细菌捕获测序平台及其设计、构建和使用方法
文献发布时间:2023-06-19 09:54:18
相关申请的交叉引用
本申请要求2018年5月24日提交的美国专利申请序列号62/675,890和2018年8月29日提交的美国专利申请序列号62/724,014的优先权,这两篇专利的全部内容均据此以引用方式并入本文。
政府支持声明
本发明是在由美国国立卫生研究院授予的AI109761下在美国政府支持下完成的。因此,美国政府对本发明具有一定权利。
技术领域
本发明涉及使用高通量测序进行多重病原菌检测、鉴定和表征的领域。
背景技术
在前抗生素时代中,自然发生的传染病是死亡的常见原因。例如,产后败血症是孕产妇死亡的常见原因。高达30%的儿童没有活过他们出生后第一年,并且社区获得性肺炎和脑膜炎分别导致了30%和70%的死亡率。细菌诊断和抗生素的出现不仅减轻了自然发生的传染病的负担,而且还通过实现临床医学创新(例如器官移植、关节置换和其他有创外科手术,免疫抑制化学疗法和烧伤管理)提高了我们的生活质量。然而,这些进展受到抗微生物抗性(AMR)出现的威胁。在2013年,世界经济论坛(collaborative World EconomicForum)估计仅在美国,每年因医院获得性感染而导致的AMR相关死亡人数就为100,000例(Golkar等人,2014)。据估计AMR的全球影响每年造成700,000例死亡,其中发展中国家负担最重。
对细菌感染进行早期、准确的鉴别诊断对于降低发病率、死亡率和医疗保健成本至关重要。它还可以减少抗生素的不当使用。常用于细菌感染的鉴别诊断的多重PCR方法可识别潜在的病原体,但不能提供对AMR基因存在或表达的洞察。此外,它们不包括仅罕有地与重大疾病相关的细菌,诸如阴道加德纳菌(G.Vaginalis),所述细菌在此参与涉及患有HIV/AIDS的个体的无法解释的败血症。此外,基于培养的方法需要两到几天才能鉴定出病原体,甚至需要更长的时间才能提供抗生素敏感性谱(Rhee等人,2017)。因此,在获取更多具体信息之前,医生通常会施用广谱抗生素(Howell和Davis,2017)。
目前尚无平台允许快速且同步地洞察实现可降低发病率、死亡率和经济负担的早期和精确的抗生素治疗所需的系统发育、致病性标记和抗微生物抗性。
因此,需要一种灵敏的、具有成本效益的捕获测序平台来检测病原菌,尤其是在临床环境中检测病原菌,以及与致病性和抗生素抗性相关的特征。本发明是用于任何类型的样品的临床诊断和细菌分析的基于灵敏且特异的高通量(HTS)的平台。
发明内容
本文描述了一种用于不仅确定样品的细菌组成,而且确定与致病性和抗生素抗性相关联的特征的存在的方法。发明人开发了一种病原菌捕获测序平台(BacCapSeq),该平台大大提高了基于序列的病原菌检测和表征的灵敏度。解决了所有已知的人类细菌病原体以及抗微生物抗性基因。该平台是使用来自Pathosystems资源整合中心(PathosystemsResource Integration Center,PATRIC)数据库的307种最重要的病原菌种类的120万种蛋白质编码序列,以及来自综合抗生素抗性数据库(Comprehensive Antibiotic ResistanceDatabase,CARD)的所有已知抗微生物抗性基因和来自毒力因子数据库(Virulence FactorDatabase,VFDB)的毒力因子设计和构建的。将这些蛋白质编码序列提取并汇集在一起作为捕获靶序列。设计了420万个探针(平均探针长度为75bp,平均探针间距为121bp)以平铺(tile)并覆盖相关靶序列。使用含有那些420万个探针的生物素化寡核苷酸探针文库,以基于溶液捕获存在于复杂样品中的病原菌核酸,该复杂样品含有不同比例的不同病原菌和宿主核酸。与传统的Illumina测序相比,使用BacCapSeq导致来自血液和脑脊液的细菌读段增加了500倍到1,000倍。
BacCapSeq平台理想地适用于基因组组成和动力学分析,并且将使得能够将高通量测序过渡到临床诊断以及研究应用。
本发明提供了用于同时检测、鉴定和/或表征已知或疑似感染脊椎动物,特别是人类的病原菌以及与致病性和抗生素抗性相关联的特征的存在的新颖方法、系统、工具和试剂盒。本文所述的方法、系统、工具和试剂盒基于细菌捕获测序平台(BacCapSeq),所述平台是由发明人开发的新颖平台。
因此,本发明是一种利用针对探针的阳性选择策略设计和/或构建细菌捕获测序平台的方法,所述探针包含来源于病原菌的核酸以及抗微生物抗性基因,所述方法包括以下步骤。
第一步骤是从细菌物种获得序列信息,所述细菌物种包括但不限于已知或疑似对脊椎动物,特别是人类有致病性的物种。表1是307种最重要的已知病原菌种类的列表。
下一步骤是从细菌基因组提取编码序列。从来自PATRIC数据库的307种最重要的已知病原菌物种,以及来自CARD数据库的所有已知的抗微生物抗性基因和来自VFDB数据库的毒力因子中提取120万个的蛋白质编码序列,并汇集在一起作为捕获靶序列。
在下一步骤中,将编码序列分解为平均长度为约75个核苷酸(nt)的片段,其中标准偏差为5.8nt。探针解链温度(Tm)平均为约82.7℃,标准偏差为约5.7℃(中值解链温度为约82.3℃,最小解链温度为约62.4℃,并且最大解链温度为约100.7℃)。
另外,将片段平铺在编码序列上,以使用约420万个探针覆盖数据库中的所有序列,这导致约100个至约150个核苷酸的间隔,其中平均间距或间隔为约120个核苷酸。如果需要更多探针,则间隔可以更小,小于约50个核苷酸,低至约1个核苷酸,甚至可为重叠的探针。如果平台中需要更少的探针,则间隔可以更大,约150个至约200个核苷酸间隔。
本发明的实施方案还提供了用于设计和/或构建细菌捕获测序平台的自动化系统和方法。由本发明的实施方案制作的模型可以被本领域技术人员用来设计和/或构建细菌捕获测序平台。
在本发明的一些实施方案中,提供了在设计模型中使用细菌和序列信息以及分析工具来设计和/或构建细菌捕获测序平台的系统、装置、方法和计算机可读介质。例如,在一些实施方案中,可以使用包含来自公开了包括所有已知的人类致病物种在内的细菌物种的表1的信息的第一分析工具来找到相关的序列信息以及来自综合抗生素抗性数据库(CARD)的所有已知的抗微生物抗性基因和来自VFDB数据库的毒力因子,并使用算法处理所述相关序列信息以提取编码序列,并且使用第二分析工具将所述编码序列分解为片段以提供具有针对所述平台的适当参数的寡核苷酸。
本发明的另一个实施方案是新颖的平台,其又被称为细菌捕获测序平台,所述平台是使用本文所述的方法设计和/或构建的。在一个实施方案中,平台包括介于约一百万个与约五百万个之间的探针,优选地约四百万个探针。在一个实施方案中,探针是寡核苷酸探针。在另一个实施方案中,寡核苷酸探针是合成的。所述平台可包括和/或来源于已知或疑似感染脊椎动物,特别是人类的病原菌的基因组,以及抗微生物抗性基因和毒力因子。在一个实施方案中,平台的探针包含和/或来源于表1中的病原菌的基因组。在另一个实施方案中,平台的探针可包含和/或来源于来自综合抗生素抗性数据库(CARD)的所有已知抗微生物抗性基因的和来自毒力因子数据库(VFDB)的毒力因子。在一个实施方案中,平台为寡核苷酸探针文库的形式。在一个实施方案中,寡核苷酸可包含DNA、RNA、连接的核酸(LNA)、桥接的核酸(BNA)或肽核酸(PNA)以及可天然获得或现在或将来合成的任何核酸。在一个实施方案中,平台为溶液的形式。在另一个实施方案中,平台为固态形式,例如微阵列或微珠。在另一个实施方案中,寡核苷酸被组合物修饰以促进与固态的结合。
本发明的一个实施方案是一种数据库,所述数据库包含所述细菌捕获测序平台上的信息,所述信息至少包括每种寡核苷酸探针的长度、核苷酸序列、解链温度和来源。另一个实施方案是具有包含信息的程序代码的计算机可读存储介质,所述程序代码为例如数据库,所述数据库包含关于所述细菌捕获测序平台的信息,所述信息至少包括每个寡核苷酸探针的长度、核苷酸序列、解链温度和来源。
此外,本发明提供了一种用于以阳性选择方案使用所述细菌捕获测序平台构建用于检测、鉴定和/或表征至少一种细菌或多种细菌的测序文库的方法。
本发明还提供了用于同时检测、鉴定和/或表征任何样品中的病原菌和/或抗微生物抗性基因或生物标记物(包括已知和未知的病原菌和/或抗微生物抗性基因或生物标记物)的系统。所述系统包括至少一个子系统,其中所述子系统包括本发明的细菌捕获测序平台。所述系统还可包括用于进一步检测、鉴定和/或表征所述细菌的子系统,包括但不限于用于从样品制备核酸、杂交、扩增、高通量测序以及鉴定和表征所述细菌的子系统。
本发明还提供了用于利用所述细菌捕获测序平台同时检测任何样品中的细菌和/或抗微生物抗性基因或生物标记物的方法。
本发明还提供了用于利用所述细菌捕获测序平台同时鉴定和表征任何样品中的细菌和/或抗微生物抗性基因或生物标记物的方法。
在前述方法的一些实施方案中,检测、鉴定和/或表征多于一种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于一百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于一百五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于两百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于两百五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于三百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征所有已知或疑似感染脊椎动物的病原菌。在前述方法的一些实施方案中,检测、鉴定和/或表征表1中所列的一些或全部细菌。
