的各种市售制剂已为本领域技术人员所知。
在另一方面,抗原包括编码抗原肽或其部分的核酸(例如多核苷酸)。多核苷酸可为例如DNA、cDNA、PNA、CNA、RNA(例如mRNA)、单链和/或双链、或原生或稳定形式的多核苷酸,例如具有硫代磷酸主链的多核苷酸,或其组合,并且其可含有或可不含内含子。另一方面提供一种能够表达多肽或其部分的表达载体。不同细胞类型的表达载体在本领域中已熟知并且无需过度实验便可选择。一般来说,DNA以适当定向插入到表达载体如质粒中且以正确阅读框进行表达。如果需要,DNA可连接于由所需宿主识别的适当转录和翻译调节控制核苷酸序列,而此类控制件一般可用于表达载体中。载体然后经由标准技术引入至宿主中。指导可见于例如Sambrook等人(1989)Molecular Cloning,A Laboratory Manual,ColdSpring Harbor Laboratory,Cold Spring Harbor,N.Y.中。
V.疫苗组合物
本文还公开了一种能够引起特异性免疫应答(例如肿瘤特异性免疫应答)的免疫原性组合物,例如疫苗组合物。疫苗组合物通常包含例如使用本文所述的方法选择的或如表A、表1.2或AACR GENIE结果中所示的一个或多个抗原。疫苗组合物也可称为疫苗。
疫苗可含有1至30个肽;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个不同的肽;6、7、8、9、10、11、12、13或14个不同的肽;或12、13或14个不同的肽。肽可包括翻译后修饰。疫苗可含有1至100或更多个核苷酸序列;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100或更多个不同的核苷酸序列;6、7、8、9、10、11、12、13或14个不同的核苷酸序列;或12、13或14个不同的核苷酸序列。疫苗可含有1至30个抗原序列;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100或更多个不同的抗原序列;6、7、8、9、10、11、12、13或14个不同的抗原序列;或12、13或14个不同的抗原序列。
在一个实施方案中,选择不同的肽和/或多肽或其编码核苷酸序列,以使得肽和/或多肽能够与不同的MHC分子(例如不同的MHC I类分子和/或不同的MHC II类分子)缔合。在一些方面,一种疫苗组合物包含能够与最常出现的MHC I类分子和/或不同的MHC II类分子缔合的肽和/或多肽的编码序列。因此,疫苗组合物可包含能够与至少2个优选的、至少3个优选的、或至少4个优选的MHC I类分子和/或不同的MHC II类分子缔合的不同片段。
疫苗组合物能够引起特异性细胞毒性T细胞反应和/或特异性辅助T细胞反应。
疫苗组合物可另外包含佐剂和/或载体。有用的佐剂和载体的实例在下文中给出。组合物可与载体缔合,例如蛋白质或抗原递呈细胞,例如能够将肽递呈于T细胞的树突状细胞(DC)。
佐剂是混合到疫苗组合物中增加或以其它方式修饰对抗原的免疫应答的任何物质。载体可为主链结构,例如能够与抗原缔合的多肽或多糖。任选地,佐剂是共价或非共价结合的。
佐剂提高对抗原的免疫应答的能力通常显现为免疫介导性反应的显著或实质性增加或疾病症状的减少。举例来说,体液免疫的提高通常显现为针对抗原所产生的抗体的效价显著增加,并且T细胞活性的增加通常显现为细胞增殖、或细胞毒性、或细胞因子分泌增加。佐剂也可改变免疫应答,例如通过将主要体液或Th反应变为主要细胞或Th反应。
适合的佐剂包括(但不限于)1018ISS、矾、铝盐、Amplivax、AS15、BCG、CP-870,893、CpG7909、CyaA、dSLIM、GM-CSF、IC30、IC31、咪喹莫特(Imiquimod)、ImuFact IMP321、ISPatch、ISS、ISCOMATRIX、JuvImmune、LipoVac、MF59、单磷酰基脂质A、Montanide IMS 1312、Montanide ISA 206、Montanide ISA 50V、Montanide ISA-51、OK-432、OM-174、OM-197-MP-EC、ONTAK、PepTel载体系统、PLG微粒、雷西莫特(resiquimod)、SRL172、病毒颗粒和其它病毒样颗粒、YF-17D、VEGF捕获剂、R848、β-葡聚糖、Pam3Cys、Aquila的源于皂素的QS21刺激子(Aquila Biotech,Worcester,Mass.,USA)、分支杆菌提取物和合成细菌细胞壁模拟物,以及其它专用佐剂,例如Ribi的Detox.Quil或Superfos。例如不完全弗氏佐剂或GM-CSF的佐剂是有用的。先前已描述特异性针对树突状细胞的数种免疫佐剂(例如MF59)及其制备(Dupuis M等人,Cell Immunol.1998;186(1):18-27;Allison A C;Dev Biol Stand.1998;92:3-11)。也可使用细胞因子。数种细胞因子已直接关联于:影响树突状细胞迁移至淋巴组织(例如TNF-α)、加速树突状细胞成熟变为T-淋巴细胞的有效抗原递呈细胞(例如GM-CSF、IL-1和IL-4)(美国专利第5,849,589号,其以全文引用的方式明确并入本文中)以及充当免疫佐剂(例如IL-12)(Gabrilovich D I等人,J Immunother Emphasis TumorImmunol.1996(6):414-418)。
还已报道CpG免疫刺激性寡核苷酸增强佐剂在疫苗环境中的效应。也可使用其它TLR结合分子,例如结合RNA的TLR 7、TLR 8和/或TLR 9。
有用佐剂的其它实例包括(但不限于)经化学修饰的CpG(例如CpR、Idera)、聚(I:C)(例如聚i:CI2U)、非CpG细菌DNA或RNA以及免疫活性小分子和抗体,例如环磷酰胺、舒尼替尼(sunitinib)、贝伐单抗(bevacizumab)、西乐葆(celebrex)、NCX-4016、西地那非(sildenafil)、他达拉非(tadalafil)、伐地那非(vardenafil)、索拉菲尼(sorafinib)、XL-999、CP-547632、帕佐泮尼(pazopanib)、ZD2171、AZD2171、伊匹单抗(ipilimumab)、曲美木单抗(tremelimumab)和SC58175,其可起治疗作用和/或充当佐剂。佐剂和添加剂的量和浓度可容易由本领域技术人员确定而无需过度实验。额外佐剂包括集落刺激因子,例如颗粒球巨噬细胞集落刺激因子(GM-CSF,沙格司亭(sargramostim))。
疫苗组合物可包含多于一种不同的佐剂。此外,治疗组合物可包含任何佐剂物质,包括以上各者中的任一者或其组合。此外预期,疫苗和佐剂可一起或以任何适当的顺序分开施用。
载体(或赋形剂)可独立于佐剂存在。载体的功能可例如为增加特定突变体的分子量以提高活性或免疫原性,赋予稳定性,增加生物活性或增加血清半衰期。此外,载体可辅助递呈肽至T细胞。载体可为本领域技术人员已知的任何适合的载体,例如蛋白质或抗原递呈细胞。载体蛋白质可为(但不限于)匙孔螺血氰蛋白、血清蛋白质(例如转铁蛋白)、牛血清白蛋白、人血清白蛋白、甲状腺球蛋白或卵白蛋白、免疫球蛋白或激素,例如胰岛素或棕榈酸。为用于人免疫接种,载体一般为生理学上可接受的载体,其为人可接受的且为安全的。然而,破伤风类毒素和/或白喉类毒素为适合的载体。或者,载体可为葡聚糖,例如琼脂糖。
细胞毒性T细胞(CTL)识别与MHC分子结合的肽形式的抗原,而非完整外来抗原本身。MHC分子本身位于抗原递呈细胞的细胞表面上。因此,如果存在肽抗原、MHC分子和APC的三聚体复合物,则可能活化CTL。相应地,如果不仅肽用于活化CTL,而且如果另外添加具有相应MHC分子的APC,则可加强免疫应答。因此,在一些实施方案中,疫苗组合物另外含有至少一种抗原递呈细胞。
抗原还可包括于基于病毒载体的疫苗平台中,例如牛痘、禽痘、自我复制甲病毒、马拉巴病毒(marabavirus)、腺病毒(参见例如Tatsis等人,Adenoviruses,MolecularTherapy(2004)10,616-629)或慢病毒,包括(但不限于)第二、第三或杂交第二/第三代慢病毒以及任一代的重组慢病毒,其经设计以靶向特定细胞类型或受体(参见例如Hu等人,Immunization Delivered by Lentiviral Vectors for Cancer and InfectiousDiseases,Immunol Rev.(2011)239(1):45-61;Sakuma等人,Lentiviral vectors:basicto translational,Biochem J.(2012)443(3):603-18;Cooper等人,Rescue of splicing-mediated intron loss maximizes expression in lentiviral vectors containingthe human ubiquitin C promoter,Nucl.Acids Res.(2015)43(1):682-690;Zufferey等人,Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo GeneDelivery,J.Virol.(1998)72(12):9873-9880)。取决于上述基于病毒载体的疫苗平台的包装能力,这种方法可递送编码一个或多个新抗原肽的一个或多个核苷酸序列。序列可侧接非突变序列,可由接头分开或可在前面有一个或多个靶向亚细胞区室的序列(参见例如Gros等人,Prospective identification of neoantigen-specific lymphocytes in theperipheral blood of melanoma patients,Nat Med.(2016)22(4):433-8;Stronen等人,Targeting of cancer neoantigens with donor-derived T cell receptorrepertoires,Science.(2016)352(6291):1337-41;Lu等人,Efficient identificationof mutated cancer antigens recognized by T cells associated with durabletumor regressions,Clin Cancer Res.(2014)20(13):3401-10)。在引入到宿主后,经感染细胞表达抗原,从而引发针对肽的宿主免疫(例如CTL)反应。用于免疫方案中的牛痘载体和方法描述于例如美国专利第4,722,848号中。另一种载体为卡介苗(Bacille CalmetteGuerin,BCG)。BCG载体描述于Stover等人(Nature 351:456-460(1991))中。根据本文描述,用于抗原的治疗性施用或免疫接种的各种其它疫苗载体,例如伤寒沙门氏菌(Salmonellatyphi)载体等对于本领域技术人员将为显而易见的。
V.A.抗原盒
鉴于本文所提供的教导内容,用于选择一个或多个抗原、克隆及构建“盒”及其插入至病毒载体中的方法在本领域的技能内。“抗原盒”意指经选择的抗原或多个抗原与转录抗原且表达转录产物所必需的其它调控元件的组合。抗原或多个抗原可以允许转录的方式可操作地连接于调控元件。此类组件包括可驱动被病毒载体转染的细胞中表达抗原的常规调控元件。因此,抗原盒也可含有经选择的启动子,其连接于抗原并且与其它任选的调控元件一起位于重组载体的经选择的病毒序列内。盒可包含表A和/或AACR GENIE结果中显示的一个或多个新抗原,和/或表1.2中显示的一个或多个抗原。
有用的启动子可以是组成型启动子或经调控(诱导型)启动子,其将能够控制待表达的抗原的量。举例来说,合乎需要的启动子是细胞巨大病毒即刻早期启动子/增强子的启动子[参见例如Boshart等人,Cell,41:521-530(1985)]。另一种合乎需要的启动子包括劳斯肉瘤(Rous sarcoma)病毒LTR启动子/增强子。另一种启动子/增强子序列是鸡细胞质β-肌动蛋白启动子[T.A.Kost等人,Nucl.Acids Res.,11(23):8287(1983)]。其它适合或合乎需要的启动子可由本领域技术人员选择。
抗原盒还可包括与病毒载体序列异源的核酸序列,包括提供转录本的有效聚腺苷酸化信号(聚(A)、聚-A或pA)的序列及具有功能性剪接供体和受体位点的内含子。本发明的示例性载体中使用的普通聚-A序列源自乳多泡病毒SV-40。聚-A序列一般可在基于抗原的序列之后及在病毒载体序列之前插入盒中。普通内含子序列也可源自SV-40,并且称为SV-40T内含子序列。抗原盒也可含有位于启动子/增强子序列与抗原之间的此类内含子。这些和其它普通载体元件的选择是常规的[参见例如Sambrook等人,“Molecular Cloning.ALaboratory Manual.”,第2版,Cold Spring Harbor Laboratory,New York(1989)和其中引用的参考文献]并且许多此类序列可从商业和工业来源以及Genbank获得。
抗原盒可具有一个或多个抗原。举例来说,给定盒可包括1-10、1-20、1-30、10-20、15-25、15-20、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个抗原。抗原可彼此直接连接。抗原也可用接头彼此连接。抗原可相对于彼此呈任一定向,包括N至C或C至N。
如上所述,抗原盒可位于病毒载体中任何经选择的缺失位点,例如E1基因区缺失或E3基因区缺失的位点等可经选择的位点。
可以使用下式描述抗原盒,以从5'到3'描述每个元件的有序序列:
(P
其中P和P2包含启动子核苷酸序列,N包含MHC I类表位编码核酸序列,L5包含5'接头序列,L3包含3'接头序列,G5包含编码氨基酸接头的核酸序列,G3包含编码氨基酸接头的至少一个核酸序列之一,U包含MHC II类抗原编码核酸序列,其中对于每个X,相应的Nc是表位编码核酸序列,其中对于每个Y,相应的Uf是抗原编码核酸序列。可以通过选择存在的元件数来进一步定义组成和有序序列,例如其中a=0或1,其中b=0或1,其中c=1,其中d=0或1,其中e=0或1,其中f=1,其中g=0或1,其中h=0或1,X=1至400,Y=0、1、2、3、4或5,Z=1至400,并且W=0、1、2、3、4或5。
在一个实例中,存在的元件包括其中a=0,b=1,d=1,e=1,g=1,h=0,X=10,Y=2,Z=1和W=1,描述了其中不存在其它启动子(即仅存在由RNA甲病毒主链提供的启动子核苷酸序列),存在20个MHC I类表位,对于每个N存在5'接头,对于每个N存在3'接头,存在2个MHC II类表位,存在连接两个MHC II类表位的接头,存在将两个MHC II类表位的5'端连接到最后一个MHC I类表位的3'端的接头,以及存在将两个MHC II类表位的3'端连接到RNA甲病毒主链的接头。将抗原盒的3'端连接到RNA甲病毒的实例包括直接连接到RNA甲病毒提供的3'UTR元件,例如3'19-nt CSE。将抗原盒的5'端连接到RNA甲病毒的实例包括直接连接到26S启动子序列、甲病毒5'UTR、51nt CSE或24nt CSE。
其它实例包括:其中a=1描述其中存在不同于RNA甲病毒主链提供的启动子核苷酸序列的启动子;其中a=1且Z为大于1,其中存在多个除RNA甲病毒主链提供的启动子核苷酸序列以外的启动子,每个驱动1个或更多个不同的MHC I类表位编码核酸序列的表达;其中h=1描述其中存在一个单独的启动子来驱动MHC II类抗原编码核酸序列的表达;并且其中g=0描述MHC II类抗原编码核酸序列(如果存在)与RNA甲病毒主链直接连接。
其它实例包括其中存在的每个MHC I类表位可以具有5'接头、3'接头、都不具有或两者都有的情况。在同一抗原盒中存在多于一个MHC I类表位的实例中,一些MHC I类表位可能同时具有5'接头和3'接头,而其它MHC I类表位可能具有5'接头、3'接头或两者都没有。在同一抗原盒中存在多于一个MHC I类表位的其它实例中,一些MHC I类表位可能具有5'接头或3'接头,而其它MHC I类表位可能具有5'接头、3'接头或两者都没有。
在同一抗原盒中存在多于一个MHC II类表位的实例中,一些MHC II类表位可能同时具有5'接头和3'接头,而其它MHC II类表位可能具有5'接头、3'接头或两者都没有。在同一抗原盒中存在多于一个MHC II类表位的其它实例中,一些MHC II类表位可能具有5'接头或3'接头,而其它MHC II类表位可能具有5'接头、3'接头或两者都没有。
启动子核苷酸序列P和/或P2可以与RNA甲病毒主链提供的启动子核苷酸序列相同。举例来说,由RNA甲病毒主链提供的启动子序列Pn和P2可各自包含26S亚基因组启动子。启动子核苷酸序列P和/或P2可以不同于RNA甲病毒主链提供的启动子核苷酸序列,也可以彼此不同。
5'接头L5可以是原生序列或非天然序列。非天然序列包括但不限于AAY、RR和DPP。3'接头L3也可以是原生序列或非天然序列。另外,L5和L3可以都是原生序列,可以都是非天然序列,或者一个可以是原生的而另一个可以是非天然的。对于每个X,氨基酸接头的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。对于每个X,氨基酸接头的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
对于每个Y,氨基酸接头G5的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。对于每个Y,氨基酸接头的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
氨基酸接头G3的长度可以是2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100个或更多个氨基酸。G3的长度也可以是至少3、至少4、至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸。
对于每个X,每个N可以编码长度为7-15个氨基酸的MHC I类表位。对于每个X,每个N也可以编码长度为5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个氨基酸的MHC I类表位。对于每个X,每个N也可以编码长度为至少5、至少6、至少7、至少8、至少9、至少10、至少11、至少12、至少13、至少14、至少15、至少16、至少17、至少18、至少19、至少20、至少21、至少22、至少23、至少24、至少25、至少26、至少27、至少28、至少29或至少30个氨基酸的MHC I类表位。
V.B.免疫检查点
本文所述的载体,例如本文所述的C68载体或本文所述的甲病毒载体,可包含编码至少一种抗原的核酸,并且同一或另一载体可包含编码至少一种免疫调节物(例如抗体,例如scFv)的核酸,其结合于免疫检查点分子且阻断免疫检查点分子的活性。载体可包含抗原盒和一个或多个编码检查点抑制剂的核酸分子。
可靶向用于阻断或抑制的说明性免疫检查点分子包括(但不限于)CTLA-4、4-1BB(CD137)、4-1BBL(CD137L)、PDL1、PDL2、PD1、B7-H3、B7-H4、BTLA、HVEM、TIM3、GAL9、LAG3、TIM3、B7H3、B7H4、VISTA、KIR、2B4(属于CD2家族的分子并且在所有NK、γδ和记忆CD8+(αβ)T细胞上表达)、CD160(也称为BY55)和CGEN-15049。免疫检查点抑制剂包括抗体或其抗原结合片段、或其它结合蛋白,其结合且阻断或抑制以下中的一个或多个的活性:CTLA-4、PDL1、PDL2、PD1、B7-H3、B7-H4、BTLA、HVEM、TIM3、GAL9、LAG3、TIM3、B7H3、B7H4、VISTA、KIR、2B4、CD160和CGEN-15049。说明性免疫检查点抑制剂包括曲美木单抗(CTLA-4阻断抗体)、抗OX40、PD-L1单克隆抗体(抗B7-H1;MEDI4736)、伊匹单抗、MK-3475(PD-1阻断剂)、纳武单抗(Nivolumamb)(抗PD1抗体)、CT-011(抗PD1抗体)、BY55单克隆抗体、AMP224(抗PDL1抗体)、BMS-936559(抗PDL1抗体)、MPLDL3280A(抗PDL 1抗体)、MSB0010718C(抗PDL1抗体)和Yervoy/伊匹单抗(抗CTLA-4检查点抑制剂)。抗体编码序列可使用本领域中的普通技能经工程改造至例如C68的载体中。示例性方法描述于Fang等人,Stable antibody expressionat therapeutic levels using the 2A peptide.Nat Biotechnol.2005年5月;23(5):584-90.电子版2005年4月17日中;其以引用的方式并入本文中用于所有目的。
V.C.疫苗设计和制造的额外考虑
V.C.1.确定覆盖所有肿瘤亚克隆的肽集合
躯干肽,意指由所有或大部分肿瘤亚克隆递呈的那些肽,可优先包括于疫苗中。
V.C.2.抗原优先排序
在应用所有上述抗原过滤器后,与疫苗技术可支持的相比,更多候选抗原仍可包含于疫苗中。另外,可保留关于抗原分析的各个方面的不确定性,并且候选疫苗抗原的不同特性之间可存在折衷。因此,可考虑整合式多维模型代替选择过程的各步骤中的预定过滤器,将候选抗原置于具有至少以下轴的空间中且使用整合方法优化选择。
1.自身免疫或耐受性的风险(生殖系的风险)(自身免疫的风险较低通常为优选的)
2.测序伪影的概率(伪影的概率较低通常为优选的)
3.免疫原性的概率(免疫原性的概率较高通常为优选的)
4.递呈的概率(递呈的概率较高通常为优选的)
5.基因表达(较高表达通常为优选的)
6.HLA基因的覆盖率(参与抗原集合递呈的HLA分子数量越大可降低肿瘤经由HLA分子的下调或突变逃避免疫攻击的概率)
7.HLA类的覆盖率(同时覆盖HLA-I和HLA-II可能会增加治疗反应的概率并降低肿瘤逃避的概率)
另外,任选地,如果预测抗原是由患者的全部或部分肿瘤中失去或失活的HLA等位基因递呈的,则可以从疫苗接种中除名(例如排除)抗原。HLA等位基因的丢失可能通过体细胞突变、杂合性丢失或基因座的纯合缺失而发生。检测HLA等位基因体细胞突变的方法是本领域中众所周知的,例如(Shukla等人,2015)。同样充分描述了检测体细胞LOH和纯合缺失(包括HLA基因座)的方法。(Carter等人,2012;McGranahan等人,2017;Van Loo等人,2010)。如果质谱数据表明预测抗原未由预测HLA等位基因递呈,也可以降低抗原的优先级。
V.D.甲病毒
V.D.1.甲病毒生物学
