一种抗炎载药缓释制剂、制备方法及其应用
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及纳米生物医药技术领域,特别是涉及一种抗炎载药缓释制剂、制备方法及其应用。
背景技术
关节炎是最常见的关节疾病之一,其发病率的增加主要是和人口老龄化有关,受到影响的主要是可动关节。原发性关节炎是以肥胖和年龄增加作为最大的危险因素的。相关的病理生理学研究依然在进行,其精确的发病机制尚未完全阐明,但是已经从单纯的限制性软骨病到了目前认识的影响整个关节的慢性复杂多因子疾病。目前为止,晚期终末的患者治疗方案可以是关节置换术,但是早期及进展期的患者尚无特别有效的治疗方法。目前使用抗炎药物控制炎症是常用的治疗办法,但是有多种局限,口服药物受到首过效应和药物分布的问题,局部给药也有维持时间短以及靶向不足的问题。
在关节炎非手术治疗中,关节腔内注射是最主要的抗炎治疗方式之一,而目前常规使用是多次多剂量的治疗方式,但频繁用药是影响患者疗效和依从性的主要因素。对于“关节腔内注射”(Intra-articular injections,IAIs)的抗炎治疗,药物作用环境(关节腔内)的特殊性,使作用药物容易出现脱靶、清除速度过快、生物利用度低等问题。这些问题使得目前IAIs的单次单剂量常规给药方式疗效不佳。
随着材料学载药技术的迅速发展,针对需要频繁用药的治疗方式,研究者在其他领域陆续找到了对应的单剂量替代方案。因此,开发出一种单次单剂量注射,可长效缓释并且长久滞留于关节腔内的抗炎载药制剂,是目前关节炎非手术治疗所急迫需要的。
发明内容
本发明提供一种抗炎载药缓释制剂、制备方法及其应用,以解决上述的问题。
第一方面,本发明实施例提供一种抗炎载药缓释制剂,所述抗炎载药缓释制剂包括载药纳米粒和水凝胶微球,其中,通过羧基修饰将所述载药纳米粒和所述水凝胶微球结合得到所述抗炎载药缓释制剂;
所述载药纳米粒为亲和素化载药纳米粒;
所述水凝胶微球为生物素化水凝胶微球;
其中,所述亲和素化载药纳米粒和所述生物素化水凝胶微球结合为生物素-亲和素系统,形成生物素-亲和素修饰的复合控释给药系统。
优选地,所述载药纳米粒包括二氧化铈嵌套的介孔硅纳米药物载体和负载的药物,其中,所述药物包括治疗关节炎的药物;
所述生物素化水凝胶微球包括明胶、甲基丙烯酸酯和1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐、2-吗啉乙基磺酸缓冲液和生物素-N-羟基磺基琥珀酰亚胺。
优选地,所述抗炎载药缓释制剂具有微米尺寸的多孔结构;
通过改变所述抗炎载药缓释制剂中的抗炎药物,实施所述抗炎药物的特定作用;
通过所述抗炎载药缓释制剂的水凝胶微球的自体溶胀特性负载载药纳米粒。
第二方面,本发明实施例提供一种抗炎载药缓释制剂的制备方法,所述制备方法包括以下步骤:
步骤1,制备亲和素化载药纳米粒;
步骤2,制备生物素化水凝胶微球;
步骤3,将所述亲和素化载药纳米粒和所述生物素化水凝胶微球混合后,制得所述抗炎载药缓释制剂;
所述抗炎载药缓释制剂包括载药纳米粒和水凝胶微球,其中,通过羧基修饰将所述载药纳米粒和所述水凝胶微球结合得到所述抗炎载药缓释制剂;所述亲和素化载药纳米粒和生物素化水凝胶微球结合为生物素-亲和素系统,形成生物素-亲和素修饰的复合控释给药系统。
优选地,在步骤1中,所述制备亲和素化载药纳米粒,包括:
将AEAPS修饰的空白亲和素化载药纳米粒加入至抗炎药物前体溶液中,采用超声振荡分散所述AEAPS修饰的空白亲和素化载药纳米粒,在磁力搅拌器上高速搅拌,将所述载药后的亲和素化载药纳米离心洗涤后,去除上清液,烘干沉淀物,得到亲和素化载药纳米粒;
