用于局部药物递送的自包埋颗粒的受控释放
文献发布时间:2023-06-19 19:33:46
背景技术
药物递送是指施用药物化合物(包括大分子药物和小分子药物,下文称为“药物”)以在人和动物中实现治疗效果。已经开发了多种药物递送途径并且包括静脉内、肌肉内、鼻内、真皮内和口服施用等。药物的吸收机制以及药物的性质是确定哪种递送途径适于实现药物最高生物活性和功效的重要因素。
口服施用药物通常在胃肠(GI)道中运输和吸收,GI道包括上GI道(例如,口、食道、胃和小肠的起始部分)和下GI道(小肠的剩余部分、大肠和直肠)。对于一些沿GI道发生的疾病,可能需要口服生物可利用的药物。沿GI道局部施用化合物类似于可治疗皮肤上的局部炎症的局部霜膏。可能受益于这种局部施用方法的疾病的示例是肠易激综合征(IBS)和炎性肠病(IBD),诸如克罗恩氏病和溃疡性结肠炎。
即使在患病条件下,GI道也可表现出多种屏障,这些屏障可抑制治疗水平的口服施用药物在疾病部位的递送。此类屏障的示例可包括以下一种或多种:胃中的酸性和酶促降解、个体之间沿小肠的pH变化、微生物区系(例如,结肠中降解药物的肠细菌)、非有机体肠内容物、影响药物吸收的上皮机能改变、胃内容物和排空/滞留时间。此外,溶解对于水溶性药物可能是一个挑战,因为当药物从升结肠向降结肠转移时,结肠中水的减少并且结肠腔内容物的粘度逐渐增加。可能需要吸收增强剂(例如,非甾体抗炎药(NSAID)、表面活性剂、脂肪酸等)来克服这些屏障。
因此,仍然需要解决这些挑战并提供改善的局部药物递送的改善的系统和方法。
发明内容
在实施方案中,提供了一种药物递送装置。该药物递送装置可包括肠溶胶囊和多个含药物颗粒。该肠溶胶囊可封闭内部体积。该多个含药物颗粒可定位在该内部体积内。该多个含药物颗粒中的每一者可进一步包括基质体和分布在该基质体内的活性药物成分(API)。该多个含药物颗粒可被配置为渗透组织。
在另一个实施方案中,该肠溶胶囊包括覆盖内层的外层。该外层可溶于胃中,并且该内层可溶于小肠或大肠中。
在另一个实施方案中,该肠溶胶囊可包括单一外层。
在另一个实施方案中,该API可选自肽、大于500Da的反义寡核苷酸、细胞因子、单克隆抗体、化疗药物、PD-1抑制剂、PD-L1抑制剂以及它们的组合中的一种或多种。
这些化疗药物可选自吉西他滨、顺铂、卡铂、氟尿嘧啶(5FU)以及它们的组合中的一种或多种。
PD-1抑制剂可选自帕博利珠单抗、纳武利尤单抗、西米单抗以及它们的组合中的一种或多种。
PD-L1抑制剂可选自阿特珠单抗、阿维鲁单抗、德瓦鲁单抗以及它们的组合中的一种或多种。
在另一个实施方案中,该基质体可由生物可降解聚合物形成。
生物可降解聚合物可选自聚(乳酸-共-乙醇酸)[PLGA]聚合物、PLGA共聚物、聚(己内酯)(PCL)、聚(氰基丙烯酸烷基酯)(PACA)、聚(原酸酯)、聚(酸酐)、聚(酰胺)、聚(酯酰胺)、聚(磷酸酯)、微生物释放聚合物以及它们的组合中的一种或多种。
在另一个实施方案中,PLGA聚合物可以是聚(乙醇酸)(PGA)或聚(D,L-乳酸)(PLA)。
在另一个实施方案中,PLGA共聚物可以是聚(D,L-乳酸-共-乙醇酸)[PLGA]的共聚物或聚酯和聚乙二醇(PEG)的共聚物。
在另一个实施方案中,该多个含药物颗粒的长径比可在约5至约100的范围内。
在另一个实施方案中,该多个含药物颗粒可具有包括至少一个顶点的形状。
在另一个实施方案中,该多个含药物颗粒可具有约1GPa至约10GPa范围内的弹性模量。
在另一个实施方案中,该多个含药物颗粒的轴向失效力可在约1N至约10N的范围内。
在另一个实施方案中,该多个含药物颗粒可具有约0.1μm至约20μm范围内的锐度。
在另一个实施方案中,这些含药物颗粒的至少一部分的表面可用粘膜粘着剂来官能化。
在实施方案中,提供了一种制备药物递送组合物的方法。该方法可包括形成多个含药物颗粒。该多个含药物颗粒中的每一者可包括基质体和分布在该基质体内的活性药物成分(API)。这些含药物颗粒可被进一步被配置为穿透组织。该方法还可包括将该多个含药物颗粒封闭在肠溶胶囊的内部体积中。该肠溶胶囊可被配置为在放置于患者的胃肠道内预先确定的时间量之后从腔释放该多个含药物颗粒。