甲病毒是披膜病毒科的成员,且为正义单链RNA病毒。成员通常分类为旧世界,例如辛德毕斯、罗斯河、马雅罗、基孔肯尼亚及塞姆利基森林病毒,或新世界,例如东部马脑炎、奥拉、摩根堡、或委内瑞拉马脑炎及其衍生病毒株TC-83(Strauss Microbrial Review1994)。天然甲病毒基因组通常约12kb长,其中前三分之二含有编码非结构蛋白(nsP)的基因,所述非结构蛋白形成用于病毒基因组自我复制的RNA复制复合物,并且最后三分之一含有编码用于病毒粒子产生的结构蛋白的亚基因组表达盒(Frolov RNA 2001)。
甲病毒的模型生命周期涉及数个不同步骤(Strauss Microbrial Review 1994,Jose Future Microbiol 2009)。在病毒附着于宿主细胞后,病毒粒子与内吞区室内的膜融合,导致基因组RNA最终释放至胞溶质中。以正链定向并包含5′甲基鸟苷酸帽和3′聚A尾部的基因组RNA经翻译以产生形成复制复合物的非结构蛋白nsP1-4。在感染早期,正链然后由复合物复制成负链模板。在当前模型中,复制复合物随着感染进展被进一步加工,使得所得经加工的复合物转换成将负链转录成全长正链基因组RNA以及含有结构基因的26S亚基因组正链RNA。甲病毒的数个保守序列元件(CSE)已鉴定为可能在各种RNA复制步骤中起作用,包括:负链模板的正链RNA复制中的5′UTR的互补序列、基因组模板的负链合成复制中的51-nt CSE、负链的亚基因组RNA转录中的在nsP与26S RNA之间的接合区中的24-nt CSE,以及正链模板的负链合成中的3′19-nt CSE。
在各种RNA物种复制后,病毒粒子然后通常在病毒的天然生命周期中组装。26SRNA经翻译且所得蛋白质经进一步加工以产生结构蛋白,其包括衣壳蛋白、糖蛋白E1和E2以及两个小多肽E3和6K(Strauss 1994)。发生病毒RNA的衣壳化,衣壳蛋白通常仅特异性针对所包装的基因组RNA,接着病毒粒子组装且在膜表面出芽。
V.D.2.甲病毒作为递送载体
甲病毒(包括甲病毒序列、特征和其他元件)可用于产生基于甲病毒的递送载体(也称为甲病毒载体、甲病毒病毒载体、甲病毒疫苗载体、自我复制RNA(srRNA)载体或自我扩增RNA(samRNA)载体)。甲病毒先前已经工程改造以用作表达载体系统(Pushko 1997,Rheme 2004)。甲病毒提供数种优势,特别是在可能需要异源抗原表达的疫苗环境中。由于在宿主胞溶质中自我复制的能力,故甲病毒载体一般能够在细胞内产生高拷贝数的表达盒,从而导致高水平异源抗原产生。另外,载体一般为瞬时的,从而使得生物安全性得以改进以及减少对载体的免疫耐受性的诱导。与其它标准病毒载体(例如人腺病毒)相比,公众一般也缺乏对甲病毒载体预先存在的免疫性。基于甲病毒的载体也一般导致对经感染细胞的细胞毒性反应。在一定程度上,细胞毒性在疫苗环境中对于适当违禁引发对所表达的异源抗原的免疫应答可为重要的。然而,所需细胞毒性的程度可为平衡作用,且因此已开发数种减毒甲病毒,包括VEE的TC-83病毒株。因此,本文所述的抗原表达载体的实例可利用甲病毒主链,其允许高水平的抗原表达,引发对抗原的稳固免疫应答,不引发对载体本身的免疫应答,并且可以安全方式使用。此外,抗原表达盒可经设计以经由优化载体使用的甲病毒序列(包括但不限于源自VEE或其减毒衍生物TC-83的序列)而引发不同水平的免疫应答。
已使用甲病毒序列工程改造数种表达载体设计策略(Pushko 1997)。在一个策略中,甲病毒载体设计包括在结构蛋白基因下游插入26S启动子序列元件的第二拷贝,接着是异源基因(Frolov 1993)。因此,除天然非结构蛋白和结构蛋白之外,也产生表达异源蛋白的额外亚基因组RNA。在此系统中,存在用于产生感染性病毒粒子的所有元件,因此可能发生在未感染细胞中反复轮表达载体的感染。
另一种表达载体设计利用辅助病毒系统(Pushko 1997)。在此策略中,结构蛋白由异源基因替代。因此,在由仍完整的非结构基因介导病毒RNA的自我复制之后,26S亚基因组RNA提供异源蛋白的表达。传统上,表达结构蛋白的额外载体然后例如通过细胞系的共转染以反式供应,以产生感染性病毒。系统详细描述于USPN 8,093,021中,其出于所有目的以全文引用的方式并入本文中。辅助载体系统提供限制形成感染性粒子的可能性的益处,因此提高生物安全性。另外,辅助载体系统减小总载体长度,潜在提高复制和表达效率。因此,本文所述的抗原表达载体的实例可利用结构蛋白由抗原盒替代的甲病毒主链,所得载体降低生物安全问题,同时由于整体表达载体尺寸减小而促进有效表达。
V.D.3.体外甲病毒产生
甲病毒递送载体一般是正义RNA多核苷酸。本领域中熟知的用于产生RNA的适宜技术为体外转录IVT。在此技术中,首先通过本领域技术人员熟知的技术产生所需载体的DNA模板,包括标准分子生物学技术,例如克隆、限制性消化、连接、基因合成及聚合酶链反应(PCR)。DNA模板在期望转录成RNA的序列的5′端处含有RNA聚合酶启动子。启动子包括(但不限于)噬菌体聚合酶启动子,例如T3、T7或SP6。DNA模板然后与适当RNA聚合酶、缓冲剂及核苷酸(NTP)一起温育。所得RNA多核苷酸可任选地经进一步修饰,包括(但不限于)添加5′帽结构,例如7-甲基鸟苷或相关结构,以及任选地修饰3′端以包括聚腺苷酸(聚A)尾部。RNA可然后使用本领域中熟知的技术纯化,例如苯酚-氯仿提取。
V.D.4.经由脂质纳米粒子递送
在疫苗载体设计中考虑的一个重要方面是针对载体本身的免疫性(Riley 2017)。这可以呈对载体本身(例如某些人腺病毒系统)预先存在的免疫性形式,或呈在疫苗施用后对载体产生免疫性的形式。如果进行相同疫苗的多次施用(例如分开的初免和加强剂量),或如果使用相同疫苗载体系统递送不同抗原盒,则后者为重要的考虑因素。
在甲病毒载体的情况下,标准递送方法是先前论述的辅助病毒系统,其以反式提供衣壳、E1和E2蛋白质以产生感染性病毒粒子。然而,重要的是注意E1和E2蛋白质通常是中和抗体的主要靶标(Strauss 1994)。因此,如果中和抗体靶向感染性粒子,则使用甲病毒载体将所关注的抗原递送到靶细胞的功效可能会降低。
病毒粒子介导的基因递送的替代方案是使用纳米材料递送表达载体(Riley2017)。重要的是,纳米材料媒介物可由非免疫原性材料制成且一般避免引发对递送载体本身的免疫性。这些材料可包括(但不限于)脂质、无机纳米材料和其它聚合材料。脂质可为阳离子、阴离子或中性的。材料可为合成或天然来源的,并且在一些情况下是可生物降解的。脂质可包括脂肪、胆固醇、磷脂、脂质缀合物,包括(但不限于)聚乙二醇(PEG)缀合物(聚乙二醇化脂质)、蜡、油、甘油酯和脂溶性维生素。
脂质纳米粒子(LNP)是有吸引力的递送系统,因为脂质的两亲性使得能够形成膜和囊泡状结构(Riley 2017)。一般来说,这些囊泡通过吸收至靶细胞的膜中并将核酸释放至胞溶质中来递送表达载体。另外,LNP可经进一步修饰或官能化以有助于靶向特定细胞类型。LNP设计中的另一考虑因素是靶向效率与细胞毒性之间的平衡。脂质组合物一般包括阳离子、中性、阴离子及两性脂质的确定的混合物。在一些情况下,包括特定脂质以防止LNP聚集,防止脂质氧化,或提供有助于额外部分附着的功能性化学基团。脂质组合物可影响整体LNP大小和稳定性。在一个实例中,脂质组合物包含二亚油基甲基-4-二甲基氨基丁酸酯(MC3)或MC3样分子。MC3和MC3样脂质组合物可经配制以包括一种或多种其它脂质,例如PEG或PEG结合的脂质、固醇或中性脂质。
直接暴露于血清的核酸载体(例如表达载体)可具有数种不期望的结果,包括核酸由血清核酸酶降解或游离核酸对免疫系统的脱靶刺激。因此,包封甲病毒载体可用于避免降解,同时也避免潜在的脱靶影响。在某些实例中,甲病毒载体完全包封在递送媒介物内,例如在LNP的水性内部。甲病毒载体包封在LNP内可通过本领域技术人员熟知的技术来进行,例如在微流体液滴生成装置上进行的微流体混合和液滴生成。这些装置包括(但不限于)标准T形接合装置或流动聚焦装置。在一个实例中,所需脂质制剂(例如含有MC3或MC3样的组合物)与甲病毒递送载体和其它所需药剂并行提供至液滴生成装置,使得递送载体和所需药剂完全包封在基于MC3或MC3样的LNP内部。在一个实例中,液滴生成装置可控制所产生的LNP的尺寸范围及尺寸分布。举例来说,LNP的尺寸可在1至1000纳米直径范围内,例如1、10、50、100、500或1000纳米。在液滴生成后,包封表达载体的递送媒介物可经进一步处理或修饰以使其准备用于施用。
V.E.黑猩猩腺病毒(ChAd)
V.E.1.用黑猩猩腺病毒递送病毒
用于递送一个或多个抗原(例如经由抗原盒,并且包括表A和/或AACR GENIE结果中所示的一个或多个新抗原,和/或表1.2中所示的一个或多个抗原)的疫苗组合物可通过提供黑猩猩来源的腺病毒核苷酸序列、多种新颖载体和表达黑猩猩腺病毒基因的细胞系来产生。黑猩猩C68腺病毒(在本文中也称为ChAdV68)的核苷酸序列可用于抗原递送的疫苗组合物中(参见SEQ ID NO:1)。源自C68腺病毒的载体的使用进一步详细描述于USPN 6,083,716中,其出于所有目的以全文引用的方式并入本文中。
在另一方面,本文提供一种重组腺病毒,其包含黑猩猩腺病毒如C68的DNA序列和可操作地连接于引导其表达的调控序列的抗原盒。重组病毒能够感染哺乳动物细胞、优选人细胞,并且能够在细胞中表达抗原盒产物。在这种载体中,原生黑猩猩E1基因和/或E3基因和/或E4基因可缺失。抗原盒可插入至这些基因缺失位点中的任一者中。抗原盒可包括抗原,针对其激活的免疫应答为所需的。
在另一方面,本文提供一种被黑猩猩腺病毒如C68感染的哺乳动物细胞。
在另一方面,提供一种新颖的哺乳动物细胞系,其表达黑猩猩腺病毒基因(例如来自C68)或其功能片段。
在另一方面,本文提供一种用于将抗原盒递送到哺乳动物细胞中的方法,其包括以下步骤:向细胞中引入有效量的已经工程改造以表达抗原盒的黑猩猩腺病毒,例如C68。
另一方面提供一种用于在哺乳动物宿主中引发免疫应答以治疗癌症的方法。所述方法可包括以下步骤:向宿主施用有效量的重组黑猩猩腺病毒如C68,其包含编码免疫应答所靶向的来自肿瘤的一个或多个抗原的抗原盒。
还公开了一种非猿猴哺乳动物细胞,其表达获自SEQ ID NO:1序列的黑猩猩腺病毒基因。该基因可选自:SEQ ID NO:1的腺病毒E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5。
还公开了一种包含黑猩猩腺病毒DNA序列的核酸分子,该黑猩猩腺病毒DNA序列包含获自SEQ ID NO:1序列的基因。该基因可选自:SEQ ID NO:1的所述黑猩猩腺病毒E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因。在一些方面,核酸分子包含SEQ ID NO:1。在一些方面,核酸分子包含SEQ ID NO:1序列,其缺少选自以下的至少一个基因:SEQ ID NO:1的E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因。
还公开了一种载体,其包含获自SEQ ID NO:1的黑猩猩腺病毒DNA序列以及可操作地连接于一个或多个调控序列的抗原盒,该一个或多个调控序列引导盒在异源宿主细胞中的表达,任选地其中所述黑猩猩腺病毒DNA序列至少包含用于复制及病毒粒子衣壳化所必需的顺式元件,所述顺式元件侧接抗原盒和调控序列。在一些方面,黑猩猩腺病毒DNA序列包含选自以下的基因:SEQ ID NO:1的E1A、E1B、E2A、E2B、E3、E4、L1、L2、L3、L4和L5基因序列。在一些方面,载体可缺乏E1A和/或E1B基因。
本文还公开经本文所公开的载体转染的宿主细胞,该载体例如经工程改造以表达抗原盒的C68载体。本文还公开经由将本文所公开的载体引入细胞中而表达其中引入的经选择的基因的人细胞。
本文还公开了一种用于将抗原盒递送到哺乳动物细胞的方法,其包括向该细胞中引入有效量的本文所公开的载体,例如经工程改造以表达抗原盒的C68载体。
本文还公开了一种用于产生抗原的方法,其包括将本文所公开的载体引入哺乳动物细胞中,在适合的条件下培养细胞以及产生抗原。
V.E.2.表达E1的互补细胞系
为了产生缺失本文所述的基因中的任一者的重组黑猩猩腺病毒(Ad),缺失基因区的功能若对于病毒的复制和感染性必不可少,则可通过辅助病毒或细胞系(即互补或包装细胞系)供应至重组病毒。举例来说,为了产生复制缺陷型黑猩猩腺病毒载体,可使用表达人或黑猩猩腺病毒的E1基因产物的细胞系;这种细胞系可包括HEK293或其变体。可遵循产生表达黑猩猩E1基因产物的细胞系的方案(USPN 6,083,716的实例3和4),以产生表达任何经选择的黑猩猩腺病毒基因的细胞系。
AAV增强分析可用于鉴定表达黑猩猩腺病毒E1的细胞系。这种分析可用于鉴定通过使用例如来自其它物种的其它未表征的腺病毒的E1基因制备的细胞系中的E1功能。该分析描述于USPN 6,083,716的实例4B中。
经选择的黑猩猩腺病毒基因(例如E1)可在启动子的转录控制下用于在经选择的亲本细胞系中表达。诱导型或组成型启动子可用于此目的。在诱导型启动子中包括可由锌诱导的绵羊金属硫蛋白启动子,或可由糖皮质激素、特别是地塞米松(dexamethasone)诱导的小鼠乳腺肿瘤病毒(MMTV)启动子。其它诱导型启动子,例如以引用的方式并入本文中的国际专利申请WO95/13392中所鉴定的那些诱导型启动子,也可用于产生包装细胞系。此外也可采用控制黑猩猩腺病毒基因表达的组成型启动子。
亲本细胞可经选择以产生表达任何所需C68基因的新颖细胞系。这种亲本细胞系可为(但不限于)HeLa[ATCC寄存编号CCL 2]、A549[ATCC寄存编号CCL 185]、KB[CCL 17]、Detroit[例如Detroit 510、CCL 72]和WI-38[CCL 75]细胞。其它适合的亲本细胞系可获自其它来源。亲本细胞系可包括CHO、HEK293或其变体、911、HeLa、A549、LP-293、PER.C6或AE1-2a。
表达E1的细胞系可用于产生重组黑猩猩腺病毒E1缺失的载体。使用基本上相同的程序构建的表达一种或多种其它黑猩猩腺病毒基因产物的细胞系可用于产生缺失编码那些产物的基因的重组黑猩猩腺病毒载体。此外,表达其它人Ad E1基因产物的细胞系也可用于产生黑猩猩重组Ad。
V.E.3.作为载体的重组病毒粒子
本文所公开的组合物可包含病毒载体,其将至少一个抗原递送至细胞。这种载体包含黑猩猩腺病毒DNA序列如C68和可操作地连接于引导盒表达的调控序列的抗原盒。C68载体能够在经感染的哺乳动物细胞中表达盒。C68载体可能功能性缺失一个或多个病毒基因。抗原盒包含在一个或多个调控序列如启动子控制下的至少一个抗原。任选的辅助病毒和/或包装细胞系可向黑猩猩病毒载体供应缺失的腺病毒基因的任何必需产物。
术语“功能性缺失”意指移除或以其它方式改变(例如通过突变或修饰)足够量的基因区,使得基因区不再能够产生一种或多种基因表达的功能性产物。可导致功能性缺失的突变或修饰包括(但不限于)无义突变,例如引入过早的终止密码子和去除典型和非典型的起始密码子,改变mRNA剪接或其它转录加工的突变,或其组合。如果需要,可去除整个基因区。
形成本文所公开的载体的核酸序列的修饰,包括序列缺失、插入和其它突变,可使用标准分子生物学技术产生并且在本发明的范围内。
V.E.4.病毒质粒载体的构建
可用于本发明的黑猩猩腺病毒C68载体包括重组缺陷型腺病毒,即在E1a或E1b基因中功能性缺失且任选地携带其它突变(例如其它基因中的温度敏感性突变或缺失)的黑猩猩腺病毒序列。预期这些黑猩猩序列也可用于形成来自其它腺病毒和/或腺相关病毒序列的杂交载体。由人腺病毒制备的同源腺病毒载体描述于公开的文献中[参见例如上文所引用的Kozarsky I和II,以及其中引用的参考文献,美国专利第5,240,846号]。
在构建可用于将抗原盒递送至人(或其它哺乳动物)细胞的黑猩猩腺病毒C68载体时,可在载体中采用一系列腺病毒核酸序列。包含最小黑猩猩C68腺病毒序列的载体可与辅助病毒结合使用以产生感染性重组病毒粒子。辅助病毒提供最小黑猩猩腺病毒载体的病毒感染性和繁殖所需的基本基因产物。当在另外的功能性病毒载体中仅产生黑猩猩腺病毒基因的一个或多个经选择的缺失时,可通过在经选择的包装细胞系中繁殖病毒而在病毒载体生产过程中供应缺失的基因产物,该包装细胞系提供反式缺失的基因功能。
V.E.5.重组最小腺病毒
最小的黑猩猩Ad C68病毒是仅含有复制及病毒粒子衣壳化所必需的腺病毒顺式元件的病毒粒子。也就是说,载体含有腺病毒的顺式作用5′和3′反向末端重复(ITR)序列(其充当复制起点)以及原生5′包装/增强子结构域(其含有用于包装线性Ad基因组所必需的序列和E1启动子的增强子元件)。参见例如在国际专利申请WO96/13597中所描述且以引用的方式并入本文中的用于制备“最小”人Ad载体的技术。
V.E.6.其它缺陷型腺病毒
重组复制缺乏型腺病毒也可比最小黑猩猩腺病毒序列含有更多。这些其它Ad载体可通过病毒基因区的各个部分的缺失以及通过任选地使用辅助病毒和/或包装细胞系形成的感染性病毒粒子来表征。
作为一个实例,适合的载体可通过使C68腺病毒立即早期基因E1a和延迟早期基因E1b的全部或足够部分缺失来形成,从而消除其正常的生物功能。当在含有提供相应反式基因产物的功能性腺病毒E1a和E1b基因的黑猩猩腺病毒转化的互补细胞系上生长时,复制缺陷型E1缺失病毒能够复制并产生感染性病毒。基于与已知腺病毒序列的同源性,预期与本领域的人重组E1缺失腺病毒一样,所得重组黑猩猩腺病毒能够感染许多细胞类型并且可表达抗原,但无法在大部分不携带黑猩猩E1区DNA的细胞中复制,除非细胞以极高感染倍率感染。
作为另一个实例,C68腺病毒延迟早期基因E3的全部或一部分可从形成重组病毒的一部分的黑猩猩腺病毒序列消除。
也可构建具有E4基因缺失的黑猩猩腺病毒C68载体。另一个载体可在延迟早期基因E2a中含有缺失。
也可在黑猩猩C68腺病毒基因组的晚期基因L1至L5中的任一者中获得缺失。类似地,中间基因IX和IVa2中的缺失可用于一些目的。可在其它结构性或非结构性腺病毒基因中获得其它缺失。
上述缺失可单独使用,即腺病毒序列可仅含有E1缺失。或者,可以任何组合使用有效破坏或降低其生物活性的完整基因或其部分的缺失。举例来说,在一个示例性载体中,腺病毒C68序列可缺失E1基因和E4基因,或缺失E1、E2a和E3基因,或缺失E1和E3基因,或在缺失或不缺失E3的情况下缺失E1、E2a和E4基因等等。如上文所论述,这些缺失可与其它突变(例如温度敏感性突变)组合使用,以达成所需结果。
将包含抗原的盒任选地插入至黑猩猩C68 Ad病毒的任何缺失区中。或者,如果需要,则可将盒插入至现有基因区中以破坏该区的功能。
V.E.7.辅助病毒
取决于用于携带抗原盒的病毒载体的黑猩猩腺病毒基因含量,可使用辅助腺病毒或非复制性病毒片段来提供足够的黑猩猩腺病毒基因序列以产生含有该盒的感染性重组病毒粒子。
有用的辅助病毒含有经选择的腺病毒基因序列,其不存在于腺病毒载体构建体中和/或不由载体转染的包装细胞系表达。辅助病毒可为复制缺陷型并且除上述序列之外,还含有多种腺病毒基因。辅助病毒可与本文所述的表达E1的细胞系组合使用。
对于C68,“辅助”病毒可以是通过用SspI剪切C68基因组的C端形成的片段,其从病毒的左端除去约1300bp。这种经剪切的病毒然后与质粒DNA共转染至表达E1的细胞系中,由此通过与质粒中的C68序列同源重组形成重组病毒。
辅助病毒也可形成聚阳离子缀合物,如Wu等人,J.Biol.Chem.,264:16985-16987(1989);K.J.Fisher和J.M.Wilson,Biochem.J.,299:49(1994年4月1日)中所述。辅助病毒可任选地含有报告基因。许多这类报告基因是本领域中已知的。与腺病毒载体上的抗原盒不同,辅助病毒上报告基因的存在允许独立地监测Ad载体和辅助病毒。这种第二报告基因用于纯化后能够将所得重组病毒与辅助病毒分离。
V.E.8.病毒粒子的组装和细胞系的感染
将经选择的腺病毒DNA序列、抗原盒和其它载体元件组装到各种中间质粒及穿梭载体中,以及使用所述质粒及载体产生重组病毒粒子都可使用常规技术实现。这些技术包括常规cDNA克隆技术、体外重组技术(例如吉布森组装(Gibson assembly))、腺病毒基因组的重叠寡核苷酸序列的使用、聚合酶链反应以及提供所需核苷酸序列的任何适合的方法。采用标准转染和共转染技术,例如CaPO4沉淀技术或脂质体介导的转染方法,例如脂染胺。所采用的其它常规方法包括病毒基因组的同源重组、琼脂覆层中病毒的蚀斑、测量信号产生的方法等。
例如,在构建并组装所需含有抗原盒的病毒载体后,可在辅助病毒存在下将载体体外转染至包装细胞系中。同源重组发生在辅助序列与载体序列之间,其允许载体中的腺病毒抗原序列复制且包装至病毒粒子衣壳中,从而产生重组病毒载体粒子。
所得重组黑猩猩C68腺病毒可用于将抗原盒转移到经选择的细胞中。在使用包装细胞系中生长的重组病毒的体内实验中,E1缺失的重组黑猩猩腺病毒在将盒转移到非黑猩猩(优选人)细胞中展现效用。
V.E.9.重组病毒载体的用途
所得含有抗原盒的重组黑猩猩C68腺病毒(如上所述,通过腺病毒载体和辅助病毒或腺病毒载体及包装细胞系的合作产生)因此提供一种有效的基因转移媒介物,其可将抗原体内或离体递送至受试者。
上述重组载体根据公开的基因疗法施用于人。携带抗原盒的黑猩猩病毒载体可施用于患者,优选悬浮于生物相容性溶液或药学上可接受的递送媒介物中。适合的媒介物包括无菌盐水。已知为药学上可接受的载体并且是本领域技术人员所熟知的其它水性和非水性等张无菌注射溶液以及水性和非水性无菌悬浮液可用于此目的。
黑猩猩腺病毒载体是以足以转导人细胞并提供足够水平的抗原转移和表达的量施用,从而提供治疗益处而无过度不良效应或具有医学上可接受的生理效应,其可由医药领域的技术人员来确定。常规和药学上可接受的施用途径包括(但不限于)直接递送至肝脏、鼻内、静脉内、肌肉内、皮下、皮内、经口和其它肠胃外施用途径。如果需要,施用途径可以组合。
病毒载体的剂量主要将取决于例如以下因素:所治疗的病状,患者的年龄、体重和健康状况,并且因此可在不同患者中变化。剂量将经调节以平衡治疗益处与任何副作用,并且这些剂量可根据采用重组载体的治疗应用而变化。可监测抗原表达水平以确定剂量施用频率。
重组复制缺陷型腺病毒可以“药学有效量”施用,也就是说,在施用途径中有效转染所需细胞并提供经选择的基因的足够表达水平以提供疫苗益处(即一些可测量的保护性免疫水平)的重组腺病毒的量。包含抗原盒的C68载体可与佐剂一起共施用。佐剂可与载体(例如矾)分开或在载体内编码,特别是在佐剂为蛋白质时。佐剂是本领域中众所周知的。
常规且药学上可接受的施用途径包括(但不限于)鼻内、肌肉内、气管内、皮下、皮内、经直肠、经口和其它肠胃外施用途径。如果需要,可组合或调整施用途径,视免疫原或疾病而定。举例来说,在狂犬病预防中,皮下、气管内和鼻内途径为优选的。施用途径主要将取决于所治疗疾病的性质。
可监测对抗原的免疫水平以确定是否需要增强剂。举例来说,在评定血清中的抗体效价后,可能需要任选的加强免疫。
VI.治疗和制造方法