本发明还提供了利用所述新颖的细菌捕获测序平台检测、鉴定和/或表征任何样品中的未知细菌和/或抗微生物抗性基因或生物标记物的方法。
本发明还提供了利用所述新颖的细菌捕获测序平台检测、鉴定和/或表征任何样品中已知和未知的AMR基因的方法。
另一个实施方案是一种用于设计和/或构建细菌捕获测序平台的试剂盒,所述试剂盒包括分析工具,所述分析工具用于选择序列信息并将编码序列分解为片段以提供具有针对所述平台的适当参数的寡核苷酸。
另一个实施方案是一种用于检测、鉴定和/或表征已知或疑似感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的试剂盒,所述试剂盒包括细菌捕获测序平台以及任选的引物、酶、试剂、和/或关于进一步检测、鉴定和/或表征样品中的至少一种细菌的用户说明。
附图说明
出于说明本发明的目的,在附图中描绘了本发明的某些实施方案。然而,本发明不限于附图中描绘的实施方案的精确布置和手段。
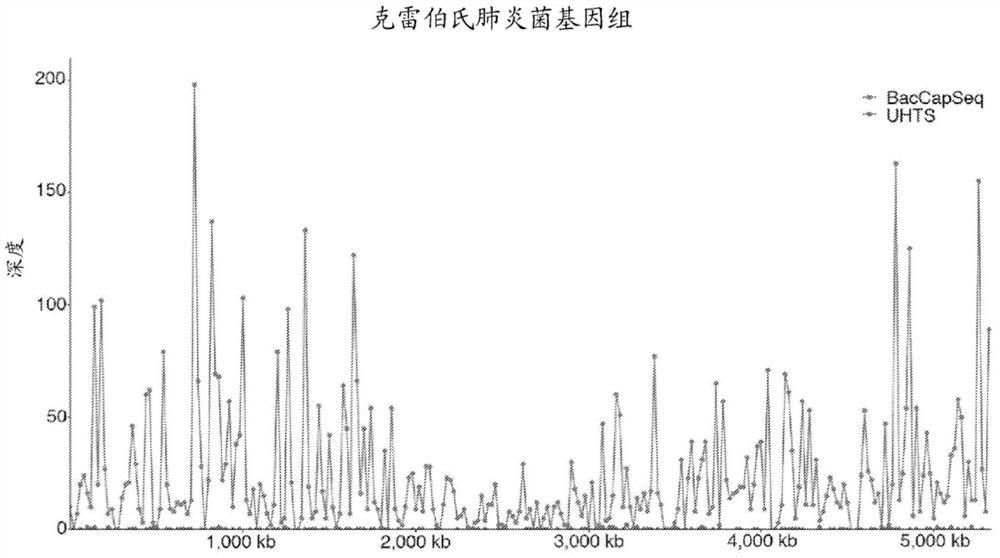
图1示出,与无偏高通量测序相比,BacCapSeq产生更多的读段和更高的基因组覆盖率。图1A是使用对克雷伯氏肺炎菌(K.pneumoniae)基因组的BacCapSeq或无偏高通量测序(UHTS)获得的读取深度的图形表示。图1B是从掺有40,000个拷贝/ml的霍乱弧菌(V.Cholerae)DNA的全血核酸获得的toxR毒力基因的代表性BacCapSeq结果。图1C是从掺有40,000个活的克雷伯氏肺炎菌细胞/ml的全血获得的bla
图2是示出掺有细菌细胞的血液中经定位的(mapped)细菌读段的图。将经定位的细菌读段归一化为通过BacCapSeq(左手侧条形)或UHTS(右手侧条形)获得的100万经质量过滤和宿主过滤的读段。示出的数据代表40,000个细胞/ml。未应用截止阈值。
图3示出了使用BacCapSeq对两名患有HIV/AIDS和无法解释的败血症的免疫抑制患者中的细菌的鉴定。图3A是示出使用BacCapSeq和UHTS鉴定肠道沙门氏菌(Salmonellaenterica)感染的图。图3B是示出使用BacCapSeq和UHTS鉴定肺炎链球菌(Streptococcuspneumoniae)和阴道加德纳菌(Gardnerella vaginalis)共感染的图。图3C示出使用BacCapSeq和UHTS对阴道加德纳菌的基因组覆盖。BacCapSeq导致回收的基因组百分比显著增加。
图4是散布图,示出了使用BacCapSeq检测抗微生物抗性(AMR)生物标记物的结果。在氨苄青霉素存在下培养45分钟、90分钟和270分钟后,测量对氨苄青霉素敏感(AMR+)耐药(AMR-)的金黄色葡萄球菌(Staphylococcus aureus)中的七种转录物的水平。箱形图表示每种基因的归一化转录物计数的对数。仅示出了使用BacCapSeq获得的结果,因为在使用UHTS时,直到以后的时间点在氨苄青霉素存在下都未检测到转录物。
具体实施方式
分子生物学
根据本发明,在本领域技术范围内可以有许多工具和技术,例如在分子免疫学、细胞免疫学、药理学和微生物学中常用的那些工具和技术。参见例如Sambrook等人,(2001)Molecular Cloning:A Laboratory Manual.第3版,Cold Spring Harbor LaboratoryPress:Cold Spring Harbor,N.Y.;Ausubel等人编著,(2005)Current Protocols inMolecular Biology.John Wiley and Sons,Inc.:Hoboken,N.J.;Bonifacino等人编著,(2005)Current Protocols in Cell Biology.John Wiley and Sons,Inc.:Hoboken,N.J.;Coligan等人编著,(2005)Current Protocols in Immunology,John Wiley andSons,Inc.Hoboken,N.J.;Coico等人编著,(2005)Current Protocols in Microbiology,John Wiley and Sons,Inc.:Hoboken,N.J.;Coligan等人编著,(2005)Current Protocolsin Protein Science,John Wiley and Sons,Inc.:Hoboken,N.J.;以及Enna等人编著,(2005)Current Protocols in Pharmacology,John Wiley and Sons,Inc.:Hoboken,N.J.。
本说明书中使用的术语通常具有它们在本领域中、在本发明的情形以及使用每个术语所处的特定情形内的普通含义。在以下或在说明书中其他地方讨论某些术语以在描述本发明方法以及如何使用它们方面对从业者提供额外指导。此外,应了解同一事物可以超过一种方式加以讲述。因此,替代性措辞和同义语可用于本文讨论的术语中的任何一者或多者,无论术语是否在本文中被详尽阐述或讨论都不附加有任何特殊意义。提供了某些术语的同义语。对一种或多种同义语的叙述不排除对其他同义语的使用。在说明书中任何地方对包括本文讨论的任何术语的示例的示例的使用都仅是说明性的,并且决不限制本发明或任何例示术语的范围和含义。同样地,本发明不限于其优选实施方案。
如本文和权利要求书中所使用的,单数形式的“一”、“一个”和“该”包括单数和复数指代物,除非上下文另外明确指出。因此,例如,对“一试剂”的提及包括单个试剂和多个此类试剂。
如本文所用,术语“细菌捕获测序平台”和“BacCapSeq”将可互换使用,并且是指本发明的新颖捕获测序平台,所述新颖捕获测序平台允许在单个高通量测序反应中同时检测、鉴定和/或表征任何单个样品中已知或疑似感染脊椎动物的病原菌。该术语表示各种形式的平台,包括但不限于合成寡核苷酸的集合(即,“探针文库”),所述合成寡核苷酸的集合代表在溶液中或附着于固相支持物的至少一种病原菌的编码序列;数据库,所述数据库包括细菌捕获测序平台上的信息,所述信息至少包括每个寡核苷酸探针的长度、核苷酸序列、解链温度和来源;以及具有程序代码的计算机可读存储介质,所述程序代码包括细菌捕获测序平台上的信息,所述信息至少包括每个寡核苷酸探针的长度、核苷酸序列、解链温度和来源。
本申请中使用的术语“受试者”是指具有免疫系统的动物,诸如禽类和哺乳动物。哺乳动物包括犬科动物、猫科动物、啮齿动物、牛科动物、马科动物、猪科动物、羊科动物和灵长类动物。禽类包括但不限于家禽、鸣禽和猛禽。因此,本发明可用于兽医学中,例如以治疗伴侣动物、农场动物、动物园中的实验室动物以及野生动物。本发明对于人类医学应用特别理想。
如本申请中所用的术语“患者”意指人受试者。
如本文所用的术语“检测(detection/detect/detecting)”等是指发现其存在或先存性。
如本文所用的术语“鉴定(identification/identify/identifying)”等是指识别来自受试者的样品中的一种或多种特定细菌和/或一种或多种基因。
如本文所用的术语“表征(characterization/characterize/characterizing)”等是指按特征描述或分类,在本文的一些情况下是按序列信息描述或分类。
如本文所用,术语“分离的”等是指所提及的材料不含在通常发现该材料的自然环境中发现的组分。具体地,分离的生物材料不含细胞组分。在核酸分子的情况下,分离的核酸包括PCR产物,分离的mRNA、cDNA,分离的基因组DNA、或限制性片段。在另一个实施方案中,优选从可在其中发现核酸的染色体切下分离的核酸。可以将分离的核酸分子插入质粒、粘粒、人工染色体等中。因此,在一个具体的实施方案中,重组核酸是分离的核酸。分离的蛋白质可以与其他蛋白质或核酸或两者缔合,在细胞中所述分离的蛋白质与这两者缔合,或者如果分离的蛋白质是膜相关的蛋白质,则所述分离的蛋白质与细胞膜缔合。分离的物质可以是纯化的,但不是必需为纯化的。
如本文所用,“核酸”、“多核苷酸”、“核酸序列”和“核苷酸序列”包括核酸、寡核苷酸、核苷酸、多核苷酸,以及它们的任何片段、变体或衍生物。所述核酸或多核苷酸可为双链、单链或三链DNA或RNA(包括cDNA),或者是遗传或合成来源的DNA-RNA杂交体,其中所述核酸含有脱氧核糖核苷酸和核糖核苷酸的任何组合,以及碱基的任何组合,所述碱基包括但不限于腺嘌呤、胸腺嘧啶、胞嘧啶、鸟嘌呤、尿嘧啶、肌苷和黄嘌呤次黄嘌呤。如本文中进一步所用的,术语“cDNA”是指分离的DNA多核苷酸或核酸分子,或它们的任何片段、衍生物或补体。它可为双链、单链或三链的,它可为重组或合成来源的,并且它可代表编码和/或非编码的5'和/或3'序列。
当关于核苷酸序列使用时,术语“片段”是指该核苷酸序列的部分。片段的大小可在5个核苷酸残基到整个核苷酸序列减去一个核酸残基的范围内。
如本文所用的术语“基因组”是指生物体的遗传信息的整体,该整体以所述生物体的主要DNA或RNA或核苷酸序列(如果适用的话则为DNA或RNA)编码。基因组包含基因和非编码序列。例如,基因组可代表病毒基因组、微生物基因组或哺乳动物基因组。
“编码序列”或“编码”表达产物(诸如RNA、多肽、蛋白质或酶)的序列,是这样的核苷酸序列,所述核苷酸序列当表达时会导致所述RNA、多肽、蛋白质、或酶的产生,即,所述核苷酸序列编码所述多肽、蛋白质或酶的氨基酸序列。蛋白质的编码序列可包含起始密码子(通常是ATG)和终止密码子。
