一种氨基四甘醇合成方法
文献发布时间:2023-06-19 19:05:50
技术领域
本发明属于化学合成技术领域,具体涉及一种氨基四甘醇合成方法。
背景技术
聚乙二醇衍生物为环氧乙烷水解产物的聚合物,无毒、无刺激性,广泛应用于各种药物制剂中,PEG及其衍生物是可用于由美国FDA认证的生物药物产品的少数聚合物之一。
氨基四甘醇性质和 PEG 极为相似,是一种重要的化工中间体,通过构建其功能基团制备适用于众多领域的聚乙二醇衍生物,尤其是在医药、生物领域的前沿技术,并且只有一个端羟基能参与反应,这些性质使得氨基四甘醇广泛用于多肽及蛋白质的结构修饰。在医药领域中,蛋白质多肽类药物具有生理活性强、疗效高、相对分子质量大、易在体内酶解、降解代谢途径多样等特点,通过键合特定功能基团的聚乙二醇醚衍生物能有效解决蛋白质多肽类药物溶解度低、稳定性差、半衰期短、存在免疫原性等不足,成为药物领域研究的新热点;在生物领域则通过不同功能基团的聚乙二醇苄醚衍生物修饰蛋白质酶可以提高其生物活性。
传统的氨基四甘醇合成方式是:以四甘醇为起始原料,与溴化苄反应以保护单端的羟基,同时另一端未保护的羟基转化为对甲苯磺酸酯或甲磺酸酯,再与叠氮化钠反应,得到叠氮化合物,然后利用合适的试剂将其还原成胺。但是叠氮化合物在加热或受到撞击时,都有爆炸的危险性,危及生命安全。因此探索一条安全的工艺路线具有重要意义。
发明内容
为解决上述问题,本发明公开了一种氨基四甘醇合成方法,以四甘醇为起始原料,与溴化苄反应以保护单端的羟基,同时另一端通过邻苯二甲酰亚胺钾盐引入邻苯二甲酰亚胺基团,然后利用合适的试剂将其还原成胺,反应条件温和,操作简单,中间体不需提纯即可直接进行下一步反应,避免了传统方法易爆炸的危险性,及难以放大生产的缺点。
为达到上述目的,本发明的技术方案如下:
一种氨基四甘醇合成方法,包括以下步骤:
(1)取四甘醇、四氢呋喃加入反应釜中,冷却至0 ℃后加入氢化钠并保温搅拌,随后滴加溴化苄并保温搅拌,向搅拌后的溶液中加入水进行淬灭反应,随后浓缩除去四氢呋喃溶剂,加入水、二氯甲烷进行萃取后分层,水相用二氯甲烷萃取一次后合并二氯甲烷层,再加入水洗涤二氯甲烷层后浓缩,得到四甘醇单苄醚粗品;
(2)将步骤(1)得到的四甘醇单苄醚粗品与二氯甲烷加入反应釜中,经搅拌后滴加30% KOH水溶液,保温搅拌,取对甲苯磺酰氯溶解于二氯甲烷得到混合溶液,将混合溶液滴加至反应釜并保温搅拌,反应结束后静止溶液分层,有机相经水洗后浓缩,得到四甘醇苄基对甲苯磺酸酯粗品;
(3)将BT4、DMF、Pht-K依次加入步骤(2)得到的四甘醇苄基对甲苯磺酸酯粗品中,经N
(4)取步骤(3)得到的四甘醇苄基邻苯二甲酰亚胺粗品与无水乙醇加入反应釜中并搅拌,与室温滴加80%水合肼,随后搅拌0.5 h,经油浴加热回流至反应结束,随后降温过滤,滤饼经无水乙醇洗涤过滤冲洗,合并滤液浓缩后加入水与HCl调节溶液pH值,再用二氯甲烷萃取,向水相中加入20% NaOH调节pH值,再加入二氯甲烷萃取后浓缩,得到氨基四甘醇单苄醚;
(5)将步骤(4)得到的氨基四甘醇单苄醚用无水乙醇溶解,进行加氢还原反应,反应结束后经过滤浓缩后得到氨基四甘醇;
作为本发明的一种改进,所述步骤(1)中四甘醇与氢化钠的摩尔比为4:1,四甘醇与溴化苄的摩尔比为2~4:1。