还提供一种通过向受试者施用一个或多个抗原(例如使用本文公开的方法鉴定的多个抗原)在受试者中诱导肿瘤特异性免疫应答,针对肿瘤接种疫苗,治疗和或缓解受试者的癌症症状的方法。
在一些方面,受试者已诊断患有癌症或具有患癌症的风险。受试者可为人、狗、猫、马或需要肿瘤特异性免疫应答的任何动物。肿瘤可为任何实体肿瘤,例如乳房肿瘤、卵巢肿瘤、前列腺肿瘤、肺肿瘤、肾脏肿瘤、胃肿瘤、结肠肿瘤、睾丸肿瘤、头颈部肿瘤、胰脏肿瘤、脑肿瘤、黑素瘤和其它组织器官肿瘤,以及血液肿瘤,例如淋巴瘤和白血病,包括急性骨髓性白血病、慢性骨髓性白血病、慢性淋巴细胞性白血病、T细胞淋巴细胞性白血病和B细胞淋巴瘤。
抗原可以足以诱导CTL反应的量施用。
抗原可以单独或与其它治疗剂组合施用。治疗剂是例如化学治疗剂、辐射或免疫疗法。可针对特定癌症施用任何适合的治疗性治疗。
另外,可向受试者进一步施用抗免疫抑制剂/免疫刺激剂,例如检查点抑制剂。举例来说,可向受试者进一步施用抗CTLA抗体或抗PD-1或抗PD-L1。通过抗体阻断CTLA-4或PD-L1可增强患者对癌细胞的免疫应答。具体来说,已显示在按照疫苗接种方案时CTLA-4阻断是有效的。
可以确定包括在疫苗组合物中的各抗原的最佳量和最佳给药方案。举例来说,可制备用于静脉内(i.v.)注射、皮下(s.c.)注射、皮内(i.d.)注射、腹膜内(i.p.)注射、肌肉内(i.m.)注射的抗原或其变体。注射方法包括s.c.、i.d.、i.p.、i.m.和i.v.。DNA或RNA注射方法包括i.d.、i.m.、s.c.、i.p.和i.v.。疫苗组合物的其它施用方法是本领域技术人员已知的。
疫苗可经编译以使得组合物中存在的抗原的选择、数量和/或量具有组织、癌症和/或患者特异性。举例来说,肽的精确选择可通过亲本蛋白质在给定组织中的表达模式来指导,或通过患者的突变状态来指导。选择可取决于癌症的具体类型、疾病状态、较早的治疗方案、患者免疫状态以及当然患者的HLA单倍型。此外,疫苗可根据特定患者的个人需要而含有个别化组分。实例包括根据抗原在特定患者中的表达改变抗原的选择或在第一轮或治疗方案之后调整二次治疗。
可以通过使用各种诊断方法(例如下面进一步描述的患者选择方法)鉴定施用抗原疫苗的患者。患者选择可以涉及鉴定一个或多个基因的突变或表达模式。在某些情况下,患者选择涉及鉴定患者的单倍型。可以并行执行各个患者选择方法,例如,测序诊断可以鉴定患者的突变和单倍型二者。可以依次执行多个患者选择方法,例如,一个诊断测试鉴定突变,并且另一个诊断测试鉴定患者的单倍型,并且其中每个测试可以是相同(例如,高通量测序)或不同(例如,一种高通量测序和另一个Sanger测序)诊断方法。
对于待用作癌症疫苗的组合物,在正常组织中大量表达的具有类似正常自身肽的抗原可以避免或以低量存在于本文所述的组合物中。另一方面,如果已知患者的肿瘤表达大量特定抗原,则用于治疗这种癌症的相应药学组合物可大量存在和/或可包括多于一种特异性针对这种特定抗原或这种抗原的途径的抗原。
可向已患有癌症的个体施用包含抗原的组合物。在治疗应用中,组合物以足以引发对肿瘤抗原的有效CTL反应且治愈或至少部分遏制症状和/或并发症的量施用于患者。足以实现此目标的量定义为“治疗有效剂量”。对这种用途有效的量将取决于例如组合物、施用方式、所治疗疾病的阶段和严重程度、患者的体重及一般健康状况,以及处方医师的判断。应记住,组合物一般可用于严重的疾病状态,也就是说,危及生命或可能危及生命的情形,尤其当癌症已转移时。在这些情况下,鉴于外来物质的最小化和抗原的相对无毒性,主治医师可能且可能感觉需要施用实质性过量的这些组合物。
对于治疗用途,可在检测或手术移除肿瘤时开始投药。此后是加强剂量,直到症状至少实质上减弱且此后持续一段时间。
用于治疗性治疗的药学组合物(例如疫苗组合物)意欲肠胃外、局部、经鼻、经口或局部施用。药学组合物可以经肠胃外施用,例如静脉内、皮下、皮内或肌肉内施用。组合物可在手术切除部位施用以诱导针对肿瘤的局部免疫应答。本文公开了用于肠胃外施用的组合物,其包含抗原和疫苗组合物溶解或悬浮于可接受的载体(例如水性载体)中的溶液。可使用多种水性载体,例如水、缓冲水、0.9%生理食盐水、0.3%甘氨酸、玻尿酸等。这些组合物可以通过常规的熟知灭菌技术灭菌,或者可经无菌过滤。所得水溶液可封装以按原样使用或冻干,冻干制剂在施用之前与无菌溶液组合。组合物可含有接近生理条件所需要的药学上可接受的辅助物质,例如pH调节剂和缓冲剂、张力调节剂、湿润剂等,例如乙酸钠、乳酸钠、氯化钠、氯化钾、氯化钙、脱水山梨糖醇单月桂酸酯、三乙醇胺油酸酯等。
抗原也可以经由脂质体施用,脂质体使其靶向特定的细胞组织,例如淋巴组织。脂质体也可用于增加半衰期。脂质体包括乳液、泡沫、胶束、不溶性单层、液晶、磷脂分散体、层状层等。在这些制剂中,有待递送的抗原作为脂质体的一部分单独或与结合于例如淋巴细胞中普遍存在的受体的分子(例如结合于CD45抗原的单克隆抗体)或与其它治疗性或免疫原性组合物一起并入。因此,用所需抗原填充的脂质体可引导至淋巴细胞的位点,在此脂质体然后递送经选择的治疗性/免疫原性组合物。脂质体可由标准的形成囊泡的脂质形成,其一般包括中性和带负电荷的磷脂和固醇(例如胆固醇)。脂质的选择一般通过考虑例如脂质体大小、脂质体在血流中的酸不稳定性和稳定性来指导。多种方法可用于制备脂质体,如例如Szoka等人,Ann.Rev.Biophys.Bioeng.9;467(1980);美国专利第4,235,871号、第4,501,728号、第4,501,728号、第4,837,028号和第5,019,369号中所述。
为了靶向免疫细胞,有待并入到脂质体中的配体可包括例如特异性针对所需免疫系统细胞的细胞表面决定子的抗体或其片段。脂质体悬浮液可以一定剂量静脉内、局部、表面等施用,该剂量尤其根据投药方式、所递送的肽以及所治疗疾病的阶段而变化。
出于治疗或免疫目的,编码肽和任选地一种或多种本文所述的肽的核酸也可施用于患者。多种方法方便地用于将核酸递送至患者。举例来说,核酸可以“裸DNA”形式直接递送。这种方法描述于例如Wolff等人,Science 247:1465-1468(1990)以及美国专利第5,580,859号和第5,589,466号中。核酸也可使用弹道式递送施用,如例如美国专利第5,204,253号中所述。可施用仅包含DNA的粒子。或者,DNA可黏附于粒子,例如金粒子。在存在或不存在电穿孔的情况下,用于递送核酸序列的方法可包括病毒载体、mRNA载体和DNA载体。
核酸也可以与阳离子化合物(例如阳离子脂质)复合递送。脂质介导的基因递送方法描述于例如9618372WOAWO 96/18372;9324640WOAWO 93/24640;Mannino和Gould-Fogerite,BioTechniques 6(7):682-691(1988);美国专利第5,279,833号;Rose美国专利第5,279,833号;9106309WOAWO 91/06309;以及Felgner等人,Proc.Natl.Acad.Sci.USA84:7413-7414(1987)中。
抗原还可以包括于基于病毒载体的疫苗平台中,例如牛痘、禽痘、自我复制甲病毒、马拉巴病毒、腺病毒(参见例如Tatsis等人,Adenoviruses,Molecular Therapy(2004)10,616-629)或慢病毒,包括(但不限于)第二、第三或杂交第二/第三代慢病毒以及任一代的重组慢病毒,其经设计以靶向特定细胞类型或受体(参见例如Hu等人,ImmunizationDelivered by Lentiviral Vectors for Cancer and Infectious Diseases,ImmunolRev.(2011)239(1):45-61;Sakuma等人,Lentiviral vectors:basic to translational,Biochem J.(2012)443(3):603-18;Cooper等人,Rescue of splicing-mediated intronloss maximizes expression in lentiviral vectors containing the humanubiquitin C promoter,Nucl.Acids Res.(2015)43(1):682-690;Zufferey等人,Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo Gene Delivery,J.Virol.(1998)72(12):9873-9880)。取决于上述基于病毒载体的疫苗平台的包装能力,这种方法可递送编码一个或多个抗原肽的一个或多个核苷酸序列。序列可侧接非突变序列,可由接头分开或可在前面有一个或多个靶向亚细胞区室的序列(参见例如Gros等人,Prospective identification of neoantigen-specific lymphocytes in theperipheral blood of melanoma patients,Nat Med.(2016)22(4):433-8;Stronen等人,Targeting of cancer neoantigens with donor-derived T cell receptorrepertoires,Science.(2016)352(6291):1337-41;Lu等人,Efficient identificationof mutated cancer antigens recognized by T cells associated with durabletumor regressions,Clin Cancer Res.(2014)20(13):3401-10)。在引入宿主中后,经感染细胞表达抗原,从而引发针对肽的宿主免疫(例如CTL)反应。可用于免疫方案中的牛痘载体和方法描述于例如美国专利第4,722,848号中。另一种载体是BCG(卡介苗)。BCG载体描述于Stover等人(Nature 351:456-460(1991))中。根据本文描述,可用于抗原的治疗性施用或免疫接种的各种其它疫苗载体,例如伤寒沙门氏菌载体等对于本领域技术人员将是显而易见的。
施用核酸的手段使用编码一个或多个表位的袖珍基因构建体。为产生编码经选择在人细胞中表达的CTL表位(袖珍基因)的DNA序列,逆翻译表位的氨基酸序列。使用人密码子使用表来指导各氨基酸的密码子选择。这些编码表位的DNA序列直接邻接,产生连续多肽序列。为了优化表达和/或免疫原性,可将额外元件并入到袖珍基因设计中。可经逆翻译且包括于袖珍基因序列中的氨基酸序列的实例包括:辅助T淋巴细胞、表位、前导(信号)序列和内质网滞留信号。另外,可通过包括相邻于CTL表位的合成(例如聚丙氨酸)或天然存在的侧接序列来改进CTL表位的MHC递呈。通过组装编码袖珍基因的正链和负链的寡核苷酸而将袖珍基因序列转化成DNA。重叠寡核苷酸(30-100个碱基长)是在适当条件下使用熟知技术合成、磷酸化、纯化和黏接的。寡核苷酸的末端使用T4 DNA连接酶连接。编码CTL表位多肽的这种合成袖珍基因可以然后克隆到所期望的表达载体中。
可制备经纯化的质粒DNA以便使用多种制剂注射。其中最简单的是冻干DNA在无菌磷酸盐缓冲生理食盐水(PBS)中复原。已描述多种方法,并且可使用新技术。如上文所指出,核酸宜用阳离子脂质配制。另外,统称为保护性、相互作用性、非缩合性(PINC)的糖脂、促融脂质体、肽和化合物也可与经纯化的质粒DNA复合以影响诸如以下变量:稳定性、肌肉内分散或运输至特定器官或细胞类型。
还公开了一种制造肿瘤疫苗的方法,其包括执行本文公开的方法的步骤;以及产生包含多个抗原或多个抗原的子集的肿瘤疫苗。
本文所公开的抗原可使用本领域中已知的方法制造。举例来说,产生本文所公开的抗原或载体(例如包括编码一个或多个抗原的至少一个序列的载体)的方法可包括在适于表达抗原或载体的条件下培养宿主细胞,其中该宿主细胞包含至少一个编码抗原或载体的多核苷酸;以及纯化抗原或载体。标准纯化方法包括色谱技术、电泳、免疫、沉淀、透析、过滤、浓缩和色谱聚焦技术。
宿主细胞可包括中国仓鼠卵巢(CHO)细胞、NS0细胞、酵母或HEK293细胞。宿主细胞可用一个或多个包含至少一个编码本文所公开的抗原或载体的核酸序列的多核苷酸转化,任选地其中该经分离的多核苷酸另外包含可操作地连接于编码抗原或载体的至少一个核酸序列的启动子序列。在某些实施方案中,经分离的多核苷酸可为cDNA。
VII.抗原使用和施用
可使用疫苗接种方案给予受试者一种或多种抗原。初免疫苗和加强疫苗可用于向受试者给药。初免疫苗可基于C68(例如SEQ ID NO:1或2中所示的序列)或srRNA(例如SEQID NO:3或4中所示的序列),并且加强疫苗可基于C68(例如SEQ ID NO:1或2中所示的序列)或srRNA(例如SEQ ID NO:3或4中所示的序列)。各载体通常包括包含抗原的盒。盒可包括约20种抗原,其由间隔子(例如通常包围各抗原的天然序列)或其它非天然间隔序列(例如AAY)分开。盒还可包括MHCII抗原,例如破伤风类毒素抗原和PADRE抗原,其可视为通用II类抗原。盒还可包括靶向序列,例如泛素靶向序列。另外,各疫苗剂量可与检查点抑制剂(CPI)结合(例如同时、之前或之后)施用于受试者。CPI可包括抑制CTLA4、PD1和/或PDL1的那些,例如抗体或其抗原结合部分。这些抗体可包括曲美木单抗或德瓦鲁单抗(durvalumab)。
初免疫苗可注射(例如肌肉内)于受试者。可使用每一剂量双侧注射。举例来说,可使用一次或多次ChAdV68(C68)注射(例如总剂量1×10
可在初免疫苗接种之后注射(例如肌肉内)疫苗加强剂(加强疫苗)。加强疫苗可在初免后约每1、2、3、4、5、6、7、8、9或10周,例如每4周和/或8周施用。可使用每一剂量双侧注射。举例来说,可使用一次或多次ChAdV68(C68)注射(例如总剂量1×10
也可向受试者施用抗CTLA-4(例如曲美木单抗)。举例来说,抗CTLA4可在肌肉内疫苗注射(ChAdV68初免或srRNA低剂量)部位附近皮下施用,以确保引流至同一淋巴结。曲美木单抗为CTLA-4的选择性人IgG2 mAb抑制剂。目标抗CTLA-4(曲美木单抗)皮下剂量通常为70-75mg(特别是75mg),其剂量范围为例如1-100mg或5-420mg。
在某些情况下,可使用抗PD-L1抗体,例如德瓦鲁单抗(MEDI 4736)。德瓦鲁单抗为一种选择性、高亲和力人IgG1 mAb,其阻断PD-L1结合于PD-1和CD80。德瓦鲁单抗一般每4周以20mg/kg i.v.施用。
可在疫苗施用之前、期间和/或之后进行免疫监测。除其它参数外,这种监测可告知安全性和功效。
为了进行免疫监测,通常使用PBMC。PBMC可在初免疫苗接种之前以及在初免疫苗接种之后(例如4周及8周)分离。PBMC可仅在加强疫苗接种之前以及在每次加强疫苗接种之后(例如4周和8周)收集。
可评定T细胞反应作为免疫监测方案的一部分。可使用本领域中已知的一种或多种方法测量T细胞反应,例如ELISpot、细胞内细胞因子染色、细胞因子分泌和细胞表面捕捉、T细胞增殖、MHC多聚体染色或通过细胞毒性分析。针对疫苗中编码的表位的T细胞反应可通过使用ELISpot分析测量细胞因子(例如IFN-γ)的诱导而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8 T细胞反应可通过使用流式细胞测量术测量胞内或胞外捕捉的细胞因子(例如IFN-γ)的诱导而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8T细胞反应可通过使用MHC多聚体染色测量表达特异性针对表位/MHC I类复合物的T细胞受体的T细胞群体而从PBMC监测。针对疫苗中编码的表位的特异性CD4或CD8 T细胞反应可通过在3H-胸苷、溴脱氧尿苷和羧基荧光素-二乙酸酯-琥珀酰亚胺酯(CFSE)并入后测量T细胞群的离体扩增而从PBMC监测。特异性针对疫苗中编码的表位的源于PBMC的T细胞的抗原识别能力和溶解活性可通过铬释放分析或替代性比色细胞毒性分析来功能性评定。
VIII.抗原鉴定
VIII.A.抗原候选物鉴定
已描述关于肿瘤及正常外显子组及转录组的NGS分析的研究方法且将其应用于抗原鉴定空间。
VIII.B.HLA肽的分离和检测
在裂解及溶解组织样品后,使用经典免疫沉淀(IP)方法进行HLA-肽分子的分离(55-58)。澄清的溶解物用于HLA特异性IP。
免疫沉淀使用与珠粒偶联的抗体来进行,其中抗体特异性针对HLA分子。对于泛I类HLA免疫沉淀,使用泛I类CR抗体,对于II类HLA-DR,使用HLA-DR抗体。在过夜温育期间,抗体共价连接于NHS-琼脂糖珠粒。在共价连接后,将珠粒洗涤并等分用于IP。(59,60)免疫沉淀也可以用未共价结合至珠粒的抗体进行。通常使用包被有蛋白A和/或蛋白G的琼脂糖或磁性珠粒将抗体固定在柱上来完成此操作。下面列出了一些可用于选择性富集MHC/肽复合物的抗体。
将澄清的组织溶解物添加至抗体珠粒中进行免疫沉淀。在免疫沉淀后,从溶解物移除珠粒且将溶解物储存用于额外实验,包括额外IP。洗涤IP珠粒以移除非特异性结合且使用标准技术从珠粒洗脱HLA/肽复合物。使用分子量旋转柱或C18分级分离从肽移除蛋白质组分。所得肽通过SpeedVac蒸发变干且在一些情况下,在MS分析之前储存在-20C下。
干燥的肽在适于反相色谱法的HPLC缓冲液中复原且装载于C-18微毛细管HPLC柱上,以便在Fusion Lumos质谱仪(Thermo)中梯度洗脱。在Orbitrap检测器中以高分辨率收集肽质量/电荷(m/z)的MS1谱,接着在经选择的离子的HCD片段化后,在离子阱检测器中收集MS2低分辨率扫描。另外,可使用CID或ETD片段化方法或三种技术的任何组合来获得MS2谱,以达到肽的更大的氨基酸覆盖率。MS2谱也可在Orbitrap检测器中以高分辨率质量精度测量。
使用Comet(61,62)对来自各分析的MS2谱进行蛋白质数据库搜索,并且使用Percolator(63-65)对肽鉴定进行评分。使用PEAKS studio(Bioinformatics SolutionsInc.)进行其它测序,并且可以使用其它搜索引擎或测序方法,包括光谱匹配和从头测序(97)。
VIII.B.1.支持综合HLA肽测序的MS检测限值研究
使用肽YVYVADVAAK,使用装载于LC柱上的不同量的肽确定检测限值。所测试的肽的量为1pmol、100fmol、10fmol、1fmol和100amol。(表1)结果示于图24A和24B中。这些结果表明,最低检测限值(LoD)在埃摩尔范围(10
表1
IX.递呈模型
递呈模型可以用于鉴定患者中肽递呈的可能性。多种递呈模型是本领域技术人员已知的,例如在国际专利申请公开WO/2017/106638、WO/2018/195357、WO/2018/208856、WO2016187508和美国专利申请US20110293637中更详细描述的递呈模型,其出于所有目的以全文引用的方式并入本文中。
X.训练模块
训练模块可用于基于训练数据集构建一个或多个递呈模型,其产生肽序列是否将由与肽序列相关的MHC等位基因递呈的可能性。多种训练模块对于本领域技术人员而言是已知的,例如在国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细描述的递呈模型,其出于所有目的以全文引用的方式并入本文中。训练模块可以构建递呈模型,以在独立等位基因的基础上预测递呈可能性。训练模块可以构建递呈模型,以在存在两个或多个MHC等位基因的多等位基因设置中预测肽的递呈可能性。
XI.预测模块
预测模块可用于接受序列数据并使用递呈模型选择序列数据中的候选抗原。具体来说,序列数据可以是从患者的肿瘤组织细胞提取的DNA序列、RNA序列和/或蛋白质序列。预测模块可以通过将从患者的正常组织细胞提取的序列数据与从患者的肿瘤组织细胞提取的序列数据相比较以鉴定含有一个或多个突变的部分,由此鉴定出呈突变肽序列的候选新抗原。预测模块可以通过将从患者的正常组织细胞提取的序列数据与从患者的肿瘤组织细胞提取的序列数据相比较来鉴定与正常细胞或组织相比在肿瘤细胞或癌组织中具有改变的表达的候选抗原,以鉴定不适当表达的候选抗原。
递呈模块可将一个或多个递呈模型应用于经处理的肽序列以估计所述肽序列的递呈可能性。具体来说,预测模块可以通过将递呈模型应用于候选抗原来选择一个或多个可能在肿瘤HLA分子上递呈的候选抗原肽序列。在一个实施方式中,递呈模块选出估计递呈可能性超过预定阈值的候选抗原序列。在另一实施方式中,递呈模型选出N个具有最高估计递呈可能性的候选抗原序列(其中N一般是可以在疫苗中递送的最大表位数量)。包括选择用于给定患者的候选抗原的疫苗可以注射至患者体内以诱导免疫应答。
XI.B.盒设计模块
XI.B.1综述
盒设计模块可用于基于选择用于注射到患者体内的候选肽来产生疫苗盒序列。多种盒设计模块对于本领域技术人员而言是已知的,例如国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的盒设计模块,其出于所有目的以全文引用的方式并入本文中。
可以基于由预测模块确定的与超过预定阈值的递呈可能性相关联的所选肽产生治疗性表位集合,其中所述递呈可能性由递呈模型测定。然而,应了解,在其它实施方案中,可以基于多种方法中的任一种或多种(单独或组合形式),例如基于针对患者的I类或II类HLA等位基因的结合亲和力或预测的结合亲和力、针对患者的I类或II类HLA等位基因的结合稳定性或预测的结合稳定性、随机取样等产生治疗性表位的集合。
治疗性表位可以对应于所选肽本身。除所选肽外,治疗性表位还可包括C端和/或N端侧接序列。N端和C端侧接序列可以是处于源蛋白质环境中的治疗性疫苗表位的原生N端和C端侧接序列。治疗性表位可以表示固定长度的表位。治疗性表位可以表示可变长度的表位,其中表位的长度可以取决于例如C-或N-侧接序列的长度而变化。举例来说,C端侧接序列和N端侧接序列各自可具有2-5个残基的变化的长度,由此产生16种可能的表位选择。
盒设计模块也可以通过考虑横跨该盒中一对治疗性表位之间的接合点的接合点表位的递呈来产生盒序列。接合点表位由于在该盒中串接治疗性表位和接头序列的过程而在该盒中产生的新颖非自身但不相关的表位序列。接合点表位的新颖序列不同于该盒的治疗性表位本身。
盒设计模块可以产生降低在患者中递呈接合点表位的可能性的盒序列。具体来说,当将盒注射至患者体内时,接合点表位有可能被患者的HLA I类或HLA II类等位基因递呈,并且分别刺激CD8或CD4 T细胞反应。由于T细胞与接合点表位的反应没有治疗益处,且可能因抗原竞争而减弱针对该盒中所选治疗性表位的免疫应答,故此类反应常常是不合需要的。
盒设计模块可以迭代一个或多个候选盒,且确定与盒序列相关联的接合点表位的递呈评分低于数字阈值的盒序列。接合点表位递呈评分是与该盒中接合点表位的递呈可能性相关联的量,并且较高的接合点表位递呈评分值指示该盒的接合点表位会被HLA I类或HLA II类或两者递呈的可能性较高。
在一个实施方案中,盒设计模块可以确定候选盒序列中与最低接合点表位递呈评分相关联的盒序列。