如本文所用的术语“测序文库”是指与下一代高通量测序仪兼容的核酸文库。
如本文所用,术语“寡核苷酸”或“寡核苷酸探针”是指可与基因组DNA分子、cDNA分子、或编码基因的mRNA分子、mRNA、cDNA或其他感兴趣的核酸杂交的通常为至少10个核苷酸,优选至少15个核苷酸,更优选至少20个核苷酸,优选不超过100个核苷酸的核酸。包含寡核苷酸的核酸包括但不限于DNA、RNA、连接的核酸(LNA)、桥接的核酸(BNA)和肽核酸(PNA)。寡核苷酸可例如用
术语“合成寡核苷酸”是指可合成的优选具有约10个至约100个碱基的单链DNA或RNA分子。通常,尽管可以合成具有相关序列并且在核苷酸序列内特定位置处具有不同核苷酸组成的分子家族,但这些合成分子被设计为具有独特或所需的核苷酸序列。术语合成寡核苷酸将用于指具有设计或所需核苷酸序列的DNA或RNA分子。
如本文所用的术语“标志物(identifier)”是指可用于鉴定核酸片段的起源基因组的任何独特的非天然存在的核酸序列。标志物功能有时可以与其他功能(例如衔接子或引物)结合使用,并且可以位于任何方便的位置。
如本文所用的术语“下一代测序平台”、“高通量测序”和“HTS”是指利用大规模并行技术的任何核酸测序设备。例如,此类平台可包括但不限于Illumina测序平台。
如本文所用,术语“互补的”或“互补性”是关于与碱基配对规则相关的“多核苷酸”和“寡核苷酸”(它们是可互换的术语,是指核苷酸序列)使用的。它也可包括可能不忠实地遵守碱基配对规则的碱基模拟物或人工碱基。例如,序列“C-A-G-T”与序列“G-T-C-A”互补。互补性可以是“部分的”或“全部的”。“部分”互补性是其中一个或多个核酸碱基根据碱基配对规则不匹配的情况。核酸之间的“全部”或“完全”互补性是其中在碱基配对规则下,各个和每一个核酸碱基与另一个碱基匹配的情况。核酸链之间的互补程度对核酸链之间杂交的效率和强度具有重大影响。这在扩增反应以及依赖于核酸之间结合的检测方法中特别重要。
术语“核酸杂交”或“杂交”是指两个单链核酸之间的反向平行氢键键合,其中A与T(或者如果是RNA核酸则为U)配对,并且C与G配对。当一个核酸分子的至少一条链在限定的严格性条件下可与另一核酸分子的互补碱基形成氢键时,所述核酸分子就是彼此“可杂交的”。杂交的严格性是例如通过(i)执行杂交和/或洗涤的温度,以及杂交溶液和洗涤溶液的(ii)离子强度和(iii)变性剂(诸如甲酰胺)的浓度,以及其他参数来确定的。杂交需要两条链包含基本上互补的序列。然而,取决于杂交的严格性,可容许一定程度的错配。在“低严格性”条件下,较大百分比的错配是可容许的(即,将不会阻止反向平行杂交体的形成)。
如本文所用,术语“杂交产物”是指借助于互补的G和C碱基之间以及互补的A和T碱基之间的氢键的形成而在两个核酸序列之间形成的复合物;这些氢键可通过碱基堆积相互作用进一步稳定化。两条互补核酸序列以反向平行构型进行氢键键合。杂交产物可以在溶液中形成,或在溶液中存在的一个核酸序列与固定到固相支持物的另一核酸序列之间形成。
如本文所用的,术语“T
如本文所用,术语“严格性”是关于进行核酸杂交的温度、离子强度和其他化合物(诸如有机溶剂)存在的条件使用的。“严格性”典型地发生在约T
“扩增”被定义为核酸序列的附加拷贝的产生,并且通常在体内或体外进行,即例如使用聚合酶链式反应进行。
如本文所用,术语“聚合酶链式反应”(“PCR”)是指美国专利号4,683,195和4,683,202中公开的方法,所述美国专利以引用方式并入本文,所述美国专利描述了在不进行克隆或纯化的情况下增加基因组DNA混合物中靶序列区段的浓度的方法。所需靶序列的扩增区段的长度由两个寡核苷酸引物相对于彼此的相对位置确定,并且因此,该长度是可控的参数。由于该方法的重复方面,该方法被称为“聚合酶链式反应”(下文称为“PCR”)。因为靶序列的所需扩增区段变成混合物中的主要序列(就浓度而言),所以称它们为“经PCR扩增的”。使用PCR,可以将基因组DNA中特定靶序列的单拷贝扩增到能够通过几种不同方法(例如,与标记探针杂交;掺入生物素化的引物,之后进行抗生物素蛋白-酶缀合物检测;将
术语“序列相似性百分比(%)”、“序列同一性百分比(%)”等通常是指可能共享或可能不共享共同进化起源的核酸分子的不同核苷酸序列之间或可能共享或可能不共享共同进化起源的蛋白质的不同氨基酸序列之间的同一性或对应性程度。序列同一性可以使用许多公开可用的序列比较算法中的任何一种来确定,所述序列比较算法为诸如BLAST、FASTA、DNA Strider和GCG(Genetics Computer Group,用于GCG软件包的程序手册,版本7,Madison,Wisconsin)。
为了确定两个氨基酸序列或两个核酸分子之间的同一性百分比,出于最佳比较目的对所述序列进行比对。两个序列之间的同一性百分比是由序列共享的相同位置的数目的函数(即,同一性百分比=相同位置的数目/位置的总数目(例如重叠位置)×100)。在一个实施方案中,两个序列具有相同的长度或大致相同的长度。两个序列之间的同一性百分比可以使用类似于下面描述的技术在允许或不允许空位的情况下来确定。在计算序列同一性百分比时,通常计数精确匹配。
本文显示的是一种平台,所述平台提高高通量测序用于检测和表征细菌、毒力决定簇和抗微生物抗性(AMR)基因的敏感性。该系统使用的探针组由基于Pathosystems资源整合中心(PATRIC)数据库、综合抗生素抗性数据库(CARD)和毒力因子数据库(VFDB)的420万个寡核苷酸组成,所述寡核苷酸代表307种细菌种类,所述307种细菌种类分别包括所有已知的人类致病物种、已知的抗微生物抗性基因和已知的毒力因子。与传统的无偏高通量测序(UHTS)相比,细菌捕获测序(BacCapSeq)的使用导致血液样品中的细菌读段增加高达1,000倍,并且将检测限降低了1-2个数量级,低到与试剂特异性实时PCR相当的水平,每个样品产生的总读段少至500万。它不仅检测AMR基因的存在,而且还检测包括组成型表达和差异表达的转录物两者的AMR的生物标记物的存在。BacCapSeq平台理想地适用于基因组组成和动力学分析,并且将使得能够将高通量测序过渡到临床诊断以及研究应用。
用掺有已知浓度的细菌DNA(实施例3)或细菌细胞(实施例4)的血液样品获得的结果表明,与无偏高通量测序(UHTS)相比,使用BacCapSeq回收的读段数和获得的基因组覆盖率呈剂量依赖的、一致的提高。在细菌载量低至每毫升40个细胞的情况下,UHTS未检测到肺结核分枝杆菌(M.tuberculosis)、克雷伯氏肺炎菌(K.pneumoniae)、脑膜炎奈瑟氏球菌(N.meningitidis)或肺炎链球菌(S.pneumoniae)的序列或仅检测到百日咳博代氏杆菌(B.pertussis)的一个读段。在这些情况中的每种情况下,BacCapSeq均检测到多个读数(肺结核分枝杆菌,6个;克雷伯氏肺炎菌,522个;脑膜炎奈瑟氏球菌,151个;肺炎链球菌,4个;百日咳博代氏杆菌,269个)(实施例4;表4)。在分析来自患有无法解释的败血症的患者的血液时也观察到了这一优势(实施例6;图3),其中对于肠道沙门氏菌(S.enterica)(3183对比132)、肺炎链球菌(419070对比130)和阴道加德纳菌(776113对比2080),使用BacCapSeq获得的读段均高于使用UHTS获得的读段。这些发现表明,在血液中细菌水平低于每毫升40个细胞的情况下,BacCapSeq可能指示存在可能被UHTS遗漏的致病性病原体。
血液培养系统中的孵育期为3天到5天(Bourbeau等人,2005;Cockerill等人,2004)。灵敏检测奈瑟氏菌属(Neisseria)、立克次体属(Rickettsia)、分支杆菌属(Mycobacterium)、细螺旋体属(Leptospira)、埃立克体属(Ehrlichia)、柯克斯氏体属(Coxiella)、弯曲杆菌属(Campylobacter)、伯克氏菌属(Burkholderia)、布鲁氏菌属(Brucella)、博代氏杆菌属(Bordetella)、和巴尔通氏体属(Bartonella)的一些病原菌种类可能需要更长的时间间隔。另一个挑战是细菌载量可为很低或断续的。Cockerill等人和Lee等人已建议,需要在至少20ml血液的四次独立收集中收集共80ml血液,才能在检测活菌时达到99%的检测灵敏度。目前对BacCapSeq灵敏度的估计(最小40个拷贝/ml)有利地与培养测试(Lee等人,2007)中推荐的80ml样品量相当。美国微生物学会和临床与实验室标准协会(American Society for Microbiology and the Clinical and LaboratoryStandards Institute,CLSI)要求假阳性率低于3%(CLSI 2007)。因为普通的消毒剂无法消除核酸,因此使用BacCapSeq时诊断微生物学中的卫生学规程将比培养时更为严格,从而减少了假阳性。
BacCapSeq还被设计用于检测CARD数据库中的所有AMR基因。在这些基因位于细菌染色体上的情况下,预期侧翼序列将允许与样品内的特定细菌缔合,即使当这些样品含有多于一种细菌种类时也是如此。BacCapSeq将使得能够发现组成型表达和诱导的转录物,这些组成型表达和诱导的转录物反映了功能性细菌特异性AMR元件的存在。