在步骤2中,所述制备生物素化水凝胶微球,包括:
步骤2-1,将称取的明胶分散于磷酸缓冲盐溶液中,水浴搅拌至所述明胶溶解澄清,用注射器抽取甲基丙烯酸酐,采用微量注射泵缓慢将所述甲基丙烯酸酐加入至溶解的明胶中,加入磷酸缓冲盐溶液终止反应,制得前体甲基丙烯酸酯明胶;
步骤2-2,在所述反应终止后,将所述前体甲基丙烯酸酯明胶放入透析袋中,透析过夜,离心除去不溶物后,将上清液继续透析直至制得甲基丙烯酸酯明胶;
步骤2-3,将所述甲基丙烯酸酯明胶与1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐在高速搅拌下混合后,放入透析袋中进行透析,制得碳二亚胺化反应水凝胶;
步骤2-4,采用2-吗啉乙基磺酸缓冲液调整所述二亚胺化反应水凝胶的pH值,加入生物素-N-羟基磺基琥珀酰亚胺,高速搅拌后透析,制得生物素化水凝胶;
步骤2-5,将所述生物素化水凝胶储存在冰箱中,备用;
步骤2-6,在微流控平台下冻干制备所述生物素化水凝胶微球。
优选地,在步骤3中,将所述亲和素化载药纳米粒和所述生物素化水凝胶微球混合,制得所述抗炎载药缓释制剂,包括:
步骤3-1,将所述亲和素化载药纳米粒加入至2-吗啉乙基磺酸缓冲液和聚乙二醇溶液,再加入1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐和所述生物素化水凝胶微球反应,直至反应结束,得到抗炎载药缓释微球;
步骤3-2,依次采用乙醇和双蒸水洗涤所述载药缓释微球,然后将洗涤后的所述抗炎载药缓释微球重悬分散在甘氨酸-氢氧化钠Good's缓冲液中,超声振荡分散,采用双蒸水洗涤后重悬于RS溶液中,得到所述抗炎载药缓释制剂。
第三方面,本发明实施例提供一种抗炎载药缓释制剂的应用,应用于制备治疗关节炎的制剂;所述抗炎载药缓释制剂包括载药纳米粒和水凝胶微球,其中,通过羧基修饰将所述载药纳米粒和所述水凝胶微球结合得到所述抗炎载药缓释制剂;
所述载药纳米粒为亲和素化载药纳米粒;
所述水凝胶微球为生物素化水凝胶微球。
优选地,所述生物素化水凝胶用于负载载药纳米粒;所述负载的载药纳米粒中的药物为治疗关节炎的药物;所述生物素化水凝胶通过溶胀特性负载所述治疗关节炎的药物,基于缓释将所述治疗关节炎的药物释放作用于关节炎部位。
优选地,所述抗炎载药缓释制剂通过局部给药,细胞通过内吞作用摄取载药纳米粒,所述抗炎载药缓释制剂能穿透整个软骨层。
优选地,所述抗炎载药缓释制剂的应用具有关节腔内滞留和双峰型药物代谢的控释特性;
其中,所述抗炎载药缓释制剂的应用通过关节腔内滞留介导炎症的代谢转移,通过调节软骨细胞中的活性氧降低炎症反应和减少分解代谢蛋白的产生。
本发明包括以下优点:
(1)本发明制备抗炎载药缓释制剂的方法简单易行、成本低、效率高,低污染且重复性好、性质稳定,可以进行大规模应用,且具有多孔结构、仅有微米尺寸,不仅能够实现可调节的降解率,还能发挥水凝胶的生物相容性,通过增加目标功能基团,来发挥其特定作用,通过自体溶胀特性来负载药物等作用物;
(2)本发明实施例制备的抗炎载药缓释制剂通过控制药物的释放速率和方式,延长药物在体内的作用时间,这种长效释放特性可以减少药物频繁给药的需求,提高治疗的便利性和患者的依从性;
(3)本发明实施例制备的抗炎载药缓释制剂可以适应不同类型的药物,包括疏水性和亲水性药物,因为凝胶结构可以调整以容纳不同性质的药物分子,这使得它在多种疾病治疗中具有广泛的应用前景;