在另一个实施方案中,该肠溶胶囊可包括覆盖内层的外层。该外层可溶于胃中,并且该内层可溶于小肠或大肠中。
在另一个实施方案中,该肠溶胶囊包括单一外层。
在另一个实施方案中,该API可选自肽、大于500Da的反义寡核苷酸、细胞因子、单克隆抗体、化疗药物、PD-1抑制剂、PD-L1抑制剂以及它们的组合中的一种或多种。
在另一个实施方案中,这些化疗药物可选自吉西他滨、顺铂、卡铂、氟尿嘧啶(5FU)以及它们的组合中的一种或多种。
在另一个实施方案中,PD-1抑制剂可选自帕博利珠单抗、纳武利尤单抗、西米单抗以及它们的组合中的一种或多种。
在另一个实施方案中,PD-L1抑制剂可选自阿特珠单抗、阿维鲁单抗、德瓦鲁单抗以及它们的组合中的一种或多种。
在另一个实施方案中,该基质体可由生物可降解聚合物形成。
生物可降解聚合物可选自聚(乳酸-共-乙醇酸)[PLGA]聚合物、PLGA共聚物、聚(己内酯)(PCL)、聚(氰基丙烯酸烷基酯)(PACA)、聚(原酸酯)、聚(酸酐)、聚(酰胺)、聚(酯酰胺)、聚(磷酸酯)、微生物释放聚合物以及它们的组合中的一种或多种。
在另一个实施方案中,PLGA聚合物可以是聚(乙醇酸)(PGA)或聚(D,L-乳酸)(PLA)。
在另一个实施方案中,PLGA共聚物可以是聚(D,L-乳酸-共-乙醇酸)[PLGA]的共聚物或聚酯和聚乙二醇(PEG)的共聚物。
在另一个实施方案中,该多个颗粒的长径比可在约5至约100的范围内。
在另一个实施方案中,该多个含药物颗粒可具有包括至少一个顶点的形状。
在另一个实施方案中,该多个含药物颗粒可具有约1GPa至约10GPa范围内的弹性模量。
在另一个实施方案中,该多个含药物颗粒的轴向失效力可在约1N至约10N的范围内。
在另一个实施方案中,该多个含药物颗粒可具有约0.1μm至约20μm范围内的锐度。
在另一个实施方案中,这些含药物颗粒的至少一部分的表面可用粘膜粘着剂来官能化。
在另一个实施方案中,形成该多个含药物颗粒可包括浇注含药物颗粒的液体前体,固化该液体前体以形成含药物颗粒的片,在模具的腔内促使该片的一部分形成离散的含药物颗粒,以及从该模具移除该离散的含药物颗粒。
在一个实施方案中,提供了一种将药物口服递送至胃肠道的方法,并且包括口服施用有效量的药物递送装置。该药物递送装置可包括肠溶胶囊和多个含药物颗粒。该肠溶胶囊可封闭内部体积。该多个含药物颗粒可定位在该内部体积内。该多个含药物颗粒中的每一者可进一步包括基质体和分布在该基质体内的活性药物成分(API)。该多个含药物颗粒可被配置为渗透组织。
在另一个实施方案中,该方法进一步包括口服施用在GI道内基本上不溶的一个或多个第二胶囊。
在另一个实施方案中,含药物颗粒的表面可用配置为促进含药物颗粒吸附到该一个或多个第二胶囊的化合物来官能化。
在另一个实施方案中,该一个或多个第二胶囊可被配置为在GI道内膨胀。
在另一个实施方案中,第二胶囊的尺寸可在约5mm至约25mm的范围内独立地选择。
在另一个实施方案中,第二胶囊可被配置用于与含药物颗粒的静电亲和力。
附图说明
根据下面结合附图的详细描述,可更全面地理解本公开的实施方案,其中:
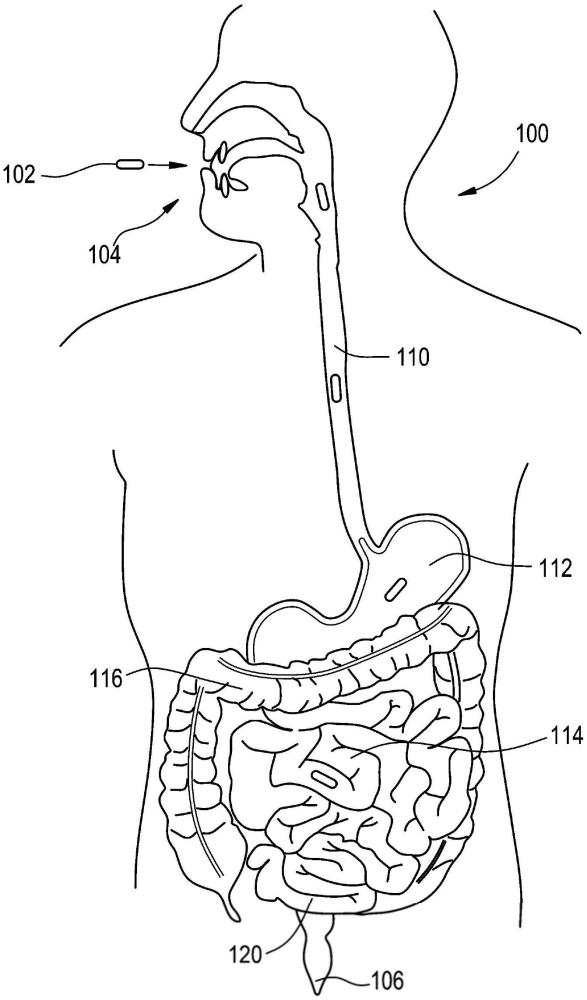
图1是示出了通过胃肠(GI)道行进的口服施用药物递送装置的一个示例性实施方案的患者的图;
图2A是示出图1的药物递送装置的一个示例性实施方案的图,该药物递送装置包括容纳在胶囊内的多个含药物颗粒,该胶囊包括外包衣层和内包衣层;
图2B是示出外包衣层溶解之后的图2A的药物递送装置的图;
图2C是示出在外包衣层溶解和内包衣层部分溶解之后的图2A的药物递送装置的示意图;
图2D是示出在外包衣层和内包衣层溶解之后含药物颗粒从图2A的药物递送装置释放的示意图;
图3A是示出图1的药物递送装置的另一个示例性实施方案的图,该药物递送装置包括容纳在胶囊内的多个含药物颗粒,该胶囊包括单个外包衣层;