盒设计模块可以迭代一个或多个候选盒序列,确定候选盒的接合点表位递呈评分,且鉴定与低于阈值的接合点表位递呈评分相关联的最佳盒序列。
盒设计模块可以进一步检查该一个或多个候选盒序列以鉴定候选盒序列中的接合点表位中的任一个是否为设计使用该疫苗的给定患者的自身表位。为实现此目的,盒设计模块针对已知数据库如BLAST检查接合点表位。在一个实施方案中,盒设计模块可以配置成设计避免接合点自身表位的盒。
盒设计模块可以执行蛮力方法且迭代全部或大部分可能的候选盒序列以选出具有最小接合点表位递呈评分的序列。然而,由于疫苗容量增加,因此这类候选盒的数量可能极大。举例来说,对于20个表位的疫苗容量,盒设计模块必须迭代约10
盒设计模块可以产生随机产生或至少伪随机产生的候选盒的子集,且选择与低于预定阈值的接合点表位递呈评分相关联的候选盒作为盒序列。另外,盒设计模块可以从该小组中选择具有最低接合点表位递呈评分的候选盒作为盒序列。举例来说,盒设计模块可以针对20个所选表位集合产生约1百万个候选盒子集,且选出具有最小接合点表位递呈评分的候选盒。尽管产生随机盒序列子集并从该子集中选出具有低接合点表位递呈评分的盒序列可能不如蛮力方法好,但其需要明显较少的计算资源,由此使其实施在技术上是可行的。另外,相对于这种更高效的技术,执行蛮力方法可能仅引起接合点表位递呈评分的微小或甚至可忽略的改进,因此,从资源分配的观点看,蛮力方法是不值得实施的。盒设计模块可以通过将盒的表位序列用非对称旅行商问题(TSP)公式表示来确定改进的盒配置。鉴于节点列表和每对节点之间的距离,TSP确定与最短总距离相关联的节点的序列以访问每个节点恰好一次且返回至原始节点。举例来说,根据城市A、B及C且已知彼此之间的距离,TSP的解决方案产生一个闭合的城市序列,对于该序列,访问每个城市恰好一次所行进的总距离系可能途径当中最短的。TSP的非对称形式确定当一对节点之间的距离不对称时节点的最佳序列。举例来说,从节点A行进至节点B的“距离”可以不同于从节点B行进至节点A的“距离”。通过使用非对称TSP解决改进的最佳盒,盒设计模块可以寻找使所有在该盒的表位之间的接合点的递呈评分降低的盒序列。非对称TSP解决方案指示对应于应当在盒中串接表位以使该盒的所有接合点的接合点表位递呈评分减到最小的次序的治疗性表位序列。相较于随机取样方法,经由此方法确定的盒序列可以产生具有明显较少接合点表位递呈的序列,同时可能需要明显较少的计算资源,尤其是在所产生的候选盒序列数量很大时。在国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述了用于优化盒设计的不同计算方法和比较的说明性实例,其出于所有目的以全文引用的方式并入本文中。
XI.B.2.共有抗原疫苗序列选择
本领域技术人员可以使用本文提供的详细公开内容选择包含在共有抗原疫苗中的共有抗原序列以及用这种疫苗治疗的合适患者。例如,表A、1.2或AACR GENIE结果可用于序列选择。在某些情况下,特定突变和HLA等位基因组合可能是优选的(例如,基于可从给定受试者获得的测序数据,其指示每个均存在于受试者中),并且随后组合在一起来使用表A或AACR GENIE结果鉴定用于包含在疫苗中的共有新抗原序列。示例性突变及其匹配的HLA等位基因显示在表32和34中。
例如,对于KRAS_G13D,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出KRAS_G13D和C0802的行来选择考虑包含的每个相关序列。
例如,对于KRAS_Q61K或NRAS_Q61K,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61K和A0101或(2)NRAS Q61K和A0101的行来选择考虑包含的每个相关序列。
例如,对于TP53_R249M,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出TP53_R249M以及B3512、B3503和B3501中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于CTNNB1_S45P,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S45P以及A0101、A0301、B5701、A6801、A0302和A1101中的至少一个的行来选择考虑包含的每个相关序列。例如,参见32中所示的相关序列。
例如,对于CTNNB1_S45F,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S45F以及A0301、A1101和A6801中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于ERBB2_Y772_A775dup,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出ERBB2_Y772_A775dup和B1801的行来选择考虑包含的每个相关序列。
例如,对于KRAS_G12D或NRAS_G12D,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_G12D以及A1101和C0802中的至少一个或(2)NRAS_G12D以及A1101和C0802中的至少一个的行来选择考虑包含的每个相关序列。例如,参见表32中所示的相关序列。
例如,对于KRAS_Q61R或NRAS_Q61R,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61R和A0101或(2)NRAS_Q61R和A0101的行来选择考虑包含的每个相关序列。
例如,对于CTNNB1_T41A,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_T41A以及A0301、A0302、A1101、B1510、C0303和C0304中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于TP53_K132N,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出TP53_K132N以及A2402和A2301中的至少一个的行来选择考虑包含的每个相关序列。例如,参见表32中所示的相关顺序。
例如,对于KRAS_G12A,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出KRAS_G12A和A0301的行来选择考虑包含的每个相关序列。例如,参见32中所示的相关顺序。
例如,对于KRAS_Q61L或NRAS_Q61L,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61L和A0101或(2)NRAS_Q61L和A0101的行来选择考虑包含的每个相关序列。
例如,对于TP53_R213L,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出TP53_R213L以及A0207、C0802和A0201中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于BRAF_G466V,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出BRAF_G466V以及B1501和B1503中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于KRAS_G12V,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出KRAS_G12V以及A0301、A1101、A3101、C0102和A0302中的至少一个的行来选择考虑包含的每个相关序列。例如,参见表32中所示的相关序列。
例如,对于KRAS_Q61H或NRAS_Q61H,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61H和A0101或(2)NRAS_Q61H和A0101的行来选择考虑包含的每个相关序列。
例如,对于CTNNB1_S37F,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S37F以及A2301、A2402、B1510、B3906、C0501、C1402和C1403中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于TP53_S127Y,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出TP53_S127Y以及A1101和A0301中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于TP53_K132E,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出TP53_K132E以及A2402、C1403和A2301中的至少一个的行来选择考虑包含的每个相关序列。
例如,对于KRAS_G12C或NRAS_G12C,可以通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原或共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_G12C和A0201或(2)NRAS_G12C和A0201的行来选择考虑包含的每个相关序列。例如,参见32中所示的相关序列。
XIII.示例计算机
计算机可以用于本文描述的任何计算方法。本领域技术人员将认识到计算机可以具有不同的体系结构。计算机的实例是本领域技术人员已知的,例如在国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中更详细地描述的计算机,其出于所有目的以全文引用的方式并入本文中。
XIV.抗原递送载体实施例
以下是执行本发明的特定实施方案的实施例。所述实施例仅出于说明目的提供,而不打算以任何方式限制本发明的范围。已作出努力来确保所使用数字(例如量、温度等)的精确性,但一些实验误差和偏差当然应是允许的。
除非另外指明,否则本发明将采用在本领域技能范围内的蛋白质化学、生物化学、重组DNA技术和药理学常规方法实行。所述技术在文献中有充分解释。参见例如T.E.Creighton,Proteins:Structures and Molecular Properties(W.H.Freeman andCompany,1993);A.L.Lehninger,Biochemistry(Worth Publishers,Inc.,现行版);Sambrook等人,Molecular Cloning:A Laboratory Manual(第2版,1989);Methods InEnzymology(S.Colowick和N.Kaplan编,Academic Press,Inc.);Remington′sPharmaceutical Sciences,第18版(Easton,Pennsylvania:Mack Publishing Company,1990);Carey和Sundberg Advanced Organic Chemistry第3版(Plenum Press)第A和B卷(1992)。
XIV.A.新抗原盒设计
可以通过疫苗接种递送刺激相应细胞免疫应答的多个I类MHC限制性肿瘤特异性新抗原(TSNA)。在一个实例中,疫苗盒经工程改造而以单一基因产物形式编码多个表位,其中所述表位系嵌入其天然的周围肽序列内或通过非天然接头序列隔开。鉴定出会潜在地影响抗原加工和递呈且因此影响TSNA特异性CD8 T细胞反应的量值和广度的若干设计参数。在本实例中,设计并构建出若干模型盒以评估:(1)是否可以针对并入单一表达盒中的多个表位产生稳定T细胞反应;(2)什么使得最佳接头置放于表达盒内的TSNA之间,引起所有表位的最佳加工和递呈;(3)所述表位在盒内的相对位置是否影响T细胞反应;(4)盒内表位的数量是否影响针对个别表位的T细胞反应的量值或质量;(5)添加细胞靶向序列是否改善T细胞反应。
产生两个读取结果以评估抗原递呈和对模型盒内的标志物表位具有特异性的T细胞反应:(1)体外基于细胞的筛选,其允许通过专门工程改造的报告T细胞的活化进行衡量,来评定抗原递呈(Aarnoudse等人,2002;Nagai等人,2012);以及(2)使用HLA-A2转基因小鼠(Vitiello等人,1991),通过其相应表位特异性T细胞反应评定盒来源的人源表位的疫苗接种后免疫原性的体内分析(Cornet等人,2006;Depla等人,2008;Ishioka等人,1999)。
XIV.B.抗原盒设计评估
XIV.B.1.方法与材料
TCR和盒设计及克隆
当通过A*0201递呈时,所选TCR识别肽NLVPMVATV(PDB#5D2N)、CLGGLLTMV(PDB#3REV)、GILGFVFTL(PDB#1OGA)、LLFGYPVYV(PDB#1AO7)。构建含有2A肽连接的TCR次单元(β然后是α)、EMCV IRES和2A连接的CD8次单元(β然后是α和嘌呤霉素抗性基因)的转移载体。对开放阅读框序列进行密码子优化且由GeneArt合成。
产生用于体外表位加工和递呈研究的细胞系
肽购自ProImmune或Genscript,在含10mM三(2-羧基乙基)膦(TCEP)的水/DMSO(2:8,v/v)中稀释至10mg/mL。除非另外指出,否则细胞培养基和补充剂是来自Gibco。热灭活胎牛血清(FBShi)来自Seradigm。QUANTI-Luc底物、吉欧霉素(Zeocin)和嘌呤霉素来自InvivoGen。将Jurkat-Lucia NFAT细胞(InvivoGen)维持在补充有10%FBShi、丙酮酸钠和100μg/mL吉欧霉素的RPMI 1640中。转导后,这些细胞立即另外接受0.3μg/mL嘌呤霉素。在伊氏培养基(Iscove's Medium,IMDM)加20%FBShi中培养T2细胞(ATCC CRL-1992)。U-87MG(ATCC HTB-14)细胞维持在补充有10%FBShi的MEM伊格尔培养基(MEM Eagles Medium)中。
Jurkat-Lucia NFAT细胞含有NFAT诱导性Lucia报告构建体。Lucia基因在通过接合T细胞受体(TCR)活化时,将利用腔肠素的荧光素酶分泌至培养基中。这种荧光素酶可使用QUANTI-Luc荧光素酶检测试剂测量。用慢病毒转导Jurkat-Lucia细胞以表达抗原特异性TCR。HIV源性慢病毒转移载体是从GeneCopoeia获得,并且表达VSV-G(pCMV-VsvG)、Rev(pRSV-Rev)和Gag-pol(pCgpV)的慢病毒支持质粒(support plasmid)是从Cell DesignLabs获得。
通过使用40μl脂染胺和20μg DNA混合物(以重量计4:2:1:1的转移质粒:pCgpV:pRSV-Rev:pCMV-VsvG),用脂染胺2000(Thermo Fisher)转染T75烧瓶中50-80%汇合的HEK293细胞,来制备慢病毒。使用Lenti-X系统(Clontech)浓缩8-10mL含病毒的培养基,且使病毒再悬浮于100-200μl新鲜培养基中。使用此体积覆盖相等体积的Jurkat-Lucia细胞(在不同实验中使用5×10E4-1×10E6个细胞)。在含0.3μg/ml嘌呤霉素的培养基中培养之后,分选细胞以获得克隆性。使用装载肽的T2细胞测试这些Jurkat-Lucia TCR克隆的活性和选择性。
体外表位加工和递呈分析
常规地使用T2细胞,通过TCR检查抗原识别。T2细胞缺乏用于抗原加工的肽转运蛋白(TAP缺陷型)并且不能在内质网中装载内源性肽以在MHC上递呈。然而,T2细胞可以容易地装载外源肽。将五个标志物肽(NLVPMVATV、CLGGLLTMV、GLCTLVAML、LLFGYPVYV、GILGFVFTL)以及两个不相关的肽(WLSLLVPFV、FLLTRICT)装载至T2细胞上。简言之,对T2细胞计数且用IMDM加1%FBShi稀释至1×106个细胞/毫升。添加肽以产生10μg肽/1×106个细胞。接着在37℃下温育细胞90分钟。用IMDM加20%FBShi洗涤细胞两次,稀释至5×10E5个细胞/毫升并将100μL涂铺至96孔Costar组织培养板中。对Jurkat-Lucia TCR克隆计数并在RPMI 1640加10%FBShi中稀释至5×10E5个细胞/毫升,且将100μL添加至T2细胞中。培养板在37℃和5%CO2下温育过夜。然后以400g离心培养板3分钟并将20μL上清液移至白色平底Greiner板中。QUANTI-Luc底物是根据说明书制备且以每孔50μL添加。在MolecularDevices SpectraMax iE3x上读取荧光素酶表达。
为了测试腺病毒盒的标志物表位递呈,使用U-87MG细胞作为替代抗原递呈细胞(APC)且用腺病毒载体转导。收集U-87MG细胞并以5×10E5个细胞/100μl涂铺于96孔Costar组织培养板中的培养基中。在37℃温育培养板约2小时。用MEM加10%FBShi将腺病毒盒稀释至MOI为100、50、10、5、1和0并将其以每孔5μl添加至U-87MG细胞中。再在37℃下温育培养板约2小时。对Jurkat-Lucia TCR克隆计数并在RPMI加10%FBShi中稀释至5×10E5个细胞/毫升,且将其以每孔100μL添加至U-87MG细胞中。接着,在37℃及5%CO2下温育培养板约24小时。以400g离心培养板3分钟并将20μL上清液移至白色平底Greiner板中。QUANTI-Luc底物是根据说明书制备且以每孔50μL添加。在Molecular Devices SpectraMax iE3x上读取荧光素酶表达。
用于免疫原性研究的小鼠品系
转基因HLA-A2.1(HLA-A2 Tg)小鼠是从Taconic Labs,Inc获得。这些小鼠携带由嵌合I类分子组成的转基因,该嵌合I类分子包含人HLA-A2.1前导序列、α1和α2结构域以及鼠科H2-Kbα3、跨膜和细胞质域(Vitiello等人,1991)。用于所述研究的小鼠是基于C57B1/6背景的野生型BALB/cAnNTac雌性和纯合HLA-A2.1Tg雄性的第一代后代(F1)。
腺病毒载体(Ad5v)免疫接种
经由两侧肌肉内注射至胫前肌中对HLA-A2 Tg小鼠免疫接种1×10
淋巴细胞分离
从新鲜收集的经免疫接种小鼠的脾和淋巴结分离淋巴细胞。使用GentleMACS组织解离器,根据制造商的说明书,在含有10%胎牛血清与青霉素和链霉素的RPMI(完全RPMI)中解离组织。
离体酶联免疫斑点(enzyme-linked immunospot,ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则(Janetzki等人,2015),利用小鼠IFNgELISpotPLUS试剂盒(MABTECH)进行。将1×10
离体细胞内细胞因子染色(ICS)和流式细胞测量术分析
将密度为2-5×10
XIV.B.2.抗原盒设计的体外评估
作为抗原盒设计评估的实例,开发体外基于细胞的分析以评定在模型疫苗盒内的所选人表位是否被抗原递呈细胞表达、加工和递呈(图1)。在识别后,经工程改造成表达五种对明确表征的肽-HLA组合具有特异性的TCR之一的Jurkat-Lucia报告T细胞变得活化且将活化T细胞核因子(NFAT)易位至核中,引起荧光素酶报告基因的转录活化。通过生物发光定量个别报告CD8 T细胞系的抗原刺激。
通过用表达构建体转导慢病毒来改进个别Jurkat-Lucia报告株,该表达构建体包括通过P2A核糖体跳跃序列(skip sequence)分离以确保等摩尔量翻译产物的抗原特异性TCRβ和TCRα链(Banu等人,2014)。将第二CD8β-P2A-CD8α元件添加至慢病毒构建体中提供亲代报告细胞系所缺乏的CD8辅助受体的表达,因为细胞表面上的CD8对于与靶pMHC分子的结合亲和力至关重要并且通过接合其胞质尾区增强信号传导(Lyons等人,2006;Yachi等人,2006)。
在慢病毒转导之后,使Jurkat-Lucia报告细胞在嘌呤霉素选择下扩增,经历单细胞荧光辅助细胞分选(FACS)且测试单克隆群的荧光素酶表达。由此得到具有功能性细胞反应的针对特定肽抗原1、2、4和5的稳定转导的报告细胞系。(表2)。
表2:体外T细胞活化分析的发展。如通过荧光素酶的诱导所测量的肽特异性T细胞识别指示疫苗盒抗原的有效加工和递呈。
*尚未产生的针对表位3的报告T细胞
在另一实例中,对于一系列短盒,所有标志物表位均并入相同位置中(图2A)且仅分隔HLA-A*0201限制性表位的接头(图2B)是变化的。将报告T细胞个别地与被表达这些短盒的腺病毒构建体感染的U-87抗原递呈细胞(APC)混合,并且相对于未感染对照组测量荧光素酶表达。通过匹配报告T细胞识别模型盒中的全部四个抗原,展示多个抗原的有效加工和递呈。T细胞反应的量值在很大程度上遵循天然和AAY-接头的类似趋势。从基于RR-接头的盒释放的抗原显示较低荧光素酶诱导(表3)。被设计以破坏抗原加工的DPP-接头制造的疫苗盒引起低表位递呈(表3)。
表3:短盒中接头序列的评估。在体外T细胞活化分析中的荧光素酶诱导指示,除基于DPP的盒外,所有接头都有助于盒抗原的有效释放。仅T细胞表位(无接头)=9AA,天然接头一侧=17AA,天然接头两侧=25AA,非天然接头=AAY、RR、DPP
*尚未产生的针对表位3的报告T细胞
在另一实例中,构建另外一系列的短盒,所述盒除人和小鼠表位外,还含有定位于该盒的N端或C端上的靶向序列如泛素(Ub)、MHC和Ig-κ信号肽(SP)和/或MHC跨膜(TM)基序。(图3)。当通过腺病毒载体递送至U-87APC时,报告T细胞再次展示多个盒源性抗原的有效加工和递呈。然而,各种靶向特征对于T细胞反应的量值无实质影响(表4)。
表4:添加至模型疫苗盒的细胞靶向序列的评估。采用体外活化分析证实,四个HLA-A*0201限制性标志物表位从模型盒有效释放并且靶向序列没有实质改善T细胞识别和活化。
*尚未产生的针对表位3的报告T细胞
XIV.B.3.抗原盒设计的体内评估
作为抗原盒设计评估的另一实例,疫苗盒经设计以含有5个已知以HLA-A*02:01限制性方式刺激CD8 T细胞的明确表征的人I类MHC表位(图2A、3、5A)。为了评估体内免疫原性,将含有这些标志物表位的疫苗盒并入腺病毒载体中并用于感染HLA-A2转基因小鼠(图4)。该小鼠模型携带的转基因部分地由人HLA-A*0201和小鼠H2-Kb组成,因此编码由人HLA-A2.1前导序列、连接至鼠类α3的α1和α2域、跨膜和细胞质H2-Kb域组成的嵌合I类MHC分子(Vitiello等人,1991)。所述嵌合分子允许HLA-A*02:01限制性抗原递呈,同时维持CD8辅助受体与MHC上的α3域的物种相配的相互作用。