(4)本发明实施例制备的抗炎载药缓释制剂具有低毒性和可降解性,能够在体内逐渐降解成生物可吸收的代谢产物,避免了长期滞留和积累的问题,这种特性有助于减轻患者的负担,并提高治疗的安全性;
(5)本发明实施例制备的抗炎载药缓释制剂中的水凝胶材料能够有效地保护药物免受外界环境的影响,如光、氧气、湿度等,有助于保持药物的稳定性和活性,延长药物的有效期。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对本发明实施例的描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例提供的制备抗炎载药缓释制剂的合成路线图;
图2为本发明实施例提供的通过微流控制备的SP@MSR-Granule的扫描电镜表征和成品示意图;
图3为本发明实施例提供的MSR-Granule的扫描电镜表征与崩解时扫描电镜表征;
图4为本发明实施例提供的SP溶液的药物标准曲线及空白载药纳米粒的载药曲线;
图5为本发明实施例提供的甲基丙烯酸酐化水凝胶-碳二亚胺功能化水凝胶-生物素化化水凝胶合成氢谱表征;
图6为本发明实施例提供的SP@MSR-Granule对C28-I2细胞的共培养CCK-8细胞毒性结果;
图7为本发明实施例提供的SP@MEND对C28-I2细胞的摄取率分析结果;
图8为本发明实施例提供的SP-Avidin@MEND对MH7A和HUVEC共培养成管影响的结果;
图9为本发明实施例提供的SP@MSR-Granule的IAIs治疗SD大鼠CIA模型的大体验证;
图10为本发明实施例提供的SD-rat CIA模型对于SP@MSR-Granule的IAIs治疗在炎性分子指标的组间对照ELISA结果;
图11为本发明实施例提供的SD-rat CIA模型对于SP@MSR-Granule的IAIs治疗在炎性分子指标的组间对照结果;
图12为本发明实施例提供的SD-rat CIA模型对于SP@MSR-Granule的IAIs治疗在免疫组化的组间对照结果;
图13为本发明实施例提供的SP@MSR-Granule对SD-rat负重关节区域的滞留效果—在体表征结果;
图14为本发明实施例提供的SD-rat CIA模型对于SP@MSR-Granule的IAIs治疗的抗氧化应激指标的组间对照结果。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂以及其他仪器未注明生产厂商者,均为可以通过市场购买获得的常规试剂产品。
甲基丙烯酸化水凝胶(gel methacrylic anhydride,GelMA)因其优良的生物相容性而被广泛用于细胞支架和载药材料的研究。反应物和药物可在紫外固化前装入甲基丙烯酸化水凝胶溶液,或被交联的甲基丙烯酸化水凝胶表面微孔和内部纳米孔吸附。然而,对于大体积的甲基丙烯酸化水凝胶来说,无法进行原位可控降解调节,此外也不适合用于注射治疗。基于此,本发明利用微流控技术,制备出微米尺寸的关节腔内注射剂型水凝胶微球,不仅能够实现可调节的降解率,还能发挥水凝胶的生物相容性。通过微流控平台,对明胶进行光固化交联,从而制备水凝胶微球,再进行冻干来造成多微孔结构。上述技术有以下两种优势:(1)通过增加目标功能基团,来发挥其特定作用;(2)通过自体溶胀特性来负载药物等作用物。该方式用途广、步骤少、容错率高、工艺成熟,应用前景广阔。