图3B是示出在外包衣层溶解之后含药物颗粒从图3A的药物递送装置释放的示意图;
图4是示出含图1的颗粒的药物的复合微结构的示意图;
图5A、图5B和图5C示出了含药物颗粒制造方法的一个示例性实施方案;(5A)将含药物颗粒的组合物浇注成片并固化;(5B)在辊之间压缩片和包含多个腔的模具,以用含药物颗粒组合物填充腔;(5C)含药物颗粒从模腔中释放出来;
图6是示出图1的药物递送装置的含药物颗粒与肠粘膜相互作用(例如,接触和/或渗透)的图;
图7A是示出图1的药物递送装置的具有倒钩形状的含药物颗粒的形状的一个示例性实施方案的显微图;
图7B是示出图1的药物递送装置的具有锥形形状的含药物颗粒的形状的另一个示例性实施方案的显微图;
图7C是示出图1的药物递送装置的具有圆柱形形状的含药物颗粒的形状的另一个示例性实施方案的显微图;
图7D是示出图1的药物递送装置的具有矩形形状的含药物颗粒的形状的另一个示例性实施方案的显微图;
图7E是示出图1的药物递送装置的具有星样形状的含药物颗粒的形状的另一个示例性实施方案的显微图;
图8A是示出图1的药物递送装置的含药物颗粒的实施方案的图,该药物递送装置包括接近从肠侧壁延伸的微绒毛的自定向臂;
图8B是示出图8A的含药物颗粒与微绒毛接合的图;
图9是示出使用与图1的药物递送装置一前一后施用的第二胶囊以促进含药物颗粒与肠粘膜相互作用的图;并且
图10是示出药物递送装置的含药物颗粒与膀胱的组织相互作用(例如,接触和/或渗透)的图。
具体实施方式
现在将描述某些示例性实施方案,以提供对本文所公开的装置和方法的结构、功能、制造和用途的原理的全面理解。这些实施方案的一个或多个示例在附图中示出。本领域的技术人员将会理解,在本文中具体描述的和在附图中示出的装置、系统和方法是非限制性的示例性实施方案,并且本发明的范围仅由权利要求书限定。结合一个示例性实施方案示出或描述的特征可与其他实施方案的特征组合。此类修改和变型旨在包括在本发明的范围之内。
此外,在本公开中,各实施方案中名称相似的部件通常具有类似的特征部,因此在具体实施方案中,不一定完整地阐述每个名称相似的部件的每个特征部。另外,在所公开的系统、装置和方法的描述中使用线性或圆形尺寸的程度上,此类尺寸并非旨在限制可结合此类系统、装置和方法使用的形状的类型。本领域中技术人员将认识到,针对任何几何形状可容易地确定此类线性和圆形尺寸的等效尺寸。系统和装置及其部件的大小和形状可至少取决于系统和装置将用于其中的受治疗者的解剖结构、系统和装置将与其一起使用的部件的大小和形状、以及系统和装置将用于其中的方法和手术。
已经开发了多种“药丸样”装置以将治疗剂递送至肠壁。然而,这些药丸样装置具有缺陷。在一个方面中,由于制造的复杂性(例如,它们包括具有移动部件的子系统)或制造成本,现有的药丸样装置可能受到限制。因此,它们的可靠性值得怀疑。在另一个方面,现有的药丸样装置可被限制在可被递送到肠道中的特定位置的药物溶液的剂量。药物装载的这种限制是由装置的结构造成的,其中装置的大部分体积被致动机构占据并且相对小的体积被治疗剂占据。在另外的方面,对这些装置的另一个限制是它们有限的治疗区域。现有的药丸样装置可与组织的非常有限的区域接合。结果,与接合未患病组织区域相比,该装置与患病组织区域局部接合的可能性小,从而阻止了该装置从真正的局部施用获得期望的治疗有益效果。
一般来讲,本公开的实施方案提供了改善的药物递送系统及其使用方法。药物递送系统可包括容纳在肠溶胶囊中的含药物颗粒。在肠溶胶囊溶解后,含药物颗粒被释放到GI道中。含药物颗粒具有使它们能够吸附到和/或包埋在GI道的内层(例如,粘膜)中的性质。此类性质的示例包括物理性质(例如,尺寸、形状)、机械性质(例如,模量)和/或化学性质(例如,表面官能度、组成)。
药物递送系统可任选地包括与肠溶胶囊相比在GI道内相对不溶的颗粒。这些不溶颗粒可物理地促使含药物颗粒朝向粘膜,从而促进含药物颗粒在粘膜内的初始渗透和/或已经渗透粘膜的含药物颗粒的进一步渗透。