本发明包括一种设计和/或构建细菌捕获测序平台的方法;所述平台本身;以及使用所述平台构建适用于以任何高通量测序技术进行测序的测序文库的方法。本发明还包括用于使用新颖的细菌捕获测序平台同时检测任何来源的单个样品中已知或疑似感染脊椎动物,包括人类的病原菌和/或抗微生物抗性基因或生物标记物的方法和系统。与现有技术中的当前方法相比,被称为细菌捕获测序平台或BacCapSeq的本发明大大提高了基于序列的细菌检测和表征的灵敏度。它使得能够检测任何复杂样品背景中的细菌序列,包括在临床标本中发现的细菌序列。本发明不仅允许检测样品的细菌组成,而且还允许检测与致病性和抗生素抗性相关联的特征的存在。
因此,本发明是一种设计和/或构建序列捕获平台或技术,也称为细菌捕获测序平台或BacCapSeq的方法。本发明是一种设计和/或构建序列捕获平台的方法,所述序列捕获平台包含选择性富集了病原菌和抗微生物抗性基因的寡核苷酸探针;以及所得的细菌捕获测序平台。因此,所述方法可包括以下步骤。
第一步骤是从病原菌以及抗微生物抗性基因和毒力因子获得序列信息。在一个实施方案中,表1中列出的细菌用于获得序列数据。在另一个实施方案中,还可以包括新细菌以及新发现的抗微生物抗性基因。
序列信息是从任何公共或私人的细菌和/或AMR基因和/或毒力因子序列信息数据库中获得的,所述数据库包括但不限于PATRIC、CARD和VFDB。
该方法的第二步骤是从数据库中提取编码序列,以用于设计寡核苷酸。
具体地,从来自PATRIC数据库的307种重要的病原菌物种,以及来自CARD数据库的所有已知的抗微生物抗性基因和来自VFDB数据库的毒力因子中提取120万个的蛋白质编码序列,并汇集在一起作为捕获靶序列。
该方法的下一步骤是将序列分解成片段,以作为寡核苷酸的基础。具体地,设计了约420万个探针,平均探针长度为约75nt,并且平均探针间间距为121nt,以平铺和覆盖所有相关靶序列。
片段的长度为约50个至约100个核苷酸,平均长度为约75nt,标准偏差为5.8nt(中值长度为约75nt,最小长度为约50nt,并且最大长度为约100nt)。寡核苷酸可以如T
例如,寡核苷酸的最终T
另外,将片段平铺在编码序列上,以便用约420万个探针覆盖数据库中的所有序列,这导致约100个至约150个核苷酸的间隔,以及约120个核苷酸的平均间距。如果需要更多探针,则间隔可以更小,小于约100个核苷酸,低至约1个核苷酸,甚至可以为重叠探针。如果平台中需要更少的探针,则间隔可以更大,约150个至约200个核苷酸。
本发明还涉及使用计算机生成的信息来设计和/或构建细菌捕获测序平台的方法和系统。例如,在一些实施方案中,使用来自公开了病原菌的表1和来自综合抗生素抗性数据库(CARD)的所有已知抗微生物抗性基因和来自毒力因子数据库(VFDB)的毒力因子的信息的第一分析工具可用于查找相关序列信息,并使用算法处理所述相关序列信息以提取编码序列,并且使用第二分析工具将编码序列片段化为具有针对所述平台的适当参数的寡核苷酸,所述适当参数包括适当的长度、解链温度、GC分布、编码序列上的寡核苷酸之间间隔的距离,以及序列同一性百分比。
在本发明的另一方面中,可以提供分析工具(例如被配置为执行对来自表1中的细菌、来自综合抗生素抗性数据库(CARD)的所有已知抗微生物抗性基因和来自毒力因子数据库(VFDB)的毒力因子的编码序列进行选择的第一模块,以及用于执行编码序列片段化的第二模块),来确定寡核苷酸的特征,诸如适当的长度、解链温度、GC分布、编码序列上的寡核苷酸之间间隔的距离以及序列同一性百分比。这些工具的结果形成了用于设计细菌捕获测序平台的寡核苷酸的模型。
用于生成设计模型的说明性系统包括分析工具,诸如被配置为包括来自表1的细菌、来自综合抗生素抗性数据库(CARD)的所有已知抗微生物抗性基因以及来自毒力因子数据库(VFDB)的毒力因子的模块;以及序列信息数据库。分析工具可包括任何合适的硬件、软件或它们的组合,以用于确定来自表1的细菌与来自数据库的序列数据之间的相关性。第二分析工具(例如模块)用于对编码序列进行片段化。该分析工具可包括任何合适的硬件、软件或组合,以用于确定细菌捕获测序平台的寡核苷酸的必要特征,包括适当的长度、解链温度、GC分布、编码序列上的寡核苷酸之间间隔的距离,以及及序列同一性百分比。在本发明的一些实施方案中,寡核苷酸的特征是长度为约50个至100个核苷酸,解链温度在约62℃至约101℃的范围内,并且在编码序列上间隔开约100个至150个核苷酸的间隔。
在获得寡核苷酸探针的序列信息后,可以通过本领域已知的任何方法合成所述寡核苷酸,所述方法包括但不限于使用亚磷酰胺方法和衍生自受保护的2'-脱氧核苷(dA、dC、dG和T)、核糖核苷(A、C、G和U)或经化学修饰的核苷(例如连接的核酸(LNA)、桥接的核酸(BNA)或肽核酸(PNA))的亚磷酰胺结构单元进行固相合成。
寡核苷酸可以如T
该平台的一个实施方案是一种文库,所述文库包含能够从已知或疑似感染脊椎动物的至少一种病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从已知或疑似感染脊椎动物的多于一种病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从已知或疑似感染脊椎动物的多于十种病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于五十种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于一百种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于一百五十种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于两百种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于两百五十种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够从多于三百种已知或疑似感染脊椎动物的病原菌中捕获核酸的寡核苷酸探针。在一些实施方案中,所述平台是一种文库,所述文库包含能够捕获来自表1中所列的细菌的核酸的寡核苷酸探针。
另一个实施方案是一种文库,所述文库还包含能够从AMR基因捕获核酸的寡核苷酸探针。另一个实施方案是一种文库,所述文库还包含能够从毒力因子捕获核酸的寡核苷酸探针。
在一个实施方案中,平台的寡核苷酸在溶液中。
在本发明的一个实施方案中,将构成细菌捕获测序平台的寡核苷酸预结合至固相支持物或底物。优选的固相支持物包括但不限于由金属、玻璃、塑料、右旋糖酐(例如,以商品名Sephadex(Pharmacia)销售的右旋糖酐珠粒)、硅胶、琼脂糖凝胶(例如以商品名Sepharose(Pharmacia)销售的那些)或纤维素)制成的珠粒(例如,磁性珠粒(即,珠粒本身是磁性的,或者珠粒易于被磁体捕获));毛细管;扁平支持物(例如,由玻璃、金属(例如钢、金、银、铝、铜或硅)或塑料(诸如聚乙烯、聚丙烯、聚酰胺或聚偏二氟乙烯)制成的过滤器、板或膜);色谱底物;微流体底物;和针头(pin)(例如,适合于组合合成或分析平坦表面(例如晶片)的凹坑中的珠粒的针头的阵列,带有或不带有滤板)。合适的固相支持物的其他示例包括但不限于琼脂糖(agarose)、纤维素、右旋糖苷、聚丙烯酰胺、聚苯乙烯、琼脂糖凝胶(sepharose)和其他不溶性有机聚合物。熟练的技术人员可容易地确定适当的结合条件(例如,温度、pH和盐浓度)。
构成细菌捕获测序平台的寡核苷酸可以共价或非共价结合至固相支持物。此外,构成细菌捕获测序平台的寡核苷酸可以直接结合到固相支持物(例如,所述寡核苷酸以直接范德华力和/或氢键和/或盐桥与固相支持物接触),或间接结合至固相支持物(例如,所述寡核苷酸不与固相支持物本身直接接触)。当构成细菌捕获测序平台的寡核苷酸间接结合至固相支持物时,捕获核酸的核苷酸与中间组合物连接,所述中间组合物本身与固相支持物直接接触。
为了促进构成细菌捕获测序平台的寡核苷酸与固相支持物的结合,可以用一种或多种适于直接结合至固相支持物和/或借助于与固相支持物结合的中间组合物或间隔物分子(例如抗体、受体、结合蛋白或酶)间接结合至固相支持物的分子修饰构成细菌捕获测序平台的寡核苷酸。此类修饰的示例包括但不限于配体(例如,有机或无机小分子、受体的配体、结合蛋白的配体或它们的结合结构域(例如生物素和地高辛))、抗原及其结合结构域、适体、肽标签、抗体和酶底物。在一个优选的实施方案中,寡核苷酸包含生物素。
适用于作为从固体表面间隔的生物学分子和其他分子(包括核酸/多核苷酸)的接头或间隔物分子是本领域中众所周知的,并且包括但不限于多肽、饱和或不饱和双官能烃和聚合物(例如,聚乙二醇)。其他有用的接头是可商购获得的。