基于此,本发明是为了提供一种具有多孔结构、仅有微米尺寸,不仅能够实现可调节的降解率,还能发挥水凝胶的生物相容性;通过增加目标功能基团,来发挥其特定作用;通过自体溶胀特性来负载药物等作用物,解决水凝胶不适用于注射的问题。
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
第一方面,本发明实施例提供一种抗炎载药缓释制剂,所述抗炎载药缓释制剂包括载药纳米粒和水凝胶微球,其中,通过羧基修饰将所述载药纳米粒和所述水凝胶微球结合得到所述抗炎载药缓释制剂;
所述载药纳米粒为亲和素化载药纳米粒;
所述水凝胶微球为生物素化水凝胶微球;
其中,所述亲和素化载药纳米粒和所述生物素化水凝胶微球结合为生物素-亲和素系统,形成生物素-亲和素修饰的复合控释给药系统。
优选地,所述载药纳米粒包括二氧化铈嵌套的介孔硅纳米药物载体和负载的药物,其中,所述药物包括治疗关节炎的药物;
所述生物素化水凝胶微球包括明胶、甲基丙烯酸酯和1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐、2-吗啉乙基磺酸缓冲液和生物素-N-羟基磺基琥珀酰亚胺。
优选地,所述抗炎载药缓释制剂具有微米尺寸的多孔结构;
通过改变所述抗炎载药缓释制剂中的药物,实施所述药物的特定作用;
通过所述抗炎载药缓释制剂的水凝胶微球的自体溶胀特性负载载药纳米粒。
第二方面,本发明实施例提供一种抗炎载药缓释制剂的制备方法,所述制备方法包括以下步骤:
步骤1,制备亲和素化载药纳米粒;
具体地,在步骤1中,所述制备亲和素化载药纳米粒,包括:
将AEAPS修饰的空白亲和素化载药纳米粒加入至抗炎药物前体溶液中,采用超声振荡分散所述AEAPS修饰的空白亲和素化载药纳米粒,在磁力搅拌器上高速搅拌,将所述载药后的亲和素化载药纳米离心洗涤后,去除上清液,烘干沉淀物,得到亲和素化载药纳米粒。
优选地,所述超声震荡的温度为65℃,所述高速搅拌为在10000r/min持续搅拌24h,并依次采用无水乙醇洗涤2次,双蒸水洗涤1次,将所述载抗炎药物-封套纳米器制剂保存在-20℃备用。
步骤2,制备生物素化水凝胶微球;
具体地,在步骤2中,所述制备生物素化水凝胶微球,包括:
步骤2-1,将称取的明胶分散于磷酸缓冲盐溶液中,水浴搅拌至所述明胶溶解澄清,用注射器抽取甲基丙烯酸酐,采用微量注射泵缓慢将所述甲基丙烯酸酐加入至溶解的明胶中,加入磷酸缓冲盐溶液终止反应,制得前体甲基丙烯酸酯明胶;
步骤2-2,在所述反应终止后,将所述前体甲基丙烯酸酯明胶放入透析袋中,透析过夜,离心除去不溶物后,取上清液继续透析直至制得甲基丙烯酸酯明胶;
步骤2-3,将所述甲基丙烯酸酯明胶与1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐在高速搅拌下混合后,放入透析袋中进行透析,制得碳二亚胺化反应水凝胶;
步骤2-4,采用2-吗啉乙基磺酸缓冲液调整所述二亚胺化反应水凝胶的pH值,加入生物素-N-羟基磺基琥珀酰亚胺,高速搅拌24小时后透析过夜,制得生物素化水凝胶;
步骤2-5,将所述生物素化水凝胶储存在冰箱中,备用;
步骤2-6,在微流控平台下冻干制备所述生物素化水凝胶微球。
优选地,在步骤2-1中,所述采用微量注射泵以0.25mL/min的速度将所述甲基丙烯酸酐加入至溶解的明胶中;
在步骤2-2中,所述反应终止15min,所述透析袋为MWCO 3500透析袋,所述透析持续2-3天;
在步骤2-3中,所述透析袋为MWCO 3500透析袋,所述透析时间为12h;
在步骤2-4中,所述溶液的pH调整至8.8-9.5,所述高速搅拌持续24h;