如下文详细讨论的,所公开的系统和方法解决了现有药物递送系统的限制,从而提供了遍及肠的更高的药物有效载荷。这些系统和方法进一步提供了克服将药物局部递送至肠粘膜的一般挑战的途径。
药物递送系统的实施方案在口服施用和药物通过GI道吸收的背景下讨论。然而,可以理解的是,药物递送系统的另外的实施方案可通过其他途径施用,以用于其他组织吸收而没有限制。可被治疗的其他组织的示例包括但不限于膀胱和生殖道。
图1是示出了通过GI道行进的口服施用药物递送装置102的一个示例性实施方案的患者100的图;GI道是一系列中空器官,形成了从嘴104延伸到肛门106的长的扭曲管。这些中空器官包括食道110、胃112、小肠114、大肠116、直肠120和肛门106。药物递送装置102被接纳在嘴104内并且穿过GI道到达期望的治疗区域。
图2A更详细地示出了药物输送装置102的一个示例性实施方案。如图所示,药物递送装置102是限定内部体积或腔200的胶囊的形式。该胶囊包括内层202和覆盖内层202的外层204,其中内层的面向内部的壁界定腔200。多个含药物颗粒206设置在腔200内。
外层204可以是配置为在GI道内选择性溶解的肠溶包衣。例如,外层204可被配置为在胃的较强酸性条件下抵抗溶解并且易于在具有较高pH的GI道部分(例如小肠114(图2B))内溶解。以举例的方式,溶解可在约4或更高的pH、约5或更高的pH、约6或更高的pH或约7或更高的pH下开始。
内层202可被进一步被配置为以受控速率在小肠114和/或大肠116内溶解(图2C)。这样,胶囊防止含药物颗粒206暴露在胃112中并延迟在肠114、116中的释放。当内层202溶解到腔200不再被内层202完全封闭的程度时,开始释放(图2D),并且溶解速率通过暴露于更高pH和/或暴露于小肠和/或大肠114、116中的微生物区系/细菌来控制。内层202可另外被配置为提供以下中的一者或多者:制造性、含药物颗粒206的分散性、含药物颗粒206的粘结性和含药物颗粒206的渗透性。
外层204和内层202可由抗胃酸化合物形成。一般来讲,存在多种聚合物可用于实现受控的抗胃酸性。示例包括脂肪酸、蜡、虫胶、塑料和植物纤维。在某些实施方案中,外层可由乙酸邻苯二甲酸纤维素、乙酸偏苯三酸纤维素、聚乙酸乙烯酞酸酯、羟丙基甲基纤维素邻苯二甲酸酯、乙酸羟丙甲纤维素琥珀酸酯、纤维素、甲基丙烯酸和丙烯酸乙酯(1∶1比例)的共聚物或甲基丙烯酸和甲基丙烯酸甲酯(1∶1或1∶2比例)的共聚物中的任一种形成。在另外的实施方案中,内层可由硬脂酸镁、硬脂酸、明胶、微晶纤维素粉末、甘油、柠檬酸、聚乙二醇或羟丙基甲基纤维素形成。
在另外的实施方案中,外层204和/或内层202可独立地选自具有在以下中讨论的官能度的材料:“Degradable Controlled-Release Polymers and PolymericNanoparticles:Mechanisms of Controlling Drug Release”(Chem.Rev.,第116卷第4期:第2602-2663页,2016年,该文献的全部内容据此以引用方式并入。
图3A示出了药物输送装置102的另选实施方案。除了省略了内层202之外,图3A的药物递送装置102类似于图2A的药物递送装置。即,图3A的药物递送装置包括限定具有单层302(例如,外层)的内部体积或腔300的胶囊。如以上所讨论的,外层302可以是肠溶包衣,该肠溶包衣被配置为在胃的酸性条件下抵抗溶解并且在大肠/下部肠的碱性pH内溶解。当外层302溶解到腔300不再被完全封闭的程度时,开始释放(图3B)。