对于短盒,如通过IFN-γELISPOT所测定,所有标志物表位产生T细胞反应,其程度比通常所报道的程度强约10-50倍(Cornet等人,2006;Depla等人,2008;Ishioka等人,1999)。在评估的所有接头中,各自含有通过天然氨基酸序列侧接的极小表位的25聚体序列多联体产生最大且最广泛的T细胞反应(表5)。细胞内细胞因子染色(ICS)和流式细胞测量术分析揭示,抗原特异性T细胞反应是源自CD8 T细胞。
表5:短盒中接头序列的体内评估。ELISPOT数据指示,HLA-A2转基因小鼠在用1e11腺病毒病毒粒子感染后17天,针对盒中的所有I类MHC限制性表位产生T细胞反应。
在另一实例中,构建一系列长疫苗盒并将其并入腺病毒载体中,其紧邻着原始的5个标志物表位含有另外16个具有已知CD8 T细胞反应性的HLA-A*02:01、A*03:01和B*44:05表位(图5A、B)。这些长盒的尺寸近似地模仿最终临床盒设计,并且只有表位相对于彼此的位置是不同的。对于长疫苗盒与短疫苗盒,CD8 T细胞反应在量值及广度方面是相当的,证实(a)添加更多表位不会实质影响针对原始表位集合的免疫应答的量值,以及(b)表位在盒中的位置不会实质影响随之而来的针对其的T细胞反应(表6)。
表6:有关长盒中表位位置的影响的体内评估。ELISPOT数据指示,对于长疫苗盒与短疫苗盒,HLA-A2转基因小鼠在用5e10腺病毒病毒粒子感染后17天,产生的T细胞反应的量值相当。
*疑似技术失误引起T细胞反应的缺乏。
XIV.B.4.用于免疫原性和毒理学研究的抗原盒设计
总体而言,有关模型盒评估的研究结果(图2-5,表2-6)证实,对于模型疫苗盒,当采用“珠串(string of beads)”法在基于腺病毒的载体的背景下编码约20个表位时,实现强大的免疫原性。所述表位通过串接25聚体序列组装,所述序列各自嵌入在两侧上通过其天然、周围肽序列(例如在每一侧上的8个氨基酸残基)侧接的极小CD8 T细胞表位(例如9个氨基酸残基)。如本文所用,“天然”或“原生”侧接序列是指给定表位在该表位处于其源蛋白质内的天然存在环境中的N端和/或C端侧接序列。举例来说,HCMV pp65 MHC I表位NLVPMVATV通过原生5'序列WQAGILAR侧接于其5'端上且通过原生3'序列QGQNLKYQ侧接于其3'端上,由此产生在HCMV pp65源蛋白质内发现的25聚体肽WQAGILARNLVPMVATVQGQNLKYQ。天然或原生序列也可指编码通过原生侧接序列侧接的表位的核苷酸序列。每个25聚体序列直接连接至后面的25聚体序列。在极小CD8 T细胞表位大于或小于9个氨基酸的实例中,侧接肽长度可以经调整以使得总长度仍为25聚体肽序列。举例来说,10个氨基酸的CD8 T细胞表位可以通过8个氨基酸的序列和7个氨基酸侧接。多联体之后是两个通用的II类MHC表位,包括所述表位是为了刺激CD4 T辅助细胞并改善疫苗盒抗原的总体体内免疫原性。(Alexander等人,1994;Panina-Bordignon等人,1989)II类表位通过GPGPG氨基酸接头(SEQID NO:56)连接至最终I类表位。该两个II类表位也通过GPGPG氨基酸接头彼此连接且通过GPGPG氨基酸接头侧接在C端上。表位的位置和数量似乎都不会实质影响T细胞识别或反应。靶向序列似乎也不会实质影响盒源性抗原的免疫原性。
作为另一实例,基于用模型盒获得的体外及体内数据(图2-5,表2-6),产生交替已知在非人灵长类动物(NHP)、小鼠和人中具有免疫原性的明确表征的T细胞表位的盒设计。全部嵌入天然25聚体序列中的20个表位之后是存在于所评估的所有模型盒中的两个通用II类MHC表位(图6)。使用这种盒设计在多个物种中研究免疫原性以及药理学和毒理学研究。
XIV.B.5.对于30、40和50抗原的抗原盒设计和评估
设计具有30(L)、40(XL)或50(XXL)个表位的大抗原盒,每个表位的长度为25个氨基酸。这些表位是人、NHP和小鼠表位的混合物,以模拟疾病抗原,包括肿瘤抗原。图29示出了来自多种物种的表位的一般组织。表37、38和39分别描述了对于人、灵长类动物和小鼠模型表位使用的模型抗原。表37、表38和表39各自描述了表位的位置、名称、最小表位描述和MHC分类。
如所描述的,将这些盒克隆到chAd68和甲病毒疫苗载体中,以评估更长的多表位盒的功效。图30显示了由ChAdV载体表达了每个大抗原盒,如通过蛋白质印迹由预期大小的至少一个主要条带所指示的。
如所描述的,对小鼠进行免疫以评估大盒的功效。对于表位AH1(上图)和SINNFEKL(下图),在用chAd68载体免疫后通过ICS和四聚体染色(分别为图31/表40和图32/表41)以及在用srRNA载体免疫后通过ICS(图33/表42)分析T细胞应答。使用表达30(L)、40(XL)或50(XXL)个表位的chAd68和srRNA疫苗载体的免疫诱导了对模型疾病表位的CD8+免疫应答。
表37-大盒中的人表位
表38-大盒中的NHP表位
表39-大盒中的小鼠表位
表40:ChAd大盒处理的小鼠中对AH1和SIINFEKL肽应答的平均IFNg+细胞。数据表示为总CD8细胞的百分比。显示的是每组的平均值和标准偏差以及通过ANOVA和Tukey检验的p值。所有p值与MAG 20抗原盒比较。
表41:ChAd大盒处理的小鼠中对于AH1和SIINFEKL抗原的平均四聚体+细胞。数据表示为总CD8细胞的百分比。显示的是每组的平均值和标准偏差以及通过ANOVA和Tukey检验的p值。所有p值与MAG 20抗原盒比较。
表42:SAM大盒处理的小鼠中对AH1和SIINFEKL肽应答的平均IFNg+细胞。数据表示为总CD8细胞的百分比。显示的是每组的平均值和标准偏差以及通过ANOVA和Tukey检验的p值。所有p值与MAG 20抗原盒比较。
XV.ChAd抗原盒递送载体
XV.A.ChAd抗原盒递送载体构建
在一个实例中,将黑猩猩腺病毒(ChAd)工程改造成用于抗原盒的递送载体。在另一实例中,基于缺失E1(nt 457至3014)和E3(nt 27,816-31,332)序列的AC_000011.1(来自专利US 6083716的序列2)合成全长ChAdV68载体。插入处于CMV启动子/增强子控制下的报告基因代替缺失的E1序列。将该克隆转染至HEK293细胞中不会产生感染性病毒。为了确定野生型C68病毒的序列,从ATCC获得分离株VR-594,传代,然后独立测序(SEQ ID NO:10)。当将AC_000011.1序列与野生型ChAdV68病毒的ATCC VR-594序列(SEQ ID NO:10)相比较时,鉴定出6个核苷酸差异。在一个实例中,基于相应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1产生经修饰的ChAdV68载体(ChAdV68.5WTnt SEQ ID NO:1)。
在另一实例中,基于缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列且相应ATCC VR-594核苷酸在四个位置被取代的AC_000011.1产生经修饰的ChAdV68载体。插入处于CMV启动子/增强子控制下的GFP报告蛋白(ChAdV68.4WTnt.GFP;SEQ ID NO:11)或模型新抗原盒(ChAdV68.4WTnt.MAG25mer;SEQ ID NO:12)代替缺失的E1序列。
在另一实例中,基于缺失E1(nt 577至3403)和E3(nt 27,125-31,825)序列且相应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1产生经修饰的ChAdV68载体。插入处于CMV启动子/增强子控制下的GFP报告蛋白(ChAdV68.5WTnt.GFP;SEQ ID NO:13)或模型新抗原盒(ChAdV68.5WTnt.MAG25mer;SEQ ID NO:2)代替缺失的E1序列。
相关载体描述如下:
-全长ChAdVC68序列“ChAdV68.5WTnt”(SEQ ID NO:1);相应ATCC VR-594核苷酸在五个位置被取代的AC_000011.1序列。
-ATCC VR-594 C68(SEQ ID NO:10);独立测序;全长C68
-ChAdV68.4WTnt.GFP(SEQ ID NO:11);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列;相应ATCC VR-594核苷酸在四个位置被取代;插入处于CMV启动子/增强子控制下的GFP报告蛋白代替缺失的E1
-ChAdV68.4WTnt.MAG25mer(SEQ ID NO:12);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,816-31,332)序列;相应ATCC VR-594核苷酸在四个位置被取代;插入处于CMV启动子/增强子控制下的模型新抗原盒代替缺失的E1
-ChAdV68.5WTnt.GFP(SEQ ID NO:13);AC_000011.1缺失E1(nt 577至3403)和E3(nt 27,125-31,825)序列;相应ATCC VR-594核苷酸在五个位置被取代;插入处于CMV启动子/增强子控制下的GFP报告蛋白代替缺失的E1
XV.B.ChAd抗原盒递送载体测试
XV.B.1.ChAd载体评估方法和材料
使用脂染胺转染HEK293A细胞
使用以下方案,制备ChAdV68构建体(ChAdV68.4WTnt.GFP、ChAdV68.5WTnt.GFP、ChAdV68.4WTnt.MAG25mer和ChAdV68.5WTnt.MAG25mer)的DNA并将其转染至HEK293A细胞中。
用PacI消化10ug质粒DNA以释放病毒基因组。然后,根据制造商的说明书,对于较长DNA片段,使用GeneJet DNA净化微型柱(DNA cleanup Micro columns;Thermo Fisher)纯化DNA,并且在20ul预热的水中洗脱;在洗脱步骤之前,使柱在37℃下保持0.5-1小时。
在转染之前,将HEK293A以10
在37℃下温育经转染的细胞培养物至少5-7天。如果在转染后第7天未见到病毒蚀斑,则将细胞以1∶4或1∶6分离,并在37℃下温育以监测蚀斑的产生。或者,收集经转染的细胞且进行3个循环的冷冻和解冻,并使用细胞溶解产物感染HEK293A细胞且温育细胞直至观察到病毒蚀斑。
使用磷酸钙将ChAdV68转染至HEK293A细胞中并产生第三代病毒原液
使用以下方案,制备ChAdV68构建体(ChAdV68.4WTnt.GFP、ChAdV 68.5WTnt.GFP、ChAdV68.4WTnt.MAG25mer、ChAdV68.5WTnt.MAG25mer)的DNA并将其转染至HEK293A细胞中。
在转染前一天,将HEK293A细胞以10
将169μL无菌水添加至微量离心管中。然后将5μL 2M CaCl
在293F细胞中的制造
在8%C0
通过CsCl离心纯化
通过CsCl离心使病毒DNA纯化。执行两次不连续梯度操作。第一次是从细胞组分中纯化出病毒并且第二次是从细胞组分进一步优化分离并将缺陷性粒子与感染性粒子分离。
将10mL的1.2(26.8g CsCl溶解于92mL的10mM Tris pH 8.0中)CsCl添加至异质同晶聚合物管中。然后,使用移液管递送至管底部,小心地添加8mL的1.4CsCl(53g CsCl溶解于87mL的10mM Tris pH 8.0中)。将澄清的病毒小心地铺在该1.2层的顶部上。必要时,再添加10mM Tris以使各管平衡。然后将所述管置放于SW-32Ti旋转器中并在10℃下离心2小时30分钟。然后将所述管移至层流柜中并使用18号针和10mL针筒抽吸病毒带。应避免取出污染性宿主细胞DNA和蛋白质。然后用10mM Tris pH 8.0将该病毒带稀释至少2倍并如前所述铺在如上文所描述的不连续梯度上。如前所述进行操作,其中例外为此时进行该操作过夜。次日,小心抽吸病毒带以避免抽吸出任何缺陷性粒子带。然后使用Slide-a-LyzerT
病毒分析
基于1.1×10
利用病毒原液的限制性稀释分析来计算感染单位(IU)滴度。病毒最初在DMEM/5%NS/1X PS中100倍稀释,随后使用10倍稀释法稀释至1x10
免疫接种
经两侧肌肉内注射向C57BL/6J雌性小鼠和Balb/c雌性小鼠注射1×10
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。通过40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5×10
XV.B.2.在DNA转染之后ChAdV68病毒递送粒子的制造
在一个实例中,将ChAdV68.4WTnt.GFP(图7)和ChAdV68.5WTnt.GFP(图8)DNA转染至HEK293A细胞中并在转染之后7-10天观察病毒复制(病毒蚀斑)。使用光学显微镜检查(图7A和8A)和荧光显微镜检查(图7B-C和图8B-C)观测ChAdV68病毒蚀斑。GFP表示产毒ChAdV68病毒递送粒子的产生。
XV.B.3.ChAdV68病毒递送粒子扩增
在一个实例中,使ChAdV68.4WTnt.GFP、ChAdV68.5WTnt.GFP和ChAdV68.5WTnt.MAG25mer病毒在HEK293F细胞中扩增并在转染之后18天,制备纯化的病毒原液(图9)。定量经纯化ChAdV68病毒原液中的病毒粒子数且与使用相同方案制造的5型腺病毒(Ad5)和ChAdVY25(密切相关的ChAdV;Dicks,2012,PloS ONE 7,e40385)病毒原液相比较。ChAdV68病毒滴度与Ad5和ChAdVY25相当(表7)。
表7.在293F悬浮细胞中产生腺病毒载体
*SD仅在执行多个制造操作情况下报道
XV.B.4.肿瘤模型中的免疫原性评估
在小鼠免疫原性研究中评估表达小鼠肿瘤抗原的C68载体以证实C68载体引起T细胞反应。在C57BL/6J雌性小鼠中测量针对I类MHC表位SIINFEKL的T细胞反应并在Balb/c小鼠中测量针对I类MHC表位AH1-A5(Slansky等人,2000,Immunity 13:529-538)的T细胞反应。如图15中所示,在对小鼠免疫接种ChAdV68.5WTnt.MAG25mer之后测量到相对于对照较强的T细胞反应。当对C57BL/6J或Balb/c小鼠免疫接种ChAdV68.5WTnt.MAG25mer时,在免疫接种之后10天,在ELISpot分析中分别观察到每10
还在CT26肿瘤模型中评估了肿瘤浸润淋巴细胞,所述肿瘤模型评估了ChAdV和抗CTLA4抗体的共同施用。向小鼠植入CT26肿瘤细胞,并在植入后7天,用ChAdV疫苗进行免疫,并用抗CTLA4抗体(克隆9D9)或IgG处理作为对照。免疫后12天分析肿瘤浸润淋巴细胞。使用gentleMACS Dissociator(Miltenyi Biotec)和小鼠肿瘤解离试剂盒(Miltenyi Biotec)解离来自每只小鼠的肿瘤。将解离的细胞通过30微米的过滤器过滤并重悬于完全RPMI中。使用碘化丙锭染色在Attune NxT流式细胞仪(Thermo Fisher)上对细胞进行计数,以排除死细胞和凋亡细胞。然后将细胞调节至合适的活细胞浓度,以进行后续分析。抗原特异性细胞通过MHC-四聚体复合物鉴定,并用抗CD8和活力标记共染色。初免后12天收集肿瘤。
在ChAdV、抗CTLA4和ChAdV+抗CTLA4处理的组中,肿瘤内的抗原特异性CD8+T细胞分别占总活细胞群体的中值3.3%、2.2%或8.1%(图41和表45)。与单独的ChAdV和单独的抗CTLA4相比,抗CTLA的处理与活性ChAdV免疫的组合导致抗原特异性CD8+T细胞频率的统计显著增加,表明抗CTLA4在与chAd68疫苗共同施用时使肿瘤中浸润性T细胞的数量增加。
表45-CT26肿瘤中的四聚体+浸润CD8 T细胞频率
XVI.甲病毒抗原盒递送载体
XVI.A.甲病毒递送载体评估材料和方法
体外转录以产生RNA
对于体外测试:通过用PmeI限制性消化使质粒DNA线性化,遵循制造商的方案(GeneJet DNA净化试剂盒,Thermo)进行柱纯化并用作模板。根据制造商的方案,使用RiboMAX大规模RNA生产系统(Promega),利用m
对于体内研究:产生RNA且由TriLInk Biotechnologies纯化并用Enzymatic Cap1封端。
RNA转染
转染前约16小时,HEK293A细胞对于96孔是以6e4个细胞/孔接种且对于24孔是以2e5个细胞/孔接种。使用MessengerMAX脂染胺(Invitrogen)且遵循制造商的方案,用mRNA转染细胞。对于96孔,每孔使用0.15uL脂染胺和10ng mRNA,且对于24孔,每孔使用0.75uL脂染胺和150ng mRNA。GFP表达mRNA(TriLink Biotechnologies)用作转染对照。
荧光素酶分析
使用ONE-Glo荧光素酶分析(Promega),遵循制造商的方案,在每种条件下于白色壁96孔板中一式三份进行荧光素酶报告蛋白分析。使用SpectraMax测量发光。
qRT-PCR
在转染后2小时,用新鲜培养基冲洗经转染的细胞并更换培养基以移除任何未经转染的mRNA。然后,在多个时间点,将细胞收集于RLT plus溶解缓冲液(Qiagen)中,使用QiaShredder(Qiagen)均质化并使用RNeasy试剂盒(Qiagen)提取RNA,所有操作都遵循制造商的方案。使用Nanodrop(Thermo Scientific)定量总RNA。根据制造商的方案,在qTower
表8.qPCR引物/探针
B16-OVA肿瘤模型
在C57BL/6J小鼠左下方侧腹部中注射10
CT26肿瘤模型
在Balb/c小鼠左下方侧腹部中注射10
免疫接种
对于srRNA疫苗,经两侧肌肉内注射向小鼠注射10ug RNA,体积100uL(每条腿50uL)。对于Ad5疫苗,经两侧肌肉内注射向小鼠注射5×10
体内生物发光成像
在每个时间点,经由腹膜内注射向小鼠注射150mg/kg荧光素底物并在注射之后10-15分钟,使用IVIS体内成像系统(PerkinElmer)测量生物发光。
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。通过40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5×10
XVI.B.甲病毒载体
XVI.B.1.甲病毒载体体外评估
在本发明的一个实施方式中,从基于委内瑞拉马脑炎(VEE)(Kinney,1986,Virology 152:400-413)的自我复制RNA(srRNA)载体产生用于抗原表达系统的RNA甲病毒主链。在一个实例中,编码位于26S亚基因组启动子3'端的VEE结构蛋白的序列缺失(VEE序列7544至11,175缺失;编号基于Kinney等人1986;SEQ ID NO:6)且被抗原序列(SEQ ID NO:14和SEQ ID NO:4)或荧光素酶报告蛋白(例如VEE-荧光素酶,SEQ ID NO:15)替换(图10)。由srRNA DNA载体体外转录RNA,将其转染至HEK293A细胞中并测量荧光素酶报告蛋白表达。此外,用编码荧光素酶的(非复制性)mRNA转染以供比较。当比较23小时测量值与2小时测量值时,对于VEE-荧光素酶srRNA观察到srRNA报告基因信号有约30,000倍增加(表9)。相比之下,在相同时间段内,mRNA报告基因展现小于10倍的信号增加(表9)。
表9.来自VEE自我复制载体的荧光素酶的表达随时间增加。在96孔中用每孔10ngVEE-荧光素酶srRNA或10ng非复制性荧光素酶mRNA(TriLink L-6307)转染HEK293A细胞。在转染后多个时间测量发光。荧光素酶表达以相对发光单位(RLU)报道。每个数据点是3个转染孔的平均值+/-SD。
在另一实例中,通过使用定量逆转录聚合酶链反应(qRT-PCR)测量编码荧光素酶的srRNA(VEE-荧光素酶)或编码多表位盒的srRNA(VEE-MAG25mer)转染之后的RNA含量来直接确定srRNA的复制。对于VEE-荧光素酶srRNA观察到约150倍的RNA增加(表10),而对于VEE-MAG25mer srRNA观察到30-50倍的RNA增加(表11)。这些数据证实,当转染至细胞中时,VEE srRNA载体复制。
表10.VEE-荧光素酶srRNA转染的细胞中RNA复制的直接测量。用VEE-荧光素酶srRNA(150ng/孔,24孔)转染HEK293A细胞并在转染之后多个时间,通过qRT-PCR定量RNA水平。基于肌动蛋白参考基因使各测量值归一化并呈现相对于2小时时间点的倍数变化。
表11.VEE-MAG25mer srRNA转染的细胞中RNA复制的直接测量。用VEE-MAG25mersrRNA(150ng/孔,24孔)转染HEK293细胞并在转染之后多个时间,通过qRT-PCR定量RNA水平。基于GusB参考基因使各测量值归一化并呈现相对于2小时时间点的倍数变化。图上的不同线表示2个不同的qPCR引物/探针集,该两个集合都检测srRNA的表位盒区。
XVI.B.2.甲病毒载体的体内评估
在另一实例中,在体内评估VEE-荧光素酶报告蛋白表达。对小鼠注射10ug封装于脂质纳米粒子(MC3)中的VEE-荧光素酶srRNA并在注射后24小时和48小时,以及7天和14天使其成像以测定生物发光信号。在注射后24小时检测到荧光素酶信号且其随时间增加,并在srRNA注射之后7天出现峰值(图11)。
XVI.B.3.甲病毒载体肿瘤模型评估
在一个实施方式中,为了确定VEE srRNA载体是否在体内引导抗原特异性免疫应答,产生表达2种不同I类MHC小鼠肿瘤表位SIINFEKL和AH1-A5的VEE srRNA载体(VEE-UbAAY,SEQ ID NO:14)(Slansky等人,2000,Immunity 13:529-538)。利用B16-OVA黑素瘤细胞系表达SFL(SIINFEKL)表位,并且AH1-A5(SPSYAYHQF;Slansky等人,2000,Immunity)表位诱导T细胞靶向由CT26结肠癌细胞系表达的相关表位(AH1/SPSYVYHQF;Huang等人,1996,Proc Natl Acad Sci USA 93:9730-9735)。在一个实例中,对于体内研究,通过使用T7聚合酶(TriLink Biotechnologies)体外转录来产生VEE-UbAAY srRNA并将其包封在脂质纳米粒子(MC3)中。
在用MC3配制的VEE-UbAAY srRNA免疫接种带有B16-OVA肿瘤的小鼠之后两周,观察到相对于对照强烈的靶向SFL的抗原特异性T细胞反应。在一个实例中,在用SFL肽刺激之后,在ELISpot分析中测量每10