在本发明的一个实施方案中,构成细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的至少一种细菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的至少一种细菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一个实施方案中,构成所述细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于一种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于一种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一实施方案中,构成细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于五十种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于五十种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一个实施方案中,构成所述细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于一百种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于一百种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一实施方案中,构成细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于一百五十种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于一百五十种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一个实施方案中,构成所述细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于两百种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于两百种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一实施方案中,构成细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于两百五十种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于两百五十种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一个实施方案中,构成所述细菌捕获测序平台的寡核苷酸的序列是具有已知或疑似感染脊椎动物的多于三百种病原菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一个实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有已知或疑似感染脊椎动物的多于三百种病原菌的基因组以及抗微生物抗性基因和毒力因子的序列杂交。
在本发明的另一实施方案中,构成细菌捕获测序平台的寡核苷酸的序列是具有表1中所列的细菌中的一些或全部细菌的基因组以及抗生素抗性基因和毒力因子的序列的补体(即,与所述序列互补)。在另一实施方案中,构成细菌捕获测序平台的寡核苷酸能够在严格条件下与具有表1中所列的细菌中的一些或全部细菌的基因组以及抗生素抗性基因和毒力因子的序列杂交。
核酸序列的“补体”在本文中是指这样的核酸分子,所述核酸分子与另一核酸完全互补或将在高严格性条件下与所述另一核酸杂交。高严格性条件是本领域中已知的。参见例如Maniatis等人,Molecular Cloning:A Laboratory Manual,第2版(Cold SpringHarbor:Cold Spring Harbor Laboratory,1989)和Ausubel等人编著,Current Protocolsin Molecular Biology(New York,N.Y.:John Wiley&Sons,Inc.,2001)。严格条件取决于序列,并且可以根据情况而变化。
在例示性实施方案中,使用可切割的可编程阵列合成构成细菌捕获测序平台的寡核苷酸,其中该阵列包含构成细菌捕获测序平台的寡核苷酸。从阵列上切下寡核苷酸,并与来自溶液中样品的核酸杂交。
本发明还包括由本发明的一种方法制成的序列捕获平台,也称为细菌捕获测序平台。该平台包含约420万个探针。寡核苷酸包含来源于表1中所列出的细菌的基因组的序列以及来源于抗微生物抗性基因和毒力因子的序列。
本发明的细菌捕获测序平台可以是寡核苷酸集合的形式,优选如上所述设计,即为探针文库。寡核苷酸可以在溶液中或附接至固态,例如阵列或珠粒。另外,寡核苷酸可以用另一个分子修饰。在一个优选的实施方案中,寡核苷酸包含生物素。
细菌捕获测序平台也可为一个或多个数据库的形式,所述数据库可包括关于每个寡核苷酸探针的序列和长度以及T
表1-BacCapSeq中靶向的细菌
本发明的另一实施方案是一种构建适用于利用新颖的细菌捕获测序平台以任何高通量测序方法进行测序的测序文库的方法。
因此,所述方法可包括以下步骤。
从样品中获得核酸。用于本发明中的样品可以是环境样品、食品样品或生物样品。优选的样品是生物样品。生物样品可以获自受试者的组织或来自受试者的体液,所述体液包括但不限于鼻咽吸出物、血液、脑脊液、唾液、血清、尿液、痰、支气管灌洗液、心包液或腹膜液;或固体,诸如粪便。生物样品也可以是细胞、细胞培养物或细胞培养基。样品可包含或含有或可不包含或含有任何细菌核酸。在一个实施方案中,样品来自脊椎动物受试者,并且在另一实施方案中,样品来自人类受试者。在另一个实施方案中,样品包括血液。在另一个优选的实施方案中,样品包含细胞、细胞培养物、细胞培养基或用于开发药物和治疗剂的任何其他组合物。在一些实施方案中,样品来自食物或食物供应。
使来自样品的核酸经受片段化,以获得核酸片段。对可以使用的核酸样品的类型没有特别限制,并且对用于执行片段化的手段没有特别限制。可以使用随机地使核酸样品片段化的任何化学或物理方法。优选地,将核酸样品片段化以获得具有约200bp至约300bp的长度或适合于相应测序平台的任何其他大小分布的核酸片段。
在获得后,可以将核酸片段连接至衔接子。在一个实施方案中,衔接子是线性衔接子。可以通过以下方式来将线性衔接子添加至片段:对片段进行末端修复,以获得经末端修复的片段;向片段的3'末端添加腺嘌呤碱基,以获得在3'末端具有腺嘌呤的片段;以及将衔接子与在3'末端具有腺嘌呤的片段连接。
在一些实施方案中,衔接子包含标志序列。在一些实施方案中,衔接子包含用于引发扩增的序列。在一些实施方案中,衔接子包含鉴定出的序列和用于引发扩增的序列两者。
将核酸片段连接至衔接子后,在允许核酸片段与细菌捕获测序平台的寡核苷酸杂交(如果所述核酸包含来自在细菌捕获测序平台中代表的细菌或基因的任何细菌序列的话)的条件下,使所述核酸片段与细菌捕获测序平台的寡核苷酸接触。根据细菌捕获测序平台的形式,该步骤可以以溶液或以固相杂交方法执行。
与细菌捕获测序平台的寡核苷酸接触后,任何杂交产物都可能受到扩增条件的影响。在一个实施方案中,用于扩增的引物存在于与核酸片段连接的衔接子中。所得的一种或多种扩增产物构成了适合于使用现在已知或以后开发的任何HTS系统进行测序的测序文库。
扩增可以通过本领域已知的任何手段进行,包括聚合酶链式反应(PCR)和等温扩增。PCR是用于体外扩增DNA碱基序列的实用系统。例如,PCR测定可使用热稳定的聚合酶和两个引物:一个引物与在要扩增的序列的一端处的(+)链互补;并且另一个引物与在另一端处的(-)链互补。因为新合成的DNA链随后可以用作相同引物序列的附加模板,所以引物退火、链延伸和解离的连续轮次可产生所需序列的快速且高度特异性扩增。PCR也可用于检测DNA样品中限定序列的存在。在本发明的一个优选实施方案中,将杂交产物与合适的PCR试剂混合。然后执行PCR反应以扩增杂交产物。
在一个实施方案中,在可切割的阵列中使用细菌捕获测序平台构建测序文库。从样品中提取核酸,并进行逆转录酶处理,然后连接至包含标志物的衔接子和用于引发扩增的序列。使用可切割的阵列平台合成构成细菌捕获测序平台的寡核苷酸,其中所述寡核苷酸是生物素化的。然后将生物素化的寡核苷酸从固体基质切割到具有来自样品的核酸的溶液中,以使构成细菌捕获测序平台的寡核苷酸与溶液中的任何细菌核酸杂交。在杂交后,通过链霉亲和素磁性珠粒收集与构成序列捕获平台的生物素化的寡核苷酸结合的来自样品的一种或多种核酸(即,一种或多种杂交产物),并使用衔接子序列作为特异性引发位点通过PCR进行扩增,从而产生扩增产物,以在任何已知的HTS系统(Ion,Illumina,454)和将来开发的任何HTS系统上进行测序。
在另一个实施方案中,可以使用本领域已知的任何方法直接对测序文库进行测序。换句话说,被平台捕获的核酸可以进行测序而不进行扩增。
本发明包括用于以下的方法和系统:利用新颖的细菌捕获测序平台同时检测任何样品中已知或疑似感染脊椎动物,包括人类的病原菌以及抗微生物抗性基因或生物标记物;鉴定和表征任何样品中存在的细菌和/或抗微生物抗性基因或生物标记物;以及鉴定任何样品中的新颖细菌和/或抗微生物抗性基因或生物标记物。
本发明的方法和系统可用于检测研究、临床、环境和食物样品中已知和新颖的细菌和/或抗微生物抗性基因或生物标记物。附加应用包括但不限于检测感染性病原体、筛查血液产品的(例如,筛查血液产品中的病原体)、生物防御、食品安全、环境污染、法医学和基因可比性研究。本发明还提供了用于检测细胞、细胞培养物、细胞培养基和其他用于开发药物和治疗剂的组合物中的细菌和/或抗微生物抗性基因或生物标记物的方法和系统。因此,本发明提供了用于多种特定应用的方法和系统,包括但不限于用于确定样品中细菌和/或抗微生物抗性基因或生物标记物的存在的方法;用于筛查血液产品的方法;用于测定食物产品的污染的方法;用于测定环境污染样品的方法;以及用于检测经遗传修饰的生物的方法。本发明还提供了所述系统在诸如抵抗生物恐怖主义的生物防御、法医学和遗传可比性研究等一般应用中的用途。
受试者可以是任何动物,特别是脊椎动物,更特别是哺乳动物,包括但不限于牛、狗、人、猴、小鼠、猪或大鼠。优选地,受试者是人。所述受试者可已知为患有病原体感染,疑似患有病原体感染或被认为没有病原体感染。