在某些实施方案中,含药物颗粒206具有复合微结构。如图4所示,复合物400包括基质体402和包埋在基质体402内的活性药物成分(API)404。API颗粒在基质体402内的分布可以是基本上均匀的或非均匀的。基质体402内的API 404的体积分数可在约0至约0.75的范围内。另选地,API 404在基质体402内的体积分数可为约0.1至约0.3、约0.1至约0.4、约0.1至约0.5、约0.1至约0.6、约0.2至约0.3、约0.2至约0.4、约0.2至约0.5或约0.2至约0.6。API 404可与基质体402内的溶剂形成溶液相,在基质体402内部分可溶,或者是在基质体402内悬浮的固体。例如,含药物颗粒206的某些实施方案可具有纳米或微粒形式的API404,其悬浮在赋形剂基质体内,其中API 404为基质体体积的约50体积%。
基质体402的实施方案可由多种不同的材料形成。示例可包括生物可降解聚合物。生物可降解聚合物可包括但不限于聚(乳酸-共-乙醇酸)或PLGA聚合物、PLGA共聚物、聚(己内酯)(PCL)、聚(氰基丙烯酸烷基酯)(PACA)、聚(原酸酯)、聚(酸酐)、聚(酰胺)、聚(酯酰胺)、聚(磷酸酯)或微生物释放聚合物。PLGA聚合物的示例可包括聚(乙醇酸)(PGA)或聚(D,L-乳酸)(PLA)。PLGA共聚物的示例可包括聚(D,L-乳酸-共-乙醇酸)(PLGA)或聚酯聚乙二醇(PEG)共聚物。在以下中讨论了可形成基质体的赋形剂材料的另外的实施方案:“Excipientselection for compounded pharmaceutical capsules:they’re only fillers,right?”。Australian Journal of Pharmacy,第98卷第1164期:第78-83页,2017年,该文献的全部内容据此以引用方式并入。
API 404的实施方案可由多种不同的化合物形成。示例可包括:
●小分子,其为了更好的生物利用度或吸收需要更长的保留时间。
●小分子,诸如大于500Da的肽和反义寡核苷酸,因此具有较差的容易通过上皮屏障的性质(例如,蛋白质,诸如细胞因子或信号分子,和单克隆抗体)
●用于膀胱癌的药物,诸如适于治疗各种肿瘤的化疗药物。此类化学治疗药物可包括但不限于吉西他滨、顺铂、卡铂、氟尿嘧啶(5FU)、PD-1抑制剂、PD-L1抑制剂。PD-1抑制剂可包括但不限于帕博利珠单抗、纳武利尤单抗、西米单抗。PD-L1抑制剂可包括但不限于阿特珠单抗、阿维鲁单抗、德瓦鲁单抗。
其他API 404的非限制性示例包括顺铂或卡铂、紫杉醇、多西他赛、TIP(紫杉醇/紫杉酚、异环磷酰胺和顺铂/顺氯氨铂)、VeIP(长春花碱、异环磷酰胺和顺铂/顺氯氨铂)、VIP(依托泊甙/VP-16、异环磷酰胺和顺铂/顺氯氨铂)、VAC(长春新碱、更生霉素和环磷酰胺)、白蛋白结合的紫杉醇、六甲蜜胺、卡培他滨、环磷酰胺、依托泊甘、吉西他滨、异环磷酰胺、伊立替康、脂质体阿霉素、美法仑、培美曲塞、拓扑替康、长春瑞滨以及它们的组合。
在某些实施方案中,含药物颗粒206可通过模制工艺形成。模制工艺在以下文献中的一者或多者中详细讨论,这些文献中的每个文献的全部内容据此以引用方式并入。
●美国专利号7,976,759,名称为“System and Method For ProducingParticlesand Patterned Films”
●“Shape-specific,Mono-disperse Nano-molding of ProteinParticles”,Kelly,J.Y.;DeSimone*,J.M.,J.Am.Chem.,Soc.2008年,第130卷:第5438-5439页。
●“Microfabricated Particles for Engineered Drug Therapies:
Elucidation into the Mechanisms of Cellular Internalization
DeSimone*,J.M.;Pharm.Res.2008年,第25卷:第2845-2852页。
●“Reductively Labile
DeSimone*,J.M.;J.Am.Chem.Soc.2008年,第130卷:第5008-5009页。
●“The Pursuit of a Scalable Nano-fabrication Platform for UseinMaterial and Life Science Applications”,Gratton,S.E.A.;