作为本发明的一种改进,所述步骤(1)与步骤(2)中保温搅拌温度为0~10 ℃,每次保温搅拌时间为1 h。
作为本发明的一种改进,所述步骤(2)中KOH与四甘醇单苄醚粗品的摩尔比为1~2:1,KOH与对甲苯磺酰氯的摩尔比为2:1。
作为本发明的一种改进,所述步骤(3)中油浴加热的油温为75~95 ℃。
作为本发明的一种改进,所述步骤(4)中油浴加热搅拌转速为210~300 r/min,油温为85~90 ℃。
作为本发明的一种改进,所述搅拌初始转速为210 r/min,当油温升至87 ℃时主键提升转速至300 r/min。
作为本发明的一种改进,所述步骤(4)中加入HCl后溶液pH值为2~3,HCl浓度为4mol/L,加入NaOH后溶液pH值为13。
本发明的有益效果为:本发明提供的一种氨基四甘醇合成方法,采用通过邻苯二甲酰亚胺钾盐引入邻苯二甲酰亚胺基团,然后利用合适的试剂将其还原成胺,反应条件温和,操作简单,中间体不需提纯即可直接进行下一步反应,避免了传统方法易爆炸的危险性,及难以放大生产的缺点。
附图说明
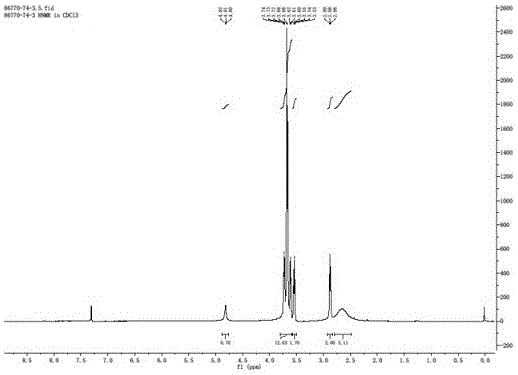
图1为本发明的实施例14氨基四甘醇的HNMR图谱。
具体实施方式
下面结合附图和具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说明本发明而不用于限制本发明的范围。
第一步:四甘醇单苄醚的合成
实施例1
取四甘醇(15.6 mol)、四氢呋喃30L加入反应釜,冷却至t<0℃,分批加入氢化钠(525g,3.9 mol),过程中保持t<10℃,加完保温搅拌1小时,之后滴加溴化苄(2.2kg,12.9mol),滴加过程中保持t<10℃,滴加完毕,保温搅拌一小时,自然升至室温反应至结束。加入水1L淬灭反应,浓缩除去四氢呋喃溶剂,加入水20L、二氯甲烷15L萃取分层,水相用二氯甲烷10L萃取一次,合并二氯甲烷层,分别使用15L水洗涤二氯甲烷层三次,浓缩二氯甲烷层,所得样品无需提纯,直接投入下一步反应。
通过外标计算样品中四甘醇单苄醚的质量为2274g,收率为62.2%。
实施例2
四甘醇用量为6.55 mol,其他同实施例1。
通过外标计算样品中四甘醇单苄醚的质量为1905g,收率为51.2%。
实施例3
四甘醇用量为9.825 mol,其他同实施例1。
通过外标计算样品中四甘醇单苄醚的质量为1905g,收率为51.2%。
实施例4
四甘醇用量为16.375 mol,其他同实施例1。
通过外标计算样品中四甘醇单苄醚的质量为2296g,收率为62.8%。
第二步:四甘醇苄基对甲苯磺酸酯的合成
实施例5
取四甘醇单苄醚、二氯甲烷10L加入反应釜,搅拌,冷却至<10℃,滴加30% KOH水溶液(896g,16mol),滴加过程中,保持t<10℃,保温搅拌一小时后,二氯甲烷5L溶解对甲苯磺酰氯(1.6kg,8.4mol),滴加,滴加过程中保持t<10℃,滴加完毕,保温搅拌一小时后,自然升至室温反应至结束。静置分层,二氯甲烷层分别使用8L水洗三次,浓缩二氯甲烷层,所得粗品无需提纯,即可投入下一步反应。
通过外标计算样品中四甘醇苄基对甲苯磺酸酯的质量为2981g,收率为85%。
实施例6