在步骤2-5中,使用所述生物素化水凝胶时,将所述生物素化水凝胶进行水浴,并加入光固化剂Photoinitiator-2959,进行冻干脱水;
在步骤2-6中,微流控平台的分散相为10%w/v GEL-B溶于ETOH的溶液,连续相为肉豆蔻酸异丙酯中加入5%聚氧乙烯脱水山梨醇单月桂酸酯的溶液,液滴以(20mW)进行波长为1480nm光固化紫外交联1min后收集微球,分别用乙醇和无菌双蒸水洗涤微球20min,然后-80℃下储存12h,再进行冷冻干燥脱水。
步骤3,将所述亲和素化载药纳米粒和所述生物素化水凝胶微球混合后,制得所述抗炎载药缓释制剂。
具体地,在步骤3中,将所述亲和素化载药纳米粒和所述生物素化水凝胶微球混合后,制得所述抗炎载药缓释制剂,包括:
步骤3-1,将所述亲和素化载药纳米粒加入至2-吗啉乙基磺酸缓冲液和聚乙二醇溶液,再加入1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐和所述生物素化水凝胶微球反应直至反应结束后,得到抗炎载药缓释微球;
步骤3-2,依次采用乙醇和双蒸水洗涤所述载药缓释微球,然后将洗涤后的所述抗炎载药缓释微球重悬分散在甘氨酸-氢氧化钠Good's缓冲液中,超声振荡分散,采用双蒸水洗涤后重悬于RS溶液中,得到所述抗炎载药缓释制剂。
优选地,在步骤3-1中,所述反应时间为30h;
在步骤3-2中,所述载药缓释微球采用乙醇洗涤1次,双蒸水洗涤3次,所述超声振荡30min,所述抗炎载药缓释制剂储存于-20℃。
第三方面,本发明实施例提供一种抗炎载药缓释制剂的应用,应用于制备治疗关节炎的制剂;所述抗炎载药缓释制剂包括载药纳米粒和水凝胶微球,其中,通过羧基修饰将所述载药纳米粒和所述水凝胶微球结合得到所述抗炎载药缓释制剂;
所述载药纳米粒为亲和素化载药纳米粒;
所述水凝胶微球为生物素化水凝胶微球;
其中,所述亲和素化载药纳米粒和所述生物素化水凝胶微球结合为生物素-亲和素系统,形成生物素-亲和素修饰的复合控释给药系统。
优选地,所述生物素化水凝胶用于负载载药纳米粒;所述负载的载药纳米粒中的药物为治疗关节炎的药物;所述生物素化水凝胶通过溶胀特性负载所述治疗关节炎的药物,基于缓释将所述治疗关节炎的药物释放作用于关节炎部位。
优选地,所述抗炎载药缓释制剂通过局部给药作用于炎症部位,炎症部位的细胞通过内吞作用摄取所述抗炎载药缓释制剂的载药纳米粒,所述抗炎载药缓释制剂能穿透整个软骨层。
优选地,所述抗炎载药缓释制剂的应用具有关节腔内滞留和双峰型药物代谢的控释特性;
其中,所述抗炎载药缓释制剂的应用通过关节腔内滞留介导炎症的代谢转移,通过调节软骨细胞中的活性氧降低炎症反应和减少分解代谢蛋白的产生。
为使本领域技术人员更好地理解本发明,以下通过具体的实施例来说明本发明提供的一种抗炎载药缓释制剂、制备方法及其应用。
实施例1:一种抗炎载药缓释制剂的制备方法
在本发明实施例中以亲和素化载药纳米粒和生物素化水凝胶微球制备的抗炎载药缓释制剂为例进行说明,具体方法如下:
步骤1:制备亲和素化载药纳米粒;
步骤1-1:将Ce(III)溶解在50mL双蒸水中,在70℃的温度下回流搅拌,制得前驱体溶液。
步骤1-2:在前驱体溶液中加入25mL 3M的一水合氨,将该溶液的pH提高至8.8-9.5之间进行沉淀反应,持续5分钟后,获得氢氧化铈。将氢氧化铈进行老化处理,将老化处理后的溶液在65℃下放置孵育20h。在老化阶段,在常温条件下进行氢氧化铈的溶解-再结晶过程,将粗产物孵育后,在室温下用10000rpm离心15分钟将溶