Williams,S.S.;Napier,M.E.;Pohlhaus,P.D.;Zhou,Z.;Wiles,K.
B.;Maynor,B.B.;Shen,C.;Olafsen,T.;Samulski,E.T.;
DeSimone*,J.M.,Accounts of Chemical Research,2008年第41卷:第1685-1695页。
●“Nanoparticle Drug Delivery Platform”,Napier,M.E.;
DeSimone*;J.M.,Polymer Reviews,2007年,第47卷:第321-327页。
●“Nanofabricated Particles for Engineered Drug Therapies:APreliminary Biodistribution Study of
DeSimone*,J.M.J.,Controlled Release,2007年,第121卷:第10-18页。
●“Imparting Size,Shape,and Composition Control of MaterialsforNanomedicine”,Eulis,L.;DuPont,J.;DeSimone*,J.M.,J.
M.Chem.Soc.Rev.,2006年,第35卷:第1095-1104页。
●“Direct Fabrication and Harvesting of Monodisperse,ShapeSpecificNano-Biomaterials”,Rolland,J.P.;Maynor,B.W.;Euliss,L.
E.;Exner,A.E.;Denison,G.M.;DeSimone*,J.M J.Am.
Chem.Soc.2005年,第127卷:第10096-10100页。
下文参考图5A至图5C提供了模制工艺的简要讨论。如图5A所示,将含药物颗粒206的液体前体500浇注在基底502上。在加热下移除溶剂以产生具有复合微结构的固态溶液膜,也称为递送片504。如图5B所示,使包括具有预先确定的几何形状的多个模腔510的模具506与递送片504接触。模具506和递送片504通过加热辊512并分裂。辊512之间的高压和热使递送片504软化,从而导致递送片504的一部分流动并填充模具腔510(填充模具506f),从而形成离散的含药物颗粒206。随后,可从模具506移除离散的药物递送颗粒206。在一个实施方案中,如图5C所示,使填充的模具506f与高能量膜或赋形剂层514接触并通过加热辊512而不分裂。在冷却之后,移除模具506以在高能量膜或赋形剂层514上露出离散的含药物颗粒206的阵列。在另一个实施方案中,离散的含药物颗粒206可在图5B的操作之后直接从模具506移除。在任一种情况下,含药物颗粒206模拟模腔510的尺寸和形状。
在另选的实施方案中,可省略图5A的膜浇注操作。相反,液体前体500可直接施加到模具506上。将模具506夹在辊512和表面之间导致液体前体500进入模具腔510。一旦液体前体500填充模具腔510,则可移除溶剂以固化液体前体500。
如图6所示,含药物颗粒206被配置为行进通过GI道内腔600并且一旦从药物递送装置102释放就与GI道壁602(诸如肠114、116的粘膜衬里604)相互作用(例如,接触和/或至少部分地嵌入其中)。因为基质体402是生物可降解的,所以当基质体402分解时,含药物颗粒206内的API 404被释放。含药物颗粒206与粘膜604相互作用为释放的API 404移动通过粘膜604并到达作为肠吸收细胞起作用的肠上皮细胞606提供了相对长的停留时间。
含药物颗粒206可具有促进含药物颗粒206与肠粘膜604接合的一种或多种物理性质、机械性质或化学性质(以任何组合)。有利地,上述模制工艺可以高精确度和可再现性提供这些性质的独立控制。