表12.在带有B16-OVA肿瘤的C57BL/6J小鼠中VEE srRNA免疫接种后14天ELISPOT和MHCI-五聚体染色分析的结果。
*应注意,从Vax组中小鼠#6得到的结果由于三个重复孔之间变化较大而从分析中排除。
在另一实施方式中,为反映临床方法,在B16-OVA和CT26小鼠肿瘤模型中进行异源初免/加强免疫,其中带有肿瘤的小鼠先用表达相同抗原盒的腺病毒载体(Ad5-UbAAY)免疫接种,接着在Ad5-UbAAY初免之后14天,用VEE-UbAAY srRNA疫苗加强免疫。在一个实例中,通过Ad5-UbAAY疫苗诱导抗原特异性免疫应答,由此在ELISpot分析中测量到每10
表13.用Ad5疫苗初免和srRNA加强免疫进行异源初免/加强免疫之后B16-OVA小鼠的免疫监测。
第14天
第28天
在另一实施方式中,在Ad5-UbAAY初免和VEE-UbAAY srRNA加强免疫之后,在CT26小鼠模型中观察到类似结果。在一个实例中,在Ad5-UbAAY初免(第14天)之后观察到AH1抗原特异性反应并且在ELISpot分析中测量到每10
表14.在CT26肿瘤小鼠模型中异源初免/加强免疫之后的免疫监测。
XVII.ChAdV/srRNA组合肿瘤模型评估
在鼠类CT26肿瘤模型中评估使用ChAdV68和自我复制RNA(srRNA)的各种给药方案。
XVII.A ChAdV/srRNA组合肿瘤模型评估方法和材料
肿瘤注入
对Balb/c小鼠注入CT26肿瘤细胞系。肿瘤细胞注入之后7天,将小鼠随机分成不同研究组(28-40只小鼠/组)并开始治疗。在Balb/c小鼠左下方侧腹部中注射10
表15-ChAdV/srRNA组合肿瘤模型评估研究组
免疫接种
对于srRNA疫苗,经两侧肌肉内注射对小鼠注射10ug VEE-MAG25mer srRNA,体积100uL(每条腿50μL)。对于C68疫苗,经两侧肌肉内注射对小鼠注射1×10
脾细胞解离
将每只小鼠的脾和淋巴结汇集于3mL完全RPMI(RPMI、10%FBS、青霉素/链霉素)中。使用gentleMACS解离器(Miltenyi Biotec),遵循制造商的方案进行机械解离。经由40微米过滤器过滤解离的细胞并用ACK溶解缓冲液(150mM NH
离体酶联免疫斑点(ELISPOT)分析
ELISPOT分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用小鼠IFNg ELISpotPLUS试剂盒(MABTECH)进行。将5×10
XVII.B CT26肿瘤模型中的ChAdV/srRNA组合评估
在CT26小鼠肿瘤模型中评估ChAdV68.5WTnt.MAG25mer/VEE-MAG25mer srRNA异源初免/加强免疫或VEE-MAG25mer srRNA同源初免/加强免疫疫苗的免疫原性和功效。对Balb/c小鼠注射CT26肿瘤细胞系。注射肿瘤细胞之后7天,将小鼠随机分成不同研究组并开始治疗。研究组详细地描述于表15中且较粗略地描述于表16中。
表16-初免/加强免疫研究组
在初免疫苗接种之后14天收集脾进行免疫监测。一周两次获取肿瘤和体重测量值并监测存活情况。在所有活性疫苗组中观察到相对于对照的强烈免疫应答。
在第一次免疫接种之后14天,在ELISpot分析中,在分别免疫接种ChAdV68.5WTnt.MAG25mer(ChAdV/第3组)、ChAdV68.5WTnt.MAG25mer+抗PD-1(ChAdV+PD-1/第4组)、VEE-MAG25mer srRNA(srRNA/第5组与第7组的组合的中值)或VEE-MAG25mer srRNA+抗PD-1(srRNA+PD-1/第6组与第8组的组合的中值)的小鼠中观察到每10
表17-CT26肿瘤模型中的细胞免疫应答
与ELISpot数据相符,在第一次免疫接种之后14天,免疫接种ChAdV68.5WTnt.MAG25mer(ChAdV/第3组)、ChAdV68.5WTnt.MAG25mer+抗PD-1(ChAdV+PD-1/第4组)、VEE-MAG25mer srRNA(srRNA/第5组与第7组的组合的中值)或VEE-MAG25mer srRNA+抗PD-1(srRNA+PD-1/第6组与第8组的组合的中值)的小鼠中分别有5.6%、7.8%、1.8%或1.9%的CD8 T细胞(中值)在细胞内细胞因子染色(ICS)分析中展现抗原特异性反应(图17和表18)。免疫接种疫苗对照或疫苗对照与抗PD-1的组合的小鼠分别显示0.2%及0.1%的抗原特异性CD8反应。
表18-CT26肿瘤模型中的CD8 T细胞反应
在CT26结肠肿瘤模型中测量所有组的肿瘤生长情况,且到开始治疗后21天(注射CT-26肿瘤细胞之后28天),出现肿瘤生长。在开始治疗后21天,基于较大肿瘤尺寸(>2500mm
表19-第21天测量的CT26模型中的肿瘤尺寸
在CT-26肿瘤模型中,在开始治疗后,监测存活情况35天(注射CT-26肿瘤细胞之后42天)。在小鼠疫苗接种4个测试组合之后,观察到存活率提高。在疫苗接种之后,利用ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mer srRNA加强免疫与抗PD-1的组合(第4组;相对于对照组1,P<0.0001)、VEE-MAG25mer srRNA初免/VEE-MAG25mer srRNA加强免疫与抗PD-1的组合(第8组;相对于对照组1,P=0.0006)、ChAdV68.5WTnt.MAG25mer初免/VEE-MAG25mer srRNA加强免疫(第3组;相对于对照组1,P=0.0003)以及VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫与抗PD-1的组合(第6组;相对于对照组1,P=0.0016)的小鼠的存活率分别为64%、46%、41%和36%(图19和表20)。其余治疗组[VEE-MAG25mer srRNA初免/ChAdV68.5WTnt.MAG25mer加强免疫(第5组)、VEE-MAG25mer srRNA初免/VEE-MAG25mer srRNA加强免疫(第7组)和单独抗PD-1(第2组)]的存活率与对照组1无显著不同(≤14%)。
表20-CT26模型中的存活率
总之,相对于对照,ChAdV68.5WTnt.MAG25mer和VEE-MAG25mer srRNA引起针对由疫苗编码的小鼠肿瘤抗原的强烈T细胞反应。向带有肿瘤的小鼠施用ChAdV68.5WTnt.MAG25mer初免和VEE-MAG25mer srRNA加强免疫并且共同施用或不共同施用抗PD-1、施用VEE-MAG25mer srRNA初免和ChAdV68.5WTnt.MAG25mer加强免疫与抗PD-1的组合或施用VEE-MAG25mer srRNA同源初免加强免疫与抗PD-1的组合使存活率提高。
XVIII.非人灵长类动物研究
在非人灵长类动物(NHP)中评估使用ChAdV68和自我复制RNA(srRNA)的各种给药方案。
材料和方法
在每个NHP中肌肉注射(IM)初免疫苗以启动研究(疫苗初免)。在每个NHP中也肌肉内注射了一种或多种加强疫苗(疫苗加强)。根据表中概述并在下面总结的组施用每剂量的双侧注射。
免疫接种
用在LNP-1或LNP-2中配制的1×10
免疫监测
在初免疫苗接种之后的指定时间,使用淋巴细胞分离培养基(LymphocyteSeparation Medium,LSM;MP Biomedicals)和LeucoSep分离管(Greiner Bio-One)分离PBMC并使其再悬浮于含有10%FBS和青霉素/链霉素的RPMI中。在Attune NxT流式细胞仪(Thermo Fisher)上使用碘化丙锭染色对细胞计数以排除死亡和凋亡的细胞。然后将细胞调整至适当活细胞浓度以供后续分析。对于研究中的每只猴,使用ELISpot或流式细胞测量方法测量T细胞反应。通过使用离体酶联免疫斑点(ELISpot)分析测量例如IFN-γ的细胞因子的诱导来监测PBMC中针对疫苗中编码的6个不同恒河猴Mamu-A*01的I类表位的T细胞反应。ELISpot分析是根据ELISPOT统一准则{DOI:10.1038/nprot.2015.068},利用猴IFNgELISpotPLUS试剂盒(MABTECH)进行。将200,000个PBMC与10μM指定肽一起在涂有IFNg抗体的96孔板中温育16小时。使用碱性磷酸酶使斑点显色。对反应定时10分钟并通过用自来水流过板终止反应。使用AID vSpot读取器谱图对斑点计数。对于ELISPOT分析,将饱和度>50%的孔记录为“太多而无法计数”。将复制孔的偏差>10%的样品从分析中排除。然后,使用下式,针对孔汇合度校正斑点计数:斑点计数+2×(斑点计数×汇合度%/[100%-汇合度%])。通过用抗原刺激的孔减去阴性肽刺激孔中的斑点计数来校正阴性背景。最后,将标记为太多而无法计数的孔设定成最高观察校正值,四舍五入至最接近的百分数。
通过使用流式细胞测量术测量例如IFN-γ的细胞内细胞因子的诱导来监测PBMC中针对疫苗中编码的6个不同恒河猴Mamu-A*01的I类表位的特异性CD4和CD8 T细胞反应。由两种方法得到的结果指示,以抗原特异性方式诱导针对表位的细胞因子。
恒河猴中的免疫原性
本研究被设计用于(a)评估30μg和100μg剂量的VEE-MAG25mer srRNA与ChAdV68.5WTnt.MAG25mer组合作为同源初免/加强免疫或异源初免/加强免疫的免疫原性和初步安全性;(b)使用LNP1与LNP2比较脂质纳米粒子中VEE-MAG25mer srRNA的免疫应答;(c)评估对VEE-MAG25mer srRNA和ChAdV68.5WTnt.MAG25mer免疫接种的T细胞反应的动力学。
研究组在Mamu-A*01印度恒河猴中进行,以证明免疫原性。本研究中使用的所选抗原仅在恒河猴中识别,特别是具有Mamu-A*01MHC I类单倍型的那些。将Mamu-A*01印度恒河猴随机分配到不同的研究组(每组6只恒河猴),并双侧IM注射施用编码包括多个Mamu-A*01限制性表位的模型抗原的ChAdV68.5WTnt.MAG25mer或VEE-MAG25mer srRNA载体。研究组如下所述。
表21:印度恒河猴中的非GLP免疫原性研究
在免疫接种之前以及初始免疫接种之后第1、2、3、4、5、6、8、9和10周收集PBMC,以进行免疫监测。
在免疫接种之前以及初始免疫接种之后第1、2、3、4、5、6、8、9和10周,针对六个不同Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答。如表21所述,动物在第4周和第8周接受剂量为30μg或100μg并用LNP1或LNP2配制的VEE-MAG25mer srRNA的加强免疫。对于每个免疫监测时间点绘制所有六个表位的组合免疫应答(图20A-D和表22-25)。
在初始VEE-MAG25mer srRNA-LNP1(30μg)初始免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每10
在初始ChAdV68.5WTnt.MAG25mer初始免疫接种后第1、2、3、4、5、6、8、9或10周,分别在每10
表22:VEE-MAG25mer srRNA-LNP1(30μg)的每个表位的每10
表23:VEE-MAG25mer srRNA-LNP1(100μg)的每个表位的每10
表24:VEE-MAG25mer srRNA-LNP2(100μg)的每个表位的每10
表25:ChAdV68.5WTnt.MAG25mer初免的每个表位的每10
印度恒河猴中的非GLP RNA剂量范围研究(较高剂量)
本研究被设计用于(a)评估300μg剂量的VEE-MAG25mer srRNA与ChAdV68.5WTnt.MAG25mer组合作为同源初免/加强免疫或异源初免/加强免疫的免疫原性;(b)使用LNP1与LNP2比较脂质纳米粒子中300μg剂量的VEE-MAG25mer srRNA的免疫应答;以及(c)评估针对VEE-MAG25mer srRNA和ChAdV68.5WTnt.MAG25mer免疫接种的T细胞反应的动力学。
研究组在Mamu-A*01印度恒河猴中进行,以证明免疫原性。非人灵长类物种如恒河猴中的疫苗免疫原性是人中疫苗效力的最佳预测指标。此外,本研究中使用的所选抗原仅在恒河猴中被识别,特别是具有Mamu-A*01MHC I类单倍型的恒河猴。将Mamu-A*01印度恒河猴随机分配到不同的研究组(每组6只恒河猴),并双侧IM注射施用编码包括多个Mamu-A*01限制性抗原的模型抗原的ChAdV68.5WTnt.MAG25mer或VEE-MAG25mer srRNA。研究组如下所述。
在免疫接种之前以及初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周收集PBMC以对第1组进行免疫监测(异源初免/加强免疫)。在免疫接种之前以及初始免疫接种后第4、5、7、8、10、11、12、13、14或15周收集PBMC以对第2组和第3组进行免疫监测(异源初免/加强免疫)。
表26:印度恒河猴中的非GLP免疫原性研究
用ChAdV68.5-WTnt.MAG25mer免疫接种Mamu-A*01印度恒河猴。在免疫接种之前以及初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周针对六个不同的Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答(图21和表27)。在第4、12和20周时,使用LNP2制剂,使动物接受VEE-MAG25mer srRNA加强免疫。在用ChAdV68.5WTnt.MAG25mer初始免疫接种后第4、5、6、7、8、10、11、12、13、14、15、16、17、18、19、20、21、22、23或24周测得组合抗原特异性免疫应答为每10
还使用两种不同的LNP制剂(LNP1和LNP2),用VEE-MAG25mer srRNA对Mamu-A*01印度恒河猴进行免疫接种。在免疫接种之前以及在初始免疫接种后第4、5、6、7、8、10、11、12、13、14或15周,针对六个不同Mamu-A*01限制性表位测量外周血单核细胞(PBMC)中的抗原特异性细胞免疫应答(图22和23,表28和29)。在第4周和第12周,分别使用LNP1或LNP2制剂,使动物接受使用VEE-MAG25mer srRNA的加强免疫。在用VEE-MAG25mer srRNA-LNP2免疫接种后第4、5、7、8、10、11、13、14、15周测得组合抗原特异性免疫应答为每106个PBMC中168、204、103、126、140、145、330、203和162个SFC(六个组合表位)(图22)。在用VEE-MAG25mer srRNA-LNP1免疫接种后第4、5、7、8、10、11、12、13、14、15周测得组合抗原特异性免疫应答为每10
表27:用ChAdV68.5WTnt.MAG25mer初免疫苗接种的每个表位的每10
表28:用VEE-MAG25mer srRNA-LNP2(300μg)初免疫苗接种的每个表位的每10
表29:用VEE-MAG25mer srRNA-LNP1(300μg)初免疫苗接种的每个表位的每10
在本发明的一种实施方式中,可以在mamu A01印度恒河猴中进行srRNA剂量范围研究,以鉴定哪个srRNA剂量可以进行NHP免疫原性研究。在一个实例中,可以通过IM注射向Mamu A01印度恒河猴施用包括多个mamu A01限制性表位的编码模型抗原的srRNA载体。在另一个实例中,抗CTLA-4单克隆抗体可以在IM疫苗注射部位附近SC施用,以靶向一组动物中的疫苗引流淋巴结。可在初始疫苗接种后每两周收集PBMC,以进行免疫监测。研究组描述如下(表30)。
表30:印度恒河猴中的非GLP RNA剂量范围研究
*在高剂量≤300μg下测定的srRNA的剂量范围。
印度恒河猴中的免疫原性研究
在mamu A01印度恒河猴(NHP)中进行了疫苗研究,以证明使用抗原载体的免疫原性。图34示出了疫苗接种策略。用ChAdV68.5-WTnt.MAG25mer免疫三组NHP,其中具有检查点抑制剂抗CTLA-4抗体伊匹单抗(第5组和第6组)或不具有检查点抑制剂(第4组)。静脉内(第5组)或皮下(第6组)施用抗体。三角形表示在第0和32周进行chAd68疫苗接种(1e12 vp/动物)。圆圈表示在第0、4、12、20、28和32周进行甲病毒疫苗接种。
对于单独chAd-MAG免疫接种(图35和表31A),chAd-MAG免疫接种和IV递送的检查点抑制剂(图36和表31B),以及chAd-MAG免疫接种和SC递送的检查点抑制剂(图37和表31C),示出了经免疫的NHP中CD8+抗表位应答的时程。结果表明chAd68载体有效地引发了灵长类动物中的CD8+应答,甲病毒载体有效地增强了chAD68疫苗的引发应答,IV或SC递送的检查点抑制剂均放大了引发和加强应答,并且在疫苗接种后重新施用chAd载体以有效增强免疫应答。
表31A:给予chAd-MAG(第4组)的恒河猴中的CD8+抗表位应答。显示了平均SFC/1e6脾细胞+/-标准误差
表31B:给予chAd-MAG加上IV递送的抗CTLA4抗体(伊匹单抗)(第5组)的恒河猴中的CD8+抗表位应答。显示了平均SFC/1e6脾细胞+/-标准误差
表31C:给予chAd-MAG加上SC递送的抗CTLA4抗体(伊匹单抗)(第6组)的恒河猴中的CD8+抗表位应答。显示了平均SFC/1e6脾细胞+/-标准误差
印度恒河猴中的记忆表型分析
在具有或不具有抗CTLA4的情况下用ChAdV68.5WTnt.MAG25mer/VEE-MAG25mersrRNA异源初免/加强方案免疫恒河猴,并且再次用ChAdV68.5WTnt.MAG25mer加强。在最终的ChAdV68施用后11个月(研究第18个月)对各组进行评估。如所描述的,进行ELISpot。图38和表43显示了免疫前(左图)和18个月后(右图)通过ELISpot测量的对六种不同的Mamu-A*01限制性表位的细胞应答。对限制性表位的应答的检出表明,ChAdV68/samRNA疫苗方案产生了抗原特异性记忆应答。
为了评估记忆,使用双色Mamu-A*01四聚体标记监测识别疫苗中编码的4种不同的恒河猴Mamu-A*01I类表位的CD8+T细胞,每种抗原由唯一双阳性组合表示,并允许鉴定单个样品中的所有4种抗原特异性群体。通过用细胞表面标记CD45RA和CCR7共染色进行记忆细胞表型分析。图39和表44显示了对于识别四种不同的Mamu-A*01限制性表位的记忆T细胞,组合的四聚体染色和CD45RA/CCR7共染色的结果。还通过流式细胞术评估了T细胞表型。图40显示了在研究第18个月时,四种Mamu-A*01四聚体+CD8+T细胞群的总和内记忆细胞类型的分布。记忆细胞表征如下:CD45RA+CCR7+=初始,CD45RA+CCR7-=效应子(Teff),CD45RA-CCR7+=中央记忆(Tcm),CD45RA-CCR7-=效应记忆(Tem)。总之,这些结果表明,在最后一次加强免疫后至少一年检测到记忆应答,表明长的持久免疫力,包括效应子、中枢记忆和效应子记忆群。
表43.在初免和记忆评估时间点(18个月),每只动物每10
ND=由于技术排斥而未确定
表44活CD8+细胞中Mamu-A*01四聚体阳性的百分比
XIX.MHC/肽靶反应性T细胞和TCR的鉴定
针对表A、AACR GENIE结果和/或表1.2中所述的一个或多个抗原/HLA肽对,鉴定了靶反应性T细胞和TCR(参见SEQ ID NO:57-29,357及以下)
可以从患者的血液、淋巴结或肿瘤中分离出T细胞。可以例如通过分选抗原-MHC四聚体结合细胞或通过分选在T细胞和抗原冲击的抗原递呈细胞的体外共培养物中刺激的活化细胞来富集T细胞中的抗原特异性T细胞。用于抗原特异性T细胞鉴定的各种试剂在本领域中是已知的,包括装载有抗原的四聚体和其它基于MHC的试剂。
抗原相关的α-β(或γ-δ)TCR二聚体可以通过抗原特异性T细胞的TCR的单细胞测序来鉴定。或者,可以使用本领域已知的TCR配对方法对抗原特异性T细胞进行批量TCR测序,并且可以确定具有高匹配概率的α-β对。
或者或此外,可以通过来自健康供体的初始T细胞的体外引发获得抗原特异性T细胞。从PBMC、淋巴结或脐带血中获得的T细胞可以被抗原冲击的抗原递呈细胞反复刺激,从而引发抗原表达T细胞的分化。然后可以类似于上文关于来自患者的抗原特异性T细胞所述来鉴定TCR。
XX.共有新抗原的鉴定
我们使用一系列步骤鉴定了共有新抗原。我们从COSMIC数据库中获得了分类为“确认的体细胞”的常见驱动突变列表。对于每个突变,我们生成了候选新表位(8至11聚体肽),使用100的TPM,并在所有建模的HLA等位基因上运行我们的EDGE预测模型(在HLA上训练的深度学习模型提供了通过MS/MS测序的肽,如国际专利申请公开WO/2017/106638、WO/2018/195357和WO/2018/208856中所述,其各自出于所有目的以全文引用的方式并入本文中)。应注意,每个肽都包含至少一个突变氨基酸,并且不是自肽。然后,我们记录了具有EDGE评分>0.001的HLA等位基因的任何肽。结果显示在表A中。因此,鉴定了总共10261个共有新抗原序列,并在SEQ ID NO:10,755-21,015中描述。显示了每个序列的相应HLA等位基因。
对表A中提供的初始列表进一步分析了患者群体中新抗原/HLA流行度的水平。“抗原/HLA流行度”计算为给定群体中抗原的频率(A)乘以给定群体中HLA等位基因的频率(B)。抗原/HLA流行度也可以称为突变/HLA流行度或新抗原/HLA流行度。作为该分析的一部分,对于每个突变,在TCGA中跨常见肿瘤类型获得其(A)频率,并以其在所有肿瘤类型中的最高频率进行记录。(B)对于EDGE中的每个HLA等位基因,记录HLA等位基因频率TCGA(主要是白种人)。HLA等位基因频率在Shukla,S.A.等人(Nat.Biotechnol.33,1152–11582015)中有更详细的描述,其出于所有目的以引用的方式并入本文中。新抗原/HLA流行度计算为(A)乘以(B)。使用这种方法,表A中>0.1%流行度的任何新表位/HLA对均鉴定为“最常见1”(2387/10261)。
另外,我们表征了代表与潜在临床研究有关的晚期癌症患者群体的一大批患者样品中癌症驱动突变的流行度。使用来自主要研究癌症中心(包括Dana-Farber、JohnsHopkins、MD Anderson、MSKCC和Vanderbilt)的公开发布的AACR Genie v4.1数据集进行EDGE预测,该数据集具有在50至500个基因的NGS癌症基因组进行测序的40,000多名患者。我们选择了肺癌、微卫星稳定结肠癌和胰腺癌中的碱基取代和插入缺失突变,并要求覆盖多个基因组。我们分析了与我们的EDGE抗原递呈预测模型中涵盖的超过90个I类HLA等位基因中的每一个配对的每种新抗原肽,并记录HLA递呈评分的EDGE概率>0.001的表位和相应的HLA等位基因。然后,我们确定了EDGE评分>0.001的那些肽的新抗原/HLA流行度,以A*B计算,其中A是三种肿瘤类型中突变的最高频率,并且B是HLA等位基因频率。通过检查来自TCGA群体的HLA等位基因并列出每个HLA等位基因的频率,我们使用了代表美国人口的HLA等位基因频率(Shukla,S.A.等人)。SEQ ID NO:21,016-29,357中描述了从分析中证明新抗原/HLA流行度>0.01%的肽和相应的HLA等位基因,并称为AACR GENIE结果。