本文所述的系统和方法支持对任何样品中的多种细菌和细菌转录物的多重检测。
因此,本发明的一个实施方案提供了一种用于同时检测任何样品中的已知或疑似感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的系统。所述系统包括至少一个子系统,其中所述子系统包括如本文所述的细菌捕获测序平台。该系统还可包括用于以下目的的附加子系统:从样品中分离和制备核酸片段;将来自样品的核酸片段与细菌捕获测序平台的寡核苷酸杂交以形成一个或多个杂交产物;扩增所述一个或多个杂交产物;以及对所述一个或多个杂交产物进行测序。
本发明还提供了一种用于同时鉴定和表征任何样品中的已知感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的系统。所述系统包括至少一个子系统,其中所述子系统包括如本文所述的细菌捕获测序平台。该系统还可包括用于以下目的附加子系统:从样品中分离和制备核酸片段;将来自样品的核酸片段与细菌捕获测序平台的寡核苷酸杂交以形成杂交产物;扩增一种或多种杂交产物;对一种或多种杂交产物进行测序;以及通过杂交产物序列与已知细菌和/或抗微生物抗性基因或生物标记物之间的比较来鉴定和表征细菌。
在前述系统的一些实施方案中,检测、鉴定和/或表征多于一种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于十种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于五十种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于一百种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于一百五十种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于两百种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于两百五十种细菌。在前述系统的一些实施方案中,检测、鉴定和/或表征多于三百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征所有已知或疑似感染脊椎动物的病原菌。在前述系统的一些实施方案中,检测、鉴定和/或表征表1中所列的一些或全部细菌。
本发明还提供了用于鉴定任何样品中的新颖细菌和/或抗微生物抗性基因或生物标记物的系统。所述系统包括至少一个子系统,其中所述子系统包括如本文所述的细菌捕获测序平台。该系统还可包括用于以下目的附加子系统:从样品中分离和制备核酸片段;将来自样品的核酸片段与细菌捕获测序平台的寡核苷酸杂交以形成杂交产物;扩增一种或多种杂交产物;对一种或多种杂交产物进行测序;以及通过比较杂交产物和已知细菌和/或抗微生物抗性基因或生物标记物的序列来鉴定出所述细菌和/或抗微生物抗性基因或生物标记物为新颖的。
另外,本发明提供了一种用于同时检测任何样品中已知或疑似感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的方法,所述方法包括以下步骤:获得所述样品;从所述样品中分离并制备核酸片段;在足以使所述核酸片段和细菌捕获测序平台的寡核苷酸杂交的条件下,使来自所述样品的所述核酸片段与所述细菌捕获测序平台的寡核苷酸接触;以及检测在所述核酸片段与所述细菌捕获测序平台的寡核苷酸之间形成的任何杂交产物。
该方法还可包括对杂交产物进行扩增和测序的步骤。
本发明提供了一种同时鉴定和表征任何样品中已知或疑似感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的方法,所述方法包括以下步骤:获得样品;从所述样品中分离并制备核酸片段;在足以使核酸片段和细菌捕获测序平台的寡核苷酸杂交的条件下,使来自所述样品的所述核酸片段与所述细菌捕获测序平台的寡核苷酸接触;对在所述核酸片段与所述细菌捕获测序平台的寡核苷酸之间形成的任何杂交产物进行测序;比较所述一个或多个杂交产物的序列与已知细菌和/或抗微生物抗性基因或生物标记物的序列;以及通过将所述一个或多个杂交产物的序列与已知的细菌和/或抗微生物抗性基因或生物标记物的序列进行比较,来确定和表征所述样品中的细菌和/或抗微生物抗性基因或生物标记物。
该方法还可包括扩增杂交产物的步骤。
在前述方法的一些实施方案中,检测、鉴定和/或表征多于一种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于一百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于一百五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于两百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于两百五十种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征多于三百种细菌。在前述方法的一些实施方案中,检测、鉴定和/或表征所有已知或疑似感染脊椎动物的病原菌。在前述方法的一些实施方案中,检测、鉴定和/或表征表1中所列的一些或全部细菌。
本发明提供了一种检测任何样品中的新颖细菌和/或抗微生物抗性基因或生物标记物的方法,所述方法包括以下步骤:获得样品;从所述样品中分离并制备核酸片段;在足以使核酸片段和细菌捕获测序平台的寡核苷酸杂交的条件下,使来自所述样品的所述核酸片段与所述细菌捕获测序平台的寡核苷酸接触;对在所述核酸片段与所述细菌捕获测序平台之间形成的任何杂交产物进行测序;比较所述一个或多个杂交产物的序列与已知的细菌和/或抗微生物抗性基因或生物标记物的序列;以及通过将所述一个或多个杂交产物的序列与已知的细菌和/或抗微生物抗性基因或生物标记物的序列进行比较来检测新颖的细菌和/或抗微生物抗性基因或生物标记物,其中如果杂交产物的序列与已知序列不同或不足够相似,则所述细菌和/或微生物抗性基因或生物标记物是新颖的。
该方法还可包括扩增杂交产物的步骤。
当实践所述用于测定和表征样品中的细菌和/或抗微生物抗性基因或生物标记物的方法以及用于检测样品中新颖的细菌和/或抗微生物抗性基因或生物标记物的存在的方法时,将杂交产物的一个或多个序列与已知的细菌和/或抗微生物抗性基因或生物标记物的核酸序列进行比较。这可以使用用于它们的用途的多种媒体形式的数据库来实现。
如上文所公开的,本发明的用于检测、鉴定和/或表征已知或疑似感染脊椎动物的病原菌和/或抗微生物抗性基因或生物标记物的方法可以对任何疑似具有细菌或细菌核酸的样品执行,所述样品包括但不限于生物样品、环境样品或食物样品。优选的样品是生物样品。生物样品可以获自受试者的组织或来自受试者的体液,所述体液包括但不限于鼻咽吸出物、血液、脑脊液、唾液、血清、尿液、痰、支气管灌洗液、心包液或腹膜液;或固体,诸如粪便。生物样品也可以是细胞、细胞培养物或细胞培养基。样品可包含或含有或可不包含或含有任何细菌核酸。
在一个优选的实施方案中,样品来自脊椎动物受试者,并且在最优选的实施方案中,样品来自人类受试者。在另一个优选的实施方案中,样品包含细胞、细胞培养物、细胞培养基或用于开发药物和治疗剂的任何其他组合物。
本发明还包括用于实践本发明方法的试剂和试剂盒。这些试剂和试剂盒可能有所不同。
一种试剂是细菌捕获测序平台。该平台可以是寡核苷酸探针的集合的形式,所述寡核苷酸探针包含来源于已知或疑似感染脊椎动物的病原菌的基因组以及抗微生物抗性基因的序列。该平台可以是寡核苷酸探针的集合的形式,所述寡核苷酸探针包括来源于表1中所列的病原菌的基因组的序列。所述寡核苷酸探针的集合可以在溶液中或附着至固态。另外,寡核苷酸探针可以经修饰以用于反应中。优选的修饰是向探针添加生物素。
平台也可以是具有关于寡核苷酸的信息的可搜索数据库的形式,所述信息至少包括序列信息、长度和解链温度以及来源。
在试剂盒中的其它试剂可包括用于从样品中分离和制备核酸的试剂、用于使来自所述样品的核酸片段与所述平台的所述寡核苷酸杂交的试剂、用于扩增所述杂交产物的试剂,以及用于获得序列信息的试剂。
本发明的试剂盒可包括上述试剂中的任何试剂,以及可用于比较例如由合适的计算装置基于序列信息的输入获得的测试序列信息的参考/对照序列。
此外,试剂盒还应包括使用说明。
另一个实施方案是一种用于设计和/或构建细菌捕获测序平台的试剂盒,所述试剂盒包括分析工具,所述分析工具用于选择序列信息并将编码序列分解为片段以提供具有针对所述平台的适当参数的寡核苷酸,所述适当参数包括适当的长度、解链温度、GC分布、编码序列上的寡核苷酸之间间隔开的距离,以及序列同一性百分比。该试剂盒还可包含有关数据库和编码序列选择的使用说明。
表2-用于qPCR的引物和探针
从307个物种的选定基因组中提取蛋白质编码序列,并将所述蛋白质编码序列与具有CARD数据库(Jia等人,2017)中的2169个抗微生物抗性基因序列和VFDB数据库(Chen等人,2016;Chen等人,2004)中的30178个毒力因子基因的完整数据集进行组合。将组合的靶序列数据集以96%的序列同一性聚类(产生1,007,426个基因),并发送到Roche-NimbleGen的生物信息学中心(Madison,WI,USA),在那里基于打印考虑对序列进行进一步过滤。通过调整探针的起始/终止位置来改进探针长度,以约束解链温度。最终文库包含4,220,566个平均长度为75nt的寡核苷酸。沿着所靶向的细菌蛋白质组、毒力和AMR靶标的探针之间的平均探针间距离为121个核苷酸。