物理性质的示例可包括尺寸和形状。含药物颗粒206的尺寸可被配置为通过与具有约1μm长度的微绒毛接合来促进与粘膜604的接合。含药物颗粒206的尺寸还可被配置为达到目标上皮和深黏膜的深度,这些深度高达约200μm至500μm。因此,非限制性实施方案中,含药物颗粒206的尺寸可独立地选自约5μm至约500μm的范围。
含药物颗粒206的形状还可通过提供高长径比(长度:直径)或“针样”几何形状(例如,类似于仙人掌针或豪猪刺)来促进与粘膜604的接合。一般来讲,含药物颗粒206的形状可采用任何几何形状的形式,该几何形状包括提供应力集中点以增强渗透的表面特征。此类几何形状的示例可包括圆锥形、具有光滑或阶梯表面的棱锥形、具有光滑或纹理表面的星样形状、或简单几何形状诸如棒或矩形的组合。示例示于图7A至图7B中。如图7A的显微照片所示,颗粒形状是细长的并且可包括面向后的倒钩。倒钩可将应力集中在较小的点上,而不是主体的整个表面区域上。结果,通过小面积的多个倒钩施加较低的压力,以在一个区域上实现与大压力相同的结果。图7B是具有带环形表面特征的锥形形状的含药物颗粒的另一个实施方案的显微照片。圆柱形、矩形和星形状的示例分别进一步示于图7C至图7E的显微照片中。
此类形状可提供具有足以渗透粘膜604的锐度的基质体402。一般来讲,锐度可表示渗透标准膜所需的力的量。因此,较高的锐度有利于渗透。在非限制性实施方案中,基质体402的纵横比可选自约5至约100的范围。在另外的非限制性实施方案中,基质体402的锐度可选自约0.1μm至约20μm的范围。
不受理论的约束,预期尖锐颗粒通过以下机制中的一种或多种(单独地或组合地)渗透粘膜604。在一个方面,渗透可通过自然分散到存在于GI粘膜604内或与之接触的局部流体中而发生。在另一个方面,当肌肉收缩和扩张时,由于肠的运动,可发生渗透。
在另外的实施方案中,含药物颗粒206可包括自取向的侧附件/臂800以控制取向。一般来讲,含药物颗粒206的取向可使得渗透特征(例如,倒钩)邻近待渗透的组织的表面定位。在某些实施方案中,该取向可将含药物颗粒206的至少一部分的纵轴定位在相对于组织表面约45度至-45度之间的角度。在一个示例中,如图8A所示,微绒毛802从肠侧壁804的表面上延伸出来,而臂800从含药物颗粒206的表面上延伸出来。臂800的构造可采用多种形状,包括直的、弯曲的、倒钩的、圆锥形的以及它们的组合。如此配置,如图8B所示,臂800可物理地接合微绒毛802。
机械性质的示例可包括模量(例如,弹性模量)。一般来讲,弹性模量是表征材料对弹性变形的抗性的材料性质。为了避免当与粘膜相互作用时显著的弹性变形,期望含药物颗粒206的基质体402和/或臂800具有大于粘膜604的弹性模量的弹性模量。在非限制性实施方案中,含药物颗粒206的弹性模量可在约1GPa至约10GPa(例如,约3GPa至约6GPa)的范围内。
机械性质的其他示例可包括机械强度。一般来讲,含药物颗粒206(例如,基质体402和/或臂800)的机械强度可足以抑制在与肠侧壁804接合期间的失效(例如,破裂)。在非限制性实施方案中,含药物颗粒206在压缩或拉伸中的轴向失效力可大于1N(例如,在约1N至约10N的范围内)。
化学性质的示例可包括表面官能度和组成。一般来讲,可使用多种化合物来使表面官能化,以便定制含药物颗粒206和粘膜604之间的相互作用。例如,基质体402的表面可被官能化以粘附到粘膜604(例如,粘膜粘着剂)上。官能化化合物可包括通过暂时破坏紧密连接而改善细胞间摄取的化合物(例如,表面活性剂诸如C10)。
在其他实施方案中,官能度可包括将粘膜粘附化学物质整合到含药物颗粒206的基质上/内。一般来讲,粘膜粘附可包括在两个表面之间形成的任何结合。在本发明的背景中,可在含药物颗粒206和胃肠表面之间提供粘膜粘附。通过粘膜粘附形成的结合可被配置为延长两个表面之间直接接触的时间。这可为含药物颗粒206的组织渗透和药物释放提供足够的时间(例如,多达约12小时)。粘膜粘着剂的实施方案可包括粘膜粘着剂聚合物,诸如亲水性聚合物和/或水凝胶。