XXI.共有新抗原递呈的验证
使用靶向质谱方法进行候选共有新抗原的质谱(MS)验证。将近500个冷冻切除的肺、结直肠和胰腺肿瘤样品均质化,并用于HLA/肽复合物的RNASeq转录组测序和免疫沉淀。通过分析转录组为每个样品生成肽靶列表,从而确定了如AACR Genie v4.1数据集中定义的复发性癌症驱动突变,并评估了RNA表达水平。然后将抗原递呈的EDGE模型应用于突变序列和表达数据,以优先选择用于靶向列表的肽。在质谱分析之前,使用尺寸排阻洗脱并收集来自HLA分子的肽,以分离出所递呈的肽。将具有相同氨基酸序列的合成重标记肽与每个样品共上样,以用于靶向质谱术。重标记肽与实验肽的共洗脱和片段化模式的分析二者被用于验证候选表位。质谱分析方法在Gillete等人(Nat Methods.2013年1月;10(1):28-34)中更详细描述,其出于所有目的以全文引用的方式并入本文中。在下表32中总结了来自以这种方式验证的具有足够流行度以用于进一步考虑的驱动突变的共有新抗原表位,以及样品肿瘤类型和相关的HLA等位基因。
表32:MS验证的新抗原新表位的表达
*当预测同一肽由患者的多个HLA等位基因递呈并且通过MS/MS检测时,则推断其由通过EDGE的最高评分HLA等位基因递呈,或者如果评分足够接近则由两个等位基因递呈。
我们进一步针对未检测到肽的突变评估了MS数据,以评估狭窄靶向具有特定HLA的患者的治疗价值,例如,要求患者具有至少一个经验证或预测的递呈疫苗盒中包含的新抗原的HLA等位基因。
例如,在KRAS的情况下,我们对其中检测到或未检测到特定HLA等位基因的KRAS表位肽的患者样品的数目进行计数。(当预测同一肽由患者的多个HLA等位基因递呈并且通过MS/MS检测时,则推断其由通过EDGE的最高评分HLA等位基因递呈,或者如果评分足够接近则由两个等位基因递呈)。结果列于表33中。基于这些结果,预期多种常见的HLA等位基因不递呈给定的KRAS新抗原,并且出于疫苗盒设计的选择标准和在这种情况下的患者选择,可以排除这些KRAS新抗原/HLA对。例如,涉及特定疫苗盒的表34(见下文第XXII节)不包括基于在17个测试样品中未检测到肽的预测新抗原/HLA对G12D/A*02:01,并且同样地不包括基于在9个测试样品中未检测到肽的G12V/A*02:01。相比之下,新抗原/HLA对G12D/A*11:01被认为是基于在1/5的测试样品中检测到肽而验证的,并且同样地G12V/A*11:01也被认为是基于在2/6的测试样品中检测到肽而验证的。
这些结果强调了在用于用共有新抗原疫苗(如表34中所描述的)进行治疗的患者选择中,鉴定相关的新抗原/HLA对正确的HLA类型选择的重要性。具体地,在MS数据表明共有新抗原疫苗不太可能用预测KRAS新抗原/HLA对为患者提供益处的情况下,出于选择标准目的而排除多种常见KRAS新抗原/HLA对(例如G12D/A*02:01或G12V/A*02:01)。
表33
XXII.A.用于疫苗盒的共有新抗原的选择
构建了包含20种共有新抗原的疫苗盒(“GO-005”)。表34描述了为盒选择的新抗原的特征。将如上文表32中所述通过质谱在肿瘤细胞表面上直接检测到的共有新抗原包含在盒中,并且将表位的HLA添加到符合条件的突变HLA列表中。如果有关于肿瘤递呈的令人信服的文献证据(例如,识别新抗原的肿瘤浸润淋巴细胞(TIL)),则在我们的测定中未被独立地验证为递呈的新抗原将被考虑为经验证并且添加到盒中。由HLA-C*08:02递呈的KRASG12D被考虑为经验证并且添加,这是基于靶向该新抗原的过继细胞疗法导致CRC患者中的肿瘤消退的文献证据(Tran等人.N Engl J Med.2016年12月8日;375(23):2255–2262)。具有经验证的HLA等位基因的新抗原占据了20个箱位中的6个。
预测由肿瘤细胞递呈但尚未通过MS验证的其他更稀有的新抗原被用于补充初始组。鉴于在EDGE评分与通过靶向质谱(MS)验证实验(参见上文第XXI节)检测到候选共有新抗原肽的可能性之间观察到强烈的依赖性,因此将具有高EDGE评分的突变优先包含作为预测的新抗原。显示EDGE评分与通过靶向MS检测到候选共有新抗原肽的可能性之间的相关性的结果显示在图25中。具体地,剩余箱位用EDGE HLA递呈评分为至少0.3并且在NSCLC、CRC和胰腺癌患者中具有最高累积新抗原/HLA流行度的预测新抗原填充。对于盒中的每个箱位,要求组合的HLA频率为至少5–10%(例如,11%的美国人口具有HLA等位基因B1501或B1503)。值得注意的是,由于KRAS和NRAS在密码子12、13和61周围具有相同的盒序列,因此引入流行的NRAS突变不需要额外的箱位。表34还列出了经验证的HLA、EDGE评分为至少0.3的预测的HLA、预测的HLA的平均EDGE评分以及三个癌症人群中新抗原/HLA流行度。
此外,我们确定了具有至少一个被鉴定(即经验证或预测)递呈共有新抗原疫苗GO-005中包含的至少一种共有新抗原的HLA等位基因的患者总群体,并且将其与具有突变的患者(不知患者是否具有鉴定的等位基因)群体进行比较。考虑到来自表34的GO-005疫苗盒,为了估计GO-005靶向的患者群体,我们从AACR Genie收集了患者突变数据。由于此类患者没有匹配的HLA等位基因,因此我们从TCGA群体中对HLA等位基因采样,并将其与AACRGenie数据集配对。然后,根据肿瘤类型,将来自AACR Genie的突变和HLA均匹配的所有患者标记为阳性,并且任何不符合标准的患者都标记为阴性。阳性百分比给出了表35中每种肿瘤类型的总体可寻址患者群体。
从表35可以容易地理解,仅携带特定突变的患者子集也携带可能将这种突变递呈为新抗原的HLA等位基因。具有突变但没有合适的HLA等位基因的患者较不可能从治疗中受益。举例来说,虽然估计约60%的胰腺癌患者携带适当的突变/新抗原,但其中三分之二以上的患者未携带相应的HLA等位基因。因此,考虑所建议的相关突变和HLA等位基因对的疫苗接种策略将仅针对可能受益的那些患者。因此,通过经验证或高评分预测的HLA来考虑表位递呈是确定共有新抗原疫苗的潜在疗效的重要步骤。
表35:目标群体中的新抗原/HLA流行度
XXII.B.共有新抗原疫苗盒序列选择
选择了用于包含在共有新抗原疫苗中的共有新抗原序列。
对于KRAS_G13D,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出KRAS_G13D和C0802的行来选择考虑的每个相关序列。
对于KRAS_Q61K或NRAS_Q61K,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61K和A0101;或(2)NRASQ61K和A0101的行来选择考虑的每个相关序列。
对于TP53_R249M,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出TP53_R249M以及B3512、B3503和B3501中的至少一个的行来选择考虑的每个相关序列。
对于CTNNB1_S45P,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S45P以及A0101、A0301、B5701、A6801、A0302和A1101中的至少一个的行来选择考虑的每个相关序列。例如,参见32中所示的相关序列。
对于CTNNB1_S45F,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S45F以及A0301、A1101和A6801中的至少一个的行来选择考虑的每个相关序列。
对于ERBB2_Y772_A775dup,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出ERBB2_Y772_A775dup和B1801的行来选择考虑的每个相关序列。
对于KRAS_G12D或NRAS_G12D,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_G12D以及A1101和C0802中的至少一个或(2)NRAS_G12D以及A1101和C0802中的至少一个的行来选择考虑的每个相关序列。例如,参见表32中所示的相关序列。
对于KRAS_Q61R或NRAS_Q61R,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61R和A0101;或(2)NRAS_Q61R和A0101的行来选择考虑的每个相关序列。
对于CTNNB1_T41A,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_T41A以及A0301、A0302、A1101、B1510、C0303和C0304中的至少一个的行来选择考虑的每个相关序列。
对于TP53_K132N,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出TP53_K132N以及A2402和A2301中的至少一个的行来选择考虑的每个相关序列。例如,参见表32中所示的相关序列。
对于KRAS_G12A,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出KRAS_G12A和A0301的行来选择考虑的每个相关序列。例如,参见表32中所示的相关序列。
对于KRAS_Q61L或NRAS_Q61L,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61L和A0101;或(2)NRAS_Q61L和A0101的行来选择考虑的每个相关序列。
对于TP53_R213L,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出TP53_R213L以及A0207、C0802和A0201中的至少一个的行来选择考虑包含的每个相关序列。
对于BRAF_G466V,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出BRAF_G466V以及B1501和B1503中的至少一个的行来选择考虑的每个相关序列。
对于KRAS_G12V,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出KRAS_G12V以及A0301、A1101、A3101、C0102和A0302中的至少一个的行来选择考虑的每个相关序列。例如,参见表32中所示的相关序列。
对于KRAS_Q61H或NRAS_Q61H,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_Q61H和A0101;或(2)NRAS_Q61H和A0101的行来选择考虑包含的每个相关序列。
对于CTNNB1_S37F,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出CTNNB1_S37F以及A2301、A2402、B1510、B3906、C0501、C1402和C1403中的至少一个的行来选择考虑的每个相关序列。
对于TP53_S127Y,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出TP53_S127Y以及A1101和A0301中的至少一个的行来选择考虑包含的每个相关序列。
对于TP53_K132E,通过参考表A或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出TP53_K132E以及A2402、C1403和A2301中的至少一个的行来选择考虑的每个相关序列。
对于KRAS_G12C或NRAS_G12C,通过参考表A 32或AACR GENIE结果选择用于包含在疫苗中的共有新抗原编码序列,其中通过鉴定所有列出(1)KRAS_G12C和A0201或(2)NRAS_G12C和A0201的行来选择考虑的每个相关序列。例如,参见表32中所示的相关序列。
XXIII.共有新抗原的T细胞识别的评估
我们评估了新抗原是否在患者中诱导免疫应答。我们从患有肺腺癌的患者获得了解离的肿瘤细胞。对肿瘤细胞进行测序,以确定患者的HLA并鉴定突变。该患者表达HLA-A*1101,并且我们在肿瘤中鉴定出KRAS G12V突变。同时,我们从肿瘤中分选并扩增了代表肿瘤浸润淋巴细胞(TIL)的CD45+细胞。用突变的肽HLA-A*11:01四聚体对扩增的TIL进行染色,以评估该突变在患者体内的免疫原性。图26显示了对CD8+细胞的流式细胞术门控策略(左图)和通过KRAS-G12V/HLA-A*11:01四聚体对CD8+细胞的染色(右图)。大部分(大于66%)的CD8+T细胞表现出与KRAS G12V:HLA*1101四聚体的结合,指示了CD8+T细胞识别新抗原的能力,并指示对新抗原的预先存在的免疫应答。
此外,我们评估了能够识别共有新抗原的健康供体的初始T细胞库中T细胞前体的存在。富集外周血单核细胞(PBMC)的初始CD8+T细胞,并用递呈疫苗盒中存在的多种共有新抗原候选物的MHC多聚体染色:GO-005:2KRAS G12V肽、KRAS G12C肽和CTNNB1_S45P肽表位。分选、扩增HLA-肽结合细胞,并确认其对新抗原的特异性。检测所有测试突变的前体(表36)。还进行了新抗原特异性T细胞的TCR测序。图27示出了一般的TCR测序策略和工作流程。图28显示了用于KRAS-G12V/HLA-A*11:01四聚体的TCR测序策略的代表性实例。TCR测序策略揭示了多克隆应答,每个肽/MHC和每个供体鉴定出中值73(范围25至987)种克隆型(表36)。因此,初始T细胞库分析表明,预期这些新抗原在通过疫苗接种施用时在选择的患者中诱导免疫应答。
表36–新抗原反应性初始T细胞前体的评估
XXIV.共有新抗原和患者群体的选择
将表34、表A、表1.2或本文描述的AACR GENIE结果中提供的一种或多种抗原(SEQID NO:57-29,357)用于配制如本文所述的疫苗组合物。将疫苗施用于患者,例如以治疗癌症。在某些情况下,选择患者,例如使用伴随诊断或常用的癌症基因组NGS测定,例如FoundationOne、FoundationOne CDx、Guardant 360、Guardant OMNI或MSK IMPACT。示例性的患者选择标准如下所述。示例性的共有新抗原疫苗组合物GO-005靶向表34中描述的突变。
患者选择
通过考虑肿瘤基因表达、体细胞突变状态和患者HLA类型来进行用于共有新抗原疫苗的患者选择。具体地,如果满足以下条件,则认为患者符合疫苗治疗的条件:
(a)患者携带预测或已知递呈疫苗中包含的表位的HLA等位基因,并且患者肿瘤表达具有该表位序列的基因,或
(b)患者携带预测或已知递呈疫苗中包含的表位的HLA等位基因,并且患者肿瘤携带产生该表位序列的突变,或
(c)与(b)相同,但还要求患者肿瘤表达具有高于某个阈值(例如1TPM或10TPM)的突变的基因,或
(d)与(b)相同,但还要求患者肿瘤表达高于某个阈值(例如,在RNA水平观察到至少1个突变读段)的突变
(e)与(b)相同,但还要求(c)和(d)二者中的附加标准
(f)以上任何一项,但还任选地要求在肿瘤中未检测到递呈HLA等位基因的缺失
通过任何已建立的方法(包括RNASeq、微阵列、PCR、Nanostring、ISH、质谱或IHC)在RNA或蛋白质水平上测量基因表达。基因表达阳性的阈值通过多种方法确定,包括:(1)在各种基因表达水平上预测由HLA等位基因递呈表位的可能性,(2)通过质谱测量的基因表达与HLA表位递呈的相关性,和/或(3)以多种水平表达基因的患者获得了疫苗接种的临床益处。进一步扩展了患者选择,以要求对于疫苗中包含的大于1种表位,例如至少2、3、4或5种表位是阳性。
通过任何已建立的方法来评估体细胞突变状态,包括外显子组测序(NGSDNASeq)、靶向外显子组测序(基因组)、转录组测序(RNASeq)、Sanger测序、基于PCR的基因分型测定(例如Taqman或液滴数字PCR)、基于质谱的方法(例如,通过Sequenom)、或本领域技术人员已知的任何其他方法。
使用所描述的任何方法(例如通过质谱法)鉴定了另外的新共有的新抗原。这些新鉴定的共有新抗原被引入本文所述的疫苗盒中。
先前验证的新抗原还被验证为由另外的HLA等位基因递呈,并告知用于疫苗盒的新抗原选择和/或扩大了潜在的可治疗群体。
包含新的新抗原使得能够扩大可寻址的肿瘤类型(例如,EGFR突变的NSCLC)或包含具有新肿瘤类型的患者。
XXV.共有抗原的鉴定
我们使用三个计算步骤鉴定了基于共有抗原基因的靶标:首先,我们使用可通过基因型组织表达(Genotype-Tissue Expression,GTEx)项目[1]可获得的数据鉴定了在大多数正常组织中低表达或无表达的基因。我们从基因型组织表达(GTEx)项目(版本V7p2)获得了汇总的基因表达数据。该数据集包含来自多于700个个体和多于50种的不同组织类型的多于11,000个验尸样品。将表达使用RNA-Seq进行测量,并根据GTEx标准线路(https://www.gtexportal.org/home/documentationPage)进行计算处理。使用用RSEM v1.2.22[2]计算的同工型表达之和计算基因表达。
接下来,我们使用来自癌症基因组图谱(TCGA)研究网络:http://cancergenome.nih.gov/的数据,鉴定了那些基因中哪些在癌症样品中异常表达。我们检查了从TCGA(Data Release 6.0)可获得的多于11,000个样品。
最后,在这些基因中,我们使用在通过MS/MS测序的HLA递呈肽上训练的深度学习模型,鉴定了哪些肽可能作为细胞表面抗原被MHC I类蛋白递呈,如国际专利申请no.PCT/US2016/067159中所述,其出于所有目的以全文引用的方式并入本文中。
为了鉴定常见的肿瘤抗原(CTA;共有抗原),我们试图定义标准以排除在正常组织中表达的基因,该标准足够严格以确保肿瘤特异性,但是将解释潜在的假象,例如读段未对准。如果基因满足以下条件,则符合纳入CTA的条件:在作为脑、心脏或肺的一部分的每个器官中,中值GTEx表达低于0.1转录本/百万(transcripts per million,TPM),且没有一个样品超过5TPM。在其他必需器官中的中值GTEx表达低于2TPM,且没有一个样品超过10TPM。忽略被分类为非必需的器官(睾丸、甲状腺和小唾液腺)的表达。如果基因在至少30个样品中在TCGA中以大于10TPM表达,则认为其在肿瘤样品中表达。根据文献综述,我们还添加了基因MAGE4和MAGGEB6。
我们还添加了基因CTAG1A/CTAG1B(NY-ESO-1)。由于无法使用TCGA Data Release6.0中的计算方法准确量化其表达,我们依靠TCGA旧版归档文件(https://portal.gdc.cancer.gov/legacy-archive)中可获得的RSEM计算来解决多重映射读段。
然后,我们检查了TCGA样品中其余基因表达的分布。当我们检查已知的CTA时,例如基因的MAGE家族,我们观察到这些基因在对数空间中的表达通常以双峰分布为特征。该分布包括在较低的表达值附近的左模式和在较高的表达水平的右模式(或粗尾巴)。这种表达方式与一种生物学模型相一致,在该生物学模型中,在许多样品中在基线处都可以检测到一些最小表达,并且在经历表观遗传失调的肿瘤子集中观察到基因的更高表达。我们审查了TCGA样品中每个基因的表达分布,并丢弃了仅观察到单峰分布而无明显右手尾巴的那些样品。通过手工策略淘汰了少量基因,例如,如果其可能在从GTEx无法获得的组织中表达的话。这产生了一组59个基因。参见表46。在表46中,X用于表示在至少1%的情况下基因以高于10TPM表达的癌症。
为了鉴定可能作为细胞表面抗原被MHC I类蛋白递呈的肽,我们使用了滑动窗将这些蛋白质中的每一个解析为其组成的8-11个氨基酸序列。我们用HLA肽递呈深度学习模型处理这些肽及其侧接序列,以计算在TCGA中对该基因观察到的99.9%百分位数表达水平下每种肽递呈的可能性。如果通过我们的模型计算得出的肽的分位数归一化的递呈概率大于0.001,则认为该肽可能被递呈(即候选靶标)。
为了优先考虑可能与给定适应症相关的基因,我们选择了基因在至少0.98%的癌症病例中以至少10TPM的水平表达的基因。
结果显示在表1.2中。鉴定出总共10698个共有抗原序列。显示了每个序列的相应HLA等位基因。
表A
参考序列表SEQ ID NO.10,755-21,015。为了清楚起见,将预测与给定的HLA等位基因以及EDGE评分>0.001的HLA等位基因相关联的每个肽分配给唯一的SEQ ID.NO。上述每个序列标识符都与以下相关:肽的氨基酸序列、HLA亚型、与肽相对应的基因名称、与肽相关的突变以及肽:HLA对的流行度是否大于0.1%(记为“1”)或小于0.1%(记为“0”)。
表A的全部内容公开在2018年5月23日提交的美国临时申请号62/675,559中,其以全文引用的方式并入本文中。
AACR GENIE结果
参考序列表SEQ ID NO.21,016-29,357。为了清楚起见,将预测与给定的HLA等位基因以及EDGE评分>0.001且流行度>0.1%的HLA等位基因相关联的每个肽分配给唯一的SEQ ID.NO。上述每个序列标识符都与以下相关:与肽相对应的基因名称和突变、HLA亚型以及肽的氨基酸序列。
表1.2
参考序列表SEQ ID NO.57-10,754。预测的共有抗原与在至少0.98%的癌症病例中以至少10TPM的水平表达的基因相关。上述每个序列标识符与基因名称、肽的氨基酸序列、Ensembl ID和相应的HLA等位基因相关。
某些序列
本文提及的载体、盒和抗体在下文进行了描述,并且通过SEQ ID NO提及。
1.Desrichard,A.,Snyder,A.&Chan,T.A.Cancer Neoantigens andApplications for Immunotherapy.Clin.Cancer Res.Off.J.Am.Assoc.Cancer Res.(2015).doi:10.1158/1078-0432.CCR-14-3175
2.Schumacher,T.N.&Schreiber,R.D.Neoantigens in cancerimmunotherapy.Science 348,69-74(2015).