对于转录分析,使用STAR读段定位包对MiSeq读段进行比对(Dobin等人,2013)。使用featureCounts(Liao等人,2014)从每个样本中提取表达数据,并将结果编译成代表每个基因的转录物计数的主数据文件。将这些数据基于每个样品的测序读段数进行归一化,然后按菌株(AMR+/AMR-)、时间点对数据进行分选,并进行抗生素处理以基于这些度量鉴定出生长方式不同的基因。
基于Pathosystems资源整合中心(PATRIC)数据库(Wattam等人,2017)装配包含420万个寡核苷酸的探针组,该探针组代表307种细菌物种,该307种细菌物种包括所有已知的人类致病物种。所述探针组还基于综合抗生素抗性数据库(CARD)(Jia等人,2016)和毒力因子数据库(VFDB)(Chen等人,2016;Chen等人,2004)中的序列代表了所有已知的抗微生物抗性基因和毒力因子。
沿着307个靶向细菌的编码序列选择探针(参见表1),其中平均长度为75个核苷酸(nt)以保持探针解链温度(Tm)的平均值为79℃。沿着捕获所靶向的带注释的蛋白质编码序列的探针之间的平均间隔为121nt。探针捕获包含与其靶标邻接的序列的片段,因此,回收了几乎完整的蛋白质编码序列。
图1A中示出了克雷伯氏肺炎菌的一个示例。基于CARD和VFDB数据库的探针确保了对AMR基因和毒力因子的覆盖,如通过检测到霍乱弧菌中的toxR毒力因子调节剂(图1B)和克雷伯氏肺炎菌中的bla
通过对使用每个样品500万个读段获得的数据进行并排比较来评定BacCapSeq对比常规无偏高通量测序(UHTS)的效率。首先评定浓度为40个至40,000个拷贝/毫升的掺有百日咳博代氏杆菌(B.pertussis)、大肠杆菌(E.coli)、脑膜炎奈瑟菌(N.meningitidis)、肠道沙门氏菌血清型伤寒沙门氏菌(S.enterica)、无乳链球菌(S.agalactiae)、肺炎链球菌(S.pneumoniae)、霍乱弧菌(V.cholerae)和空肠弯曲菌(C.jeuni)的DNA的全血的提取物。当与UHTS相比时,对于所有测试的细菌靶标,BacCapSeq产生高达100倍的读段和更高的基因组覆盖率(表3)。BacCapSeq的性能增强在较低的复制浓度下尤其明显。
表3-使用BacCapSeq和UHTS获得的掺有细菌DNA的全血提取物中的读段计数和基因组覆盖率
a-显示了在不应用截止阈值的情况下,每100万个读段中的细菌读段。
用掺有克雷伯氏肺炎菌(K.pneumoniae)、百日咳博代氏杆菌、脑膜炎奈瑟菌、肺炎链球菌和结核分枝杆菌(M.tuberculosis)细菌细胞的全血测试性能。从经掺加的样品中提取核酸并进行处理以供进行BacCapSeq或UHTS。与实施例3相似,与无偏HTS相比,BacCapSeq产生更多的读段和更高的基因组覆盖率,其中读段计数增大了高达1500倍(表4和图2)。
表4-使用BacCapSeq和UHTS获得的掺有细菌细胞的全血提取物中的读段计数和基因组覆盖率
a–显示了在不应用截止阈值的情况下,每100万个读段中的细菌读段。
b NA–不适用,因为未针对具有少于1个读段的结果计算增加倍数。
在对从纽约长老会医院/哥伦比亚大学医学中心(NewYork-PresbyterianHospital/Columbia University Medical Center)的临床微生物实验室(ClinicalMicrobiology Laboratory)获得的血液培养样品的分析中测试BacCapSeq的实用性。将患者的血液收集到常规的BacTec血液培养瓶中,并孵育直至被BD BacTec自动血液培养系统(Becton Dickinson)标记为生长阳性。BacCapSeq的使用恢复了几乎完全的基因组序列,并且鉴定出了与标准微生物学实验室抗微生物敏感性测试(AST)谱相匹配的抗微生物抗性基因(表5和表6)。
表5-培养血样中病原菌和抗微生物抗性基因的检测
a-抗微生物药物敏感性测试(AST)谱:AMP,氨苄西林;AZT,氨曲南;CEF,头孢西丁;CEPH,头孢唑啉/头孢他啶/头孢曲松钠;MERO,美罗培南;TET,四环素。R,抗性;I,中级;NA,不适用。
表6-在培养的血液样品中检测到的抗微生物抗性基因
样品1,铜绿假单胞菌(鉴定出的细菌)
样品2,大肠杆菌(鉴定出的细菌)
样品3,摩氏摩根菌(Morganella morganii)(鉴定出的细菌)
样品4,流感嗜血杆菌(Haemophilus influenzae)(鉴定出的细菌)
a仅显示了高于<10/百万个读段的阳性阈值的读段计数。
从两名患有HIV/AIDS和未知原因的败血症的免疫抑制个体提取血液样品并进行处理,以供进行并行的BacCapSeq和UHTS分析。两种方法都鉴定出了病原体,然而BacCapSeq产生了更多数量的相关读段和更好的基因组覆盖率(图3)。在一名患者中检测到了肠道沙门氏菌。另一位患者具有被肺炎链球菌和阴道加德纳菌两者共感染的迹象。
当前的探针组特异地捕获CARD数据库中存在的所有AMR基因。证明AMR基因的存在并不等于找到其功能表达的证据。为了应对这一挑战,使用BacCapSeq来在暴露于抗生素的细菌中寻找生物标记物。将接种量为1000CFU/ml的氨苄青霉素敏感和耐药的金黄色葡萄球菌(Staphylococcus aureus)菌株在存在或不存在抗生素的情况下培养45分钟、90分钟和270分钟。然后提取RNA以供BacCapSeq和UHTS执行转录组学分析,以找到区分氨苄青霉素敏感和耐氨苄青霉素的金黄色葡萄球菌(S.aureus)的生物标记物。
BacCapSeq而非UHTS,使得能够发现在介于90分钟与270分钟之间的抗生素暴露中差异表达的转录物(图4)。这些生物标记物不仅包括反映细菌复制的组成型基因,而且还包括菌株和物种特异性的标记,诸如16S RNA和23S RNA、延伸因子TU(tuf)和G(fusA)、蛋白质A(spa)、凝集因子B(clfB)、或核糖体蛋白S12(rpsL)。
Bourbeau et al.2005.Routine incubation of BacT/ALERT FA and FN bloodculture bottles for more than 3 days may not be necessary.J Clin Microbiol43:2506-2509.
Chen et al.2016.VFDB 2016:hierarchical and refined dataset for bigdata analysis-10years on.Nucleic Acids Res 44:D694-D697.
Chen et al.2004.VFDB:a reference database for bacterial virulencefactors.Nucleic Acids Res 33:D325-D328.
Clark et al.2016.GenBank.Nucleic Acids Res 44:D67-D72.34.
CLSI.2007.Principles and procedures for blood cultures;approvedguideline.CLSI document M47-A.Clinical and Laboratory Standards Institute,Wayne,PA.
Cockerill et al.2004.Optimal testing parameters for bloodcultures.Clin Infect Dis 38:1724-1730.
Dobin et al.2013.STAR:ultrafast universal RNA-seqaligner.Bioinformatics 29:15-21.
Golkar et al.2014.Bacteriophage therapy:a potential solution for theantibiotic resistance crisis.J Infect Dev Ctries 8:129-136.
Howell and Davis.2017.Management of sepsis and septic shock.JAMA 317:847-848.
Jia et al.2016.CARD 2017:expansion and model-centric curation of thecomprehensive antibiotic resistance database.Nucleic Acids Res 45:D566-D573.
Langmead and Salzberg 2012.Fast gapped-read alignment with Bowtie2.Nat Methods 9:357.
Lee et al.2007.Detection of bloodstream infections in adults:how manyblood cultures are needed?J Clin Microbiol 45:3546-3548.
Li et al.2015.MEGAHIT:an ultra-fast single-node solution for largeand complex metagenomics assembly via succinct de Bruijn graph.Bioinformatics31:1674-1676.