粘性亲水性聚合物可包括含有羧基基团的那些,示例可包括但不限于聚乙烯吡咯烷酮(PVP)、甲基纤维素(MC)、羧甲基纤维素钠(SCMC)、羟丙基纤维素(HPC)和其他纤维素衍生物。水凝胶的示例可包括但不限于基于阴离子的凝胶诸如卡波普、聚丙烯酸酯和交联改性的聚丙烯酸酯,基于阳离子的凝胶诸如壳聚糖和衍生物,以及基于中性的凝胶诸如丙烯酸树脂-NE30D。
根据图9所示的实施方案,多个第二胶囊900可任选地与药物递送装置102一前一后施用。第二胶囊900在GI道内基本上不溶,并且可被配置为通过移动穿过GI道并且放大作用于释放在其中的含药物颗粒206上的力F来帮助含药物颗粒206与粘膜604的相互作用(图9)。
例如,第二胶囊900可提供机械辅助。一般来讲,第二胶囊900的尺寸可小于其行进的通道的预先确定的量。这样,第二胶囊900可促进第二胶囊900和含药物颗粒206之间的接触并且有效地将含药物颗粒206推入粘膜604中。在一个方面,第二胶囊900可以是相对刚性的(例如,具有比含药物颗粒206或GI道内组织的弹性模量大的弹性模量)。在另一个方面中,第二胶囊900可被配置为膨胀(例如,由于从局部环境吸收水分)以便将颗粒推入粘膜604中。在刚性构型或膨胀构型中,第二胶囊900的尺寸可独立地选自约5mm至约25mm。
又如,第二胶囊900可提供化学辅助。在一个方面,化学辅助可采取将含药物颗粒206吸附到粘膜604的形式。在另一个方面,第二胶囊900可表现出与含药物颗粒206的亲和力(例如,静电)。类似于上述关于粘膜粘着剂的机制,诸如聚合物链的静电和/或机械缠结可促进第二囊900的表面和组织之间的暂时结合,从而提供第二胶囊900与组织侧壁的机械耦合。这种相互作用可在与组织表面紧密接触的含药物颗粒206上施加更大的力。这种施加的力可进一步增强含药物颗粒206向靶组织中的渗透以用于药物递送。这些相互作用力可通过胃肠道的自然肌肉运动进一步增强。
药物递送装置102的实施方案已经在口服递送和在胃肠道内释放的背景中进行了讨论。然而,设想了使用药物递送装置102递送和释放到身体的其他区域。例如,图10是示出药物递送装置102的含药物颗粒206与膀胱1000的组织相互作用(例如,接触和/或渗透)的图。围绕膀胱内腔1002的膀胱壁的组织包括尿道上皮1004、固有层1006、肌层粘膜1010和脂肪层1012。尿道上皮1004是膀胱1000的最内层,其厚度为约1mm至约3mm。固有层1006是位于尿道上皮1004和肌层粘膜1010之间的层并且厚度为约2mm至约4mm。肌层粘膜1010是定位在固有层1006和脂肪层1012之间的膀胱1000的外肌层并且厚度为约5mm至约8mm。脂肪层1012是围绕整个膀胱1000的脂肪的层。
膀胱内灌注任何装置或自包埋颗粒需要将它们自身附着于尿道上皮1004。尿道上皮1004可被认为是覆盖整个膀胱1000的动态涂层。该层是动态的,因为其吸收性质(由于高血管分布)在理解膀胱内递送和药物递送装置102在递送期望的API 404“负荷”中有效的能力中起重要作用。
由于在尿道上皮1004中的尿路上皮细胞之间的紧密连接阻止高水平的全身化学吸收的发生,因此灌注到膀胱1000中的任何化学品具有进入血流的最小吸收。因此,药物递送装置102可被定制用于穿透尿道上皮1004和递送API 404以治疗膀胱1000。如上所讨论的,受益于局部递送的API 404可包括但不限于用于各种肿瘤的任何化疗。在另外的实施方案中,API 404可以是影响膀胱收缩性的任何种类的药物和/或抗感染药物。
已在上文在本文提供的整体公开内容的上下文中仅以举例的方式描述了本公开。应当理解,在不脱离本公开的总体范围的情况下,可以在权利要求书的实质和范围内进行修改。
- 用于向软骨中受控递送被囊封的药剂的二元自组装凝胶
- 通过传输纳米包埋药物经设计用于此用途和在身体pH下于人动脉中释放所述纳米包埋药物的医疗设备来重建人阻塞动脉的血流
- 提供可变释放速率的受控药物递送系统