3.Gubin,M.M.,Artyomov,M.N.,Mardis,E.R.&Schreiber,R.D.Tumorneoantigens:building a framework for personalized cancerimmunotherapy.J.Clin.Invest.125,3413-3421(2015).
4.Rizvi,N.A.et al.Cancer immunology.Mutational landscape determinessensitivity to PD-1 blockade in non-small cell lung cancer.Science 348,124-128(2015).
5.Snyder,A.et al.Genetic basis for clinical response to CTLA-4blockade in melanoma.N.Engl.J.Med.371,2189-2199(2014).
6.Carreno,B.M.et al.Cancer immunotherapy.A dendritic cell vaccineincreases the breadth and diversity of melanoma neoantigen-specific Tcells.Science 348,803-808(2015).
7.Tran,E.et al.Cancer immunotherapy based on mutation-specific CD4+Tcells in a patient with epithelial cancer.Science 344,641-645(2014).
8.Hacohen,N.&Wu,C.J.-Y.United States Patent Application:20110293637-COMPOSITIONS AND METHODS OF IDENTIFYING TUMOR SPECIFIC NEOANTIGENS.(A1).at
9.Lundegaard,C.,Hoof,I.,Lund,O.&Nielsen,M.State of the art andchalenges in sequence based T-cell epitope preodiction.Immunome Res.6 Suppl2,S3(2010).
10.Yadav,M.et al.Predicting immunogenic tumour mutations by combiningmass spectrometry and exome sequencing.Nature 515,572-576(2014).
11.Bassani-Sternberg,M.,Pletscher-Frankild,S.,Jensen,L.J.&Mann,M.Massspectrometry of human leukocyte antigen class I peptidomes reveals strongeffects of protein abundance and turnover on antigen presentation.Mol.Cell.Proteomics MCP 14,658-673(2015).
12.Van Allen,E.M.et al.Genomic correlates of response to CTLA-4blockade in metastatic melanoma.Science 350,207-211(2015).
13.Yoshida,K.&Ogawa,S.Splicing factor mutations and cancer.WileyInterdiscip.Rev.RNA 5,445-459(2014).
14.Cancer Genome Atlas Research Network.Comprehensive molecularprofiling of lung adenocarcinoma.Nature 511,543-550(2014).
15.Rajasagi,M.et al.Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemiaBlood 124,453-462(2014).
16.Downing,S.R.et al.United States Patent Application:0120208706-OPTIMIZATION OF MULTIGENE ANALYSIS OF TUMOR SAMPLES.(A1).at
17.Target Capture for NextGen Sequencing-IDT.at
18.Shukla,S.A.et al.Comprehensive analysis of cancer-associatedsomatic mutations in class I HLA genes.Nat.Biotechnol.33,1152-1158(2015).
19.Cieslik,M.et al.The use of exome capture RNA-seq for highlydegraded RNA with application to clinical cancer sequencing.Genome Res.25,1372-1381(2015).
20.Bodini,M.et al.The hidden genomic landscape of acute myeloidleukemia:subclonal structure revealed by undetected mutations.Blood 125,600-605(2015).
21.Saunders,C.T.et al.Strelka:accurate somatic small-variant callingfrom sequenced tumor-normal sample pairs.Bioinforma.Oxf.Engl.28,1811-1817(2012).
22.Cibulskis,K.et al.Sensitive detection of somatic point mutationsin impure and heterogeneous cancer samples.Nat.Biotechnol.31,213-219(2013).
23.Wilkerson,M.D.et al.Integrated RNA and DNA sequencing improvesmutation detetion in low purity tumors.Nucleic Acids Res.42,e107(2014).
24.Mose,L.E.,Wilkerson,M.D.,Hayes,D.N.,Perou,C.M.&Parker,J.S.ABRA:improved coding indel detection via assembly-based realignment.Bioinforma.Oxf.Engl.30,2813-2815(2014).
25.Ye,K.,Schulz,M.H.,Long,Q.,Apweiler,R.&Ning,Z.Pindel:a patterngrowth approach to detect break points of large deletions and medium sizedinsertions from paired-end short reads.Bioinforma.Oxf.Engl.25,2865-2871(2009).
26.Lam,H.Y.K.et al.Nucleotide-resolution analysis of structuralvariants using BreakSeq and a breakpoint library.Nat.Biotechnol.28,47-55(2010).
27.Frampton,G.M.et al.Development and validation of a clinical cancergenomic profiling test based on massively parallel DNAsequencing.Nat.Biotechnol.31,1023-1031(2013).
28.Boegel,S.et al.HLA typing from RNA-Seq sequence reads.GenomeMed.4,102(2012).
29.Liu,C.et al.ATHLATES:accurate typing of human leukocyte antigenthrough exome sequencing.Nucleic Acids Res.41,e142(2013).
30.Mayor,N.P.et al.HLA Typing for the Next Generation.PloS One 10,e0127153(2015).
31.Roy,C.K.,Olson,S.,Graveley,B.R.,Zamore,P.D.&Moore,M.J.AssessinglonR-distance RNA sequence connectivity via RNA-templated DNA-DNAligation.eLife 4,(2015).
32.Song,L.&Florea,L.CLASS:constrained transcript assembly of RNA-seqreads.BMC Bioinformatics 14 Suppl 5,S14(2013).
33.Maretty,L.,Sibbesen,J.A.&Krogh,A.Bayesian transcriptomeassembly.Genome Biol.15,501(2014).
34.Pertea,M.et al.StringTie enables improved reconstruction of atranscriptome from RNA-seq reads.Nat.Biotechnol.33,290-295(2015).
35.Roberts,A.,Pimentel,H.,Trapnell,C.&Pachter,L.Identification ofnovel transcripts in annotated genomes using RNA-Seq.Bioinforma.Oxf.Engl.(2011).doi:10.1093/bioinformatics/btr355
36.Vitting-Seerup,K.,Porse,B.T.,Sandelin,A.&Waage,J.spliceR:an Rpackage for classification of alternative splicing and prediction of codingpotential from RNA-seq data.BMC Bioinformatics 15,81(2014).
37.Rivas,M.A.et al.Human genomics.Effect of predicted protein-truncating genetic variants on the human transcriptome.Science 348,666-669(2015).
38.Skelly,D.A.,Johansson,M.,Madeoy,J.,Wakefield,J.&Akey,J.M.Apowerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data.Genome Res.21,1728-1737(2011).
39.Anders,S.,Pyl,P.T.&Huber,W.HTSeq--a Python framework to work withhigh-throughput sequencing data.Bioinforma.Oxf.Engl.31,166-169(2015).
40.Furney,S.J.et al.SF3B1 mutations are associated with alternativesplicing in uveal melanoma.Cancer Discov.(2013).doi:10.1158/2159-8290.CD-13-0330
41.Zhou,Q.et al.A chemical genetics approach for the functionalassessment of novel cancer genes.Cancer Res.(2015).doi:10.1158/0008-5472.CAN-14-2930
42.Maguire,S.L.et al.SF3B1 mutations constitute a novel therapeutictarget in breast cancer.J.Pathol.235,571-580(2015).
43.Carithers,L.J.et al.A Novel Approach to High-Quality PostmortemTissue Procurement:The GTEx Project.Biopreservation Biobanking 13,311-319(2015).
44.Xu,G.et al.RNA CoMPASS:a dual approach for pathogen and hosttranscriptome analysis of RNA-seq datasets.PloS One 9,e89445(2014).
45.Andreatta,M.&Nielsen,M.Gapped sequence alignment using artificialneural networks:application to the MHC class I system.Bioinforma.Oxf.Engl.(2015).doi:10.1093/bioinformatics/btv639
46.
47.Larsen,M.V.et al.An integrative approach to CTL epitopeprediction:a combined algorithm integrating MHC class I binding,TAP transportefficiency,and proteasomal cleavage predictions.Eur.J.Immunol.35,2295-2303(2005).
48.Nielsen,M.,Lundegaard,C.,Lund,O.&Kesmir,C.The role oftheproteasome in generating cytotoxic T-cell epitopes:insights obtained fromimproved predictions of proteasomal cleavage.Immunogenetics 57,33-41(2005).
49.Boisvert,F.-M.et al.A Quantitative Spatial Proteomics Analysis ofProteome Turnover in Human Cells.Mol.Cell.Proteomics 11,M111.011429-M111.011429(2012).
50.Duan,F.et al.Genomic and bioinformatic profiling of mutationalneoepitopes reveals new rules to predict anticancerimmunogenicity.J.Exp.Med.211,2231-2248(2014).
51.Janeway’s Immanobiology:9780815345312:Medicine&Health ScienceBooks @Amazon.com.at
52.Calis,J.J.A.et al.Properties of MHC Class I Presented PeptidesThat Enhance Immunogenicity.PLoS Comput.Biol.9,e1003266(2013).
53.Zhang,J.et al.Intratumor heterogeneity in localized lungadenocarcinomas delineated by multiregion sequencing.Science 346,256-259(2014)
54.Walter,M.J.et al.Clonal architecture of secondary acute myeloidleukemia.N.Engl.J.Med.366,1090-1098(2012).
55.Hunt DF,Henderson RA,Shabanowitz J,Sakaguchi K,Michel H,Sevilir N,Cox AL,Appella E,Engelhard VH.Characterization of peptides bound to the classI MHC molecule HLA-A2.1 by mass spectrometry.Science 1992.255:1261-1263.
56.Zarling AL,Polefrone JM,Evans AM,Mikesh LM,Shabanowitz J,Lewis ST,Engelhard VH,Hunt DF.Identification of class I MHC-associated phosphopeptidesas targets for cancer immunotherapy.Proc Natl Acad Sci USA.2006 Oct 3;103(40):14889-94.
57.Bassani-Stemberg M,Pletscher-Ftankild S,Jensen LJ,Mann M.Massspectrometry of human leukocyte antigen class I peptidomes reveals strongeffects of protein abundance and turnover on antigen presentation.Mol CellProteomics.2015 Mar;14(3):658-73.doi:10.1074/mcp.M114.042812.
58.Abelin JG,Trantham PD,Penny SA,Patterson AM,Ward ST,Hildebrand WH,Cobbold M,Bai DL,Shabanowitz J,Hunt DF.Complementary IMAC enrichment methodsfor HLA-associated phosphopeptide identification by mass spectrometry.NatProtoc.2015 Sep;10(9):1308-18.doi:10.1038/nprot.2015.086.Epub 2015 Aug 6
59.Barnstable CJ,Bodmer WF,Brown G,Galfre G,Milstein C,Williams AF,Ziegler A.Production of monoclonal antibodies to group A erythrocytes,HLA andother human cell surface antigens-new tools for genetic analysis.Cell.1978May;14(1):9-20.
60.Goldman JM,Hibbin J,Kearney L,Orchard K,Th′ng KH.HLA-DR monoclonalantibodies inhibit the proliferation of normal and chronic granulocyticleukaemia myeloid progenitor cells.Br J Haematol.1982 Nov;52(3):411-20.
61.Eng JK,Jahan TA,Hoopmann MR.Comet:an open-source MS/MS sequencedatabase search tool.Proteomics.2013 Jan;13(1):22-4.doi:10.1002/pmic.201200439.Epub 2012 Dec 4.
62.Eng JK,Hoopmann MR,Jahan TA,Egertson JD,Noble WS,MacCoss MJ.Adeeper look into Comet--implementation and features.J Am Soc MassSpectrom.2015 Nov;26(11):1865-74.doi:10.1007/s13361-015-1179-x.Epub 2015 Jun27.
63.Lukas
64.Lukas
65.Lukas
66.Kinney RM,BJ Johnson,VL Brown,DW Trent,Nucleotide Sequence of the26 S mRNA of the Virulent Trinidad Donkey Strain of Venezuelan EquineEncephalitis Virus and Deduced Sequence of the Encoded StructuralProteins.Virology 152(2),400-413.1986 Jul 30.
67.Jill E Slansky,Frédérique M Rattis,Lisa F Boyd,Tarek Fahmy,Elizabeth M Jaffee,Jonathan P Schneck,David H Margulies,Drew MPardoll.Enhanced Antigen-Specific Antitumor Immunity with Altered PeptideLigands that Stabilize the MHC-Peptide-TCR Complex.Immunity,Volume 13,Issue4,1 October 2000,Pages 529-538.
68.A Y Huang,P H Gulden,A S Woods,M C Thomas,C D Tong,W Wang,V HEngelhard,G Pasternack,R Cotter,D Hunt,D M Pardoll,and E M Jaffee.Theimmunodominant major histocompatibility complex class I-restricted antigen ofa murine colon tumor derives from an endogenous retroviral gene product.ProcNatl Acad Sci USA.;93(18):9730-9735,1996 Sep 3.
69.JOHNSON,BARBARA J.B.,RICHARD M.KINNEY,CRYSTLE L.KOST AND DENNISW.TRENT.Molecular Determinants of Alphavirus Neurovirulence:Nucleotide andDeduced Protein Sequence Changes during Attenuation of Venezuelan EquineEncephalitis Virus.J Gen Virol 67:1951-1960,1986.
70.Aarnoudse,C.A.,Krüse,M.,Konopitzky,R.,Brouwenstijn,N.,and Schrier,P.I.(2002).TCR reconstitution in Jurkat reporter cells facilitates theidentification of novel tumor antigens by cDNA expression cloning.Int JCancer 99,7-13.
71.Alexander,J.,Sidney,J.,Southwood,S.,Ruppert,J.,Oseroff,C.,Maewal,A.,Snoke,K.,Serra,H.M.,Kubo,R.T.,and Sette,A.(1994).Development of highpotency universal DR-restricted helper epitopes by modification of highaffinity DR-blocking peptides.Immunity 1,751-761.
72.Banu,N.,Chia,A.,Ho,Z.Z.,Garcia,A.T.,Paravasivam,K.,Grotenbreg,G.M.,Bertoletti,A.,and Gehring,A.J.(2014).Building and optimizing a virus-specific T cell receptor library for targeted immunotherapy inviralinfections.Scientific Reports 4,4166.
73.Cornet,S.,Miconnet,I.,Menez,J.,Lemonnier,F.,and Kosmatopoulos,K.(2006).Optimal organization of a polypeptide-based candidate cancer vaccinecomposed of cryptic tumor peptides with enhanced immunogenicity.Vaccine 24,2102-2109.
74.Depla,E.,van der Aa,A.,Livingston,B.D.,Crimi,C.,Allosery,K.,deBrabandere,V.,Krakover,J.,Murthy,S.,Huang,M.,Power,S.,et al.(2008).Rationaldesign of a multiepitope vaccine encoding T-lymphocyte epitopes for treatmentof chronic hepatitis B virus infections.Journal of Virology 82,435-450.
75.Ishioka,G.Y.,Fikes,J.,Hermanson,G.,Livingston,B.,Crimi,C.,Qin,M.,del Guercio,M.F.,Oseroff,C.,Dahlberg,C.,Alexander,J.,et al.(1999).Utilizationof MHC class I transgenic mice for development of minigene DNA vaccinesencoding multiple HLA-restricted CTL epitopes J Immunol 162,3915-3925.
76.Janetzki,S.,Price,L.,Schroeder,H.,Britten,C.M.,Welters,M.J.P.,andHoos,A.(2015).Guidelines for the automated evaluation of Elispot assays.NatProtoc 10,1098-1115.
77.Lyons,G.E.,Moore,T.,Brasic,N.,Li,M.,Roszkowski,J.J.,and Nishimura,M.I.(2006).Influence of human CD8 on antigen recognition by T-cell receptor-transduced cells.Cancer Res 66,11455-11461.
78.Nagai,K.,Ochi,T.,Fujiwara,H.,An,J.,Shirakata,T.,Mineno,J.,Kuzushima,K.,Shiku,H.,Melenhorst,J.J.,Gostick,E.,et al.(2012).Aurora kinaseA-specific T-cell receptor gene transfer redirects T lymphocytes to displayeffective antileukemia reactivity.Blood 119,368-376.
79.Panina-Bordignon,P.,Tan,A.,Termijtelen,A.,Demotz,S.,Corradin,G.,and Lanzavecchia,A(1989).Universally immunogenic T cell epitopes:promiscuousbinding to human MHC class II and promiscuous recognition by T cells.Eur JImmunol 19,2237-2242.
80.Vitiello,A.,Marchesini,D.,Furze,J.,Sherman,L.A,and Chesnut,R.W.(1991).Analysis of the HLA-restricted influenza-specific cytotoxic Tlymphocyte response in transgenic mice carrying a chimeric human-mouse classI major histocompatibility complex.J Exp Med 173,1007-1015.
81.Yachi,P.P.,Ampudia,J.,Zal,T.,and Gascoigne,N.R.J.(2006).Alteredpeptide ligands induce delayed CD8-T cell receptor interaction--a role forCD8 in distinguishing antigen quality.Immunity 25,203-211.
82.Pushko P,Parker M,Ludwig GV,Davis NL,Johnston RE,SmithJF.Replicon-helper systems from attenuated Venezuelan equine encephalitisvirus:expression of heterologous genes in vitro and immunization againstheterologous pathogens in vivo.Virology.1997 Dec 22;239(2):389-401.
83.Strauss,JH and E G Strauss.The alphaviruses:gene expression,replication,and evolution.Microbiol Rev.1994 Sep;58(3):491-562.
84.
85.Riley,Michael K.II.and Wilfred Vermerris.Recent Advances inNanomaterials for Gene Delivery-A Review.Nanomaterials 2017,7(5),94.
86.Frolov I,Hardy R,Rice CM.Cis-acting RNA elements at the 5′end ofSindbis virus genome RNA regulate minus-and plus-strand RNAsynthesis.RNA.2001 Nov;7(11):1638-51.
87.Jose J,Snyder JE,Kuhn RJ.A structural and functional perspectiveof alphavirus replication and assembly.Future Microbiol.2009 Sep;4(7):837-56.
88.Bo Li and C.olin N.Dewey.RSEM:accurate transcript quantificationfrom RNA-Seq data with or without a referenfe genome.BMC Bioinformatics,12:323,August 2011
89.Hillary Pearson,Tariq Daouda,Diana Paola Granados,Chantal Durette,Eric Bonneil,Mathieu Courcelles,Anja Rodenbrock,Jean-Philippe Laverdure,Caroline
90.Juliane Liepe,Fabio Marino,John Sidney,Anita Jeko,DanielE.Bunting,Alessandro Sette,Peter M.Kloetzel,Michael P.H.Stumpf,AlbertJ.R.Heck,Michele Mishto.A large fraction of HLA class I ligands areproteasome-generated spliced peptides.Science,21,October 2016.
91.Mommen GP.,Marino,F.,Meiring HD.,Poelen,MC.,van Gaans-van denBrink,JA.,Mohammed S.,Heck AJ.,and van Els CA.Sampling From the Proteome tothe Human Leukocyte Antigen-DR(HLA-DR)Ligandome Proceeds Via HighSpecificity.Mol Cell Proteomics 15(4):1412-1423,April 2016.
92.Sebastian Kreiter,Mathias Vormehr,Niels van de Roemer,MustafaDiken,Martin
93.Tran E.,Turcotte S.,Gros A.,Robbins P.F.,Lu Y.C.,Dudley M.E.,Wunderlich J.R.,Somerville R.P.,Hogan K.,Hinrichs C.S.,Parkhurst M.R.,YangJ.C.,Rosenberg S.A.Cancer immunotherapy based on mutation-specific CD4+Tcells in apatient with epithelial cancer.Science 344(6184)641-645,May 2014.
94.Andreatta M.,Karosiene E.,Rasmussen M.,Stryhn A.,Buus S.,NielsenM.Accurate pan-specific prediction of peptide-MHC class II binding affinitywith improved binding core identification.Immunogenetics 67(11-12)641-650,November 2015.
95.Nielsen,M.,Lund,O.NN-align.An artificial neural network-basedalignment algorithm for MHC class II peptide binding prediction.BMCBioinformatics 10:296,September 2009.
96.Nielsen,M.,Lundegaard,C.,Lund,O.Prediction of MHC class II bindingaffinity using SMM-align,a novel stabilization matrix alignment method.BMCBioinformatics 8:238,July 2007.
97.Zhang,J.,et al.PEAKS DB:de novo sequencing assisted databasesearch for sensitive and accurate peptide identification.Molecular&CellularProteomics.11(4):1-8.1/2/2012.
98.Jensen,Kamilla Kjaergaard,et al.“Improved Methods for PreditingPeptide Binding Affinity to MHC Class II Molecules.”Immunology,2018,doi:10.1111/imm.12889.
99.Carter,S.L.,Cibulskis,K.,Helman,E.,McKenna,A.,Shen,H.,Zack,T.,Laird,P.W.,Onofrio,R.C.,Winckler,W.,Weir,B.A.,et al.(2012).Absolutequantification of somatic DNA alterations in human cancer.Nat.Biotechnol.30,413-421
100.McGranahan,N.,Rosenthal,R.,Hiley,C.T.,Rowan,A.J.,Watkins,T.B.K.,Wilson,G.A.,Birkbak,N.J.,Veeriah,S.,Van Loo,P.,Herrero,J.,et al.(2017).Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution.Cell171,1259-1271.e11.
101.Shukla,S.A.,Rooney,M.S.,Rajasagi,M.,Tiao,G.,Dixon,P.M.,Lawrence,M.S.,Stevens,J.,Lane,W.J.,Dellagatta,J.L.,Steelman,S.,et al.(2015).Comprehensive analysis of cancer-associated somatic mutations in class I HLAgenes.Nat.Biotechnol.33,1152-1158.
102.Van Loo,P.,Nordgard,S.H.,Lingjaerde,O.C.,Russnes,H.G.,Rye,I.H.,Sun,W.,Weigman,V.J.,Marynen,P.,Zetterberg,A.,Naume,B.,et al.(2010).Allele-specific copy number analysis of tumors.Proc.Natl.Acad.Sci.U.S.A.107,16910-16915.
103.Van Loo,P.,Nordgard,S.H.,Lingjaerde,O.C.,Russnes,H.G.,Rye,I.H.,Sun,W.,Weigman,V.J.,Marynen,P.,Zetterberg,A.,Naume,B.,et al.(2010)Allele-sppecific copy number analysis of tumors.Proc.Natl.Acad.Sci.U.S.A.107,16910-16915.
- HERV-K衍生抗原作为共有肿瘤抗原用于抗癌疫苗
- 共有前列腺抗原、编码所述抗原的核酸分子以及包含所述核酸分子的疫苗及其用途