Liao et al.2014.featureCounts:an efficient general purpose programfor assigning sequence reads to genomic features.Bioinformatics 30:923-930.
Mac Vane and Nolte.2016.Benefits of adding a rapid PCR-based bloodculture identification panel to an established antimicrobial stewardshipprogram.J Clin Microbiol 54:2455-2463.
Martin 2011.Cutadapt removes adapter sequences from highthroughputsequencing reads.EMBnet J 17:10-12.
Rhee et al.2017.Incidence and trends of sepsis in US hospitals usingclinical vs claims data,2009-2014.JAMA 318:1241-1249.
Robinson et al.2011.Integrative genomics viewer.Nat Biotechnol 29:24.
Schmieder and Edwards 2011.Quality control and preprocessing ofmetagenomic datasets.Bioinformatics 27:863-864.
Thorvaldsdóttir et al.2013.Integrative Genomics Viewer(IGV):high-performance genomics data visualization and exploration.BriefBioinform 14:178-192.
Wattam et al.2017.Improvements to PATRIC,the all-bacterialbioinformatics database and analysis resource center.Nucleic Acids Res 45:D535-D542.
序列表
<110> 纽约市哥伦比亚大学理事会(The Trustees of Columbia University in theCity of New York)
<120> 细菌捕获测序平台及其设计、构建和使用方法
<130> 01001/006659-WO0
<140> PCT/US 19/033922
<141> 2019-05-24
<150> 62/675,890
<151> 2018-05-24
<150> 62/724,014
<151> 2018-08-29
<160> 30
<170> PatentIn第3.5版
<210> 1
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 1
tctcggccag gatgaatttg 20
<210> 2
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 2
tttgaaggtg gggcgcacga 20
<210> 3
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 3
cgctaccacc atttcttcga 20
<210> 4
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 4
aaacggctat ctctggaagc 20
<210> 5
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 5
cccaccacca gcagacgaac tt 22
<210> 6
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 6
tgtacttctt gttggcctcg 20
<210> 7
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 7
tgccccgttg agtattgatg 20
<210> 8
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 8
agcccccgtg ataccagtac ca 22
<210> 9
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 9
gcctgtagct taacctgacc 20
<210> 10
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 10
aacagctacc aacgacagtc 20
<210> 11
<211> 24
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 11
tccactacga gaagtgctcc agga 24
<210> 12
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 12
atcaaccgca agaagagtgg 20
<210> 13
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 13
ataggaaaaa caggcgttgt 20
<210> 14
<211> 25
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 14
aggcaaagca tccatatctg cacga 25
<210> 15
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 15
accacaagca tgcattacat 20
<210> 16
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 16
cggcagaacg tcaggataaa 20
<210> 17
<211> 21
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 17
ggcagtgagg cagagattcc a 21
<210> 18
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 18
atgcgcatca gccatattca 20
<210> 19
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 19
tgcgttttga tggtgcctat 20
<210> 20
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 20
cggtaccatc gcgcgacttt 20
<210> 21
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 21
caatccaaca cggcatgaac 20
<210> 22
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 22
gtcgatcacg ttgtagaagg 20
<210> 23
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 人工探针
<400> 23
tgcctgagcg cgaagggtat 20
<210> 24
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 24
gttctgtgtc gttgaaggaa 20
<210> 25
<211> 25
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 25
catttgttct ggagcaggct gacgg 25
<210> 26
<211> 22
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 26
cgcgaagtca gagtcgacat ag 22
<210> 27
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 27
aagacctcaa cgccgatcac 20
<210> 28
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 28
gctttaagaa aagagcccgt 20
<210> 29
<211> 27
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成探针
<400> 29
tgcatatcac tcgctacaaa atgcact 27
<210> 30
<211> 20
<212> DNA
<213> 人工序列(Artificial Sequence)
<220>
<223> 合成引物
<400> 30
cttctgctaa aaatggcggt 20
- 细菌捕获测序平台及其设计、构建和使用方法
- 病毒组捕获测序平台、设计和构建方法以及使用方法