氢氧化钾的用量为9.6 mol,其他同实施例5。
通过外标计算样品中四甘醇苄基对甲苯磺酸酯的质量为2770g,收率为79%。
实施例7
氢氧化钾的用量为12mol,其他同实施例5。
通过外标计算样品中四甘醇苄基对甲苯磺酸酯的质量为2876g,收率为82%。
实施例8
氢氧化钾的用量为20mol,其他同实施例5。
通过外标计算样品中四甘醇苄基对甲苯磺酸酯的质量为2995g,收率为85.4%。
第三步:四甘醇苄基邻苯二甲酰亚胺的合成
实施例9
取1 eq BT4、8 L DMF、1512 g Pht-K、四甘醇苄基对甲苯磺酸酯依次加入反应釜(20L),N
通过外标计算样品中四甘醇苄基邻苯二甲酰亚胺的质量为2231g,收率为79.7%。
实施例10
油温为75 ℃,其他为实施例9。
通过外标计算样品中四甘醇苄基邻苯二甲酰亚胺的质量为2043g,收率为73%。
实施例11
油温为80 ℃,其他为实施例9。
通过外标计算样品中四甘醇苄基邻苯二甲酰亚胺的质量为2138g,收率为76.4%
实施例12
油温为95 ℃,其他为实施例9。
通过外标计算样品中四甘醇苄基邻苯二甲酰亚胺的质量为2236g,收率为79.9%。
第四步:氨基四甘醇单苄醚的合成
实施例13
取2345 g四甘醇苄基邻苯二甲酰亚胺粗品与33 L无水乙醇加入反应釜中并搅拌,于室温滴加80%水合肼1920g(5.4eq),随后搅拌0.5 h,经油浴加热回流至反应结束,降温后,搅拌着放出过滤,滤饼加8 L无水乙醇洗涤过滤,并用3 L无水乙醇冲洗滤饼,合并滤液浓缩后粗品加2-2.5倍体积水,4M HCl调pH至2-3,用一半体积二氯甲烷萃取三遍杂质,之后水相用20% NaOH调pH至13,一半体积二氯甲烷萃两次,萃出产品,浓缩(若最后有盐析出,则加二氯甲烷稀释过滤,浓缩即可)。
通过外标计算样品中氨基四甘醇单苄醚的质量为1356g,收率为88.7%。
随后降温过滤,滤饼经无水乙醇洗涤过滤冲洗,合并滤液浓缩后加入水与HCl调节溶液pH值,再用二氯甲烷萃取,向水相中加入20% NaOH调节pH值,再加入二氯甲烷萃取后浓缩,得到氨基四甘醇单苄醚。
第五步:氨基四甘醇的合成
实施例14
取氨基四甘醇单苄醚(900g,3.18mol)用5 L无水乙醇溶解,进行加氢还原反应,反应结束后经过滤浓缩后得到氨基四甘醇614g,收率为94%。
对比实施例1至实施例4可知,随着四甘醇的摩尔量的增加,四甘醇单苄醚的收率提高,但是当四甘醇与溴化苄的摩尔比增加至4:1以上时,对收率的提高影响较小,结合四甘醇导致的成本上升,故将四甘醇与溴化苄的摩尔比优化为4:1。
对比实施例5至实施例8可知,随着氢氧化钾的摩尔量的增加,四甘醇苄基对甲苯磺酸酯的收率提高,但是当氢氧化钾与四甘醇单苄醚的摩尔比增加至2:1以上时,对收率的提高影响较小,结合氢氧化钾导致的成本上升,故将氢氧化钾与四甘醇单苄醚的摩尔比优化为2:1。
对比实施例9至实施例12可知,合适的反应温度为85-95℃。
需要说明的是,上述仅仅是本发明的较佳实施例,并非用来限定本发明的保护范围,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,在上述实施例的基础上还可以做出若干改进和润饰,这些改进和润饰均落入本发明权利要求书的保护范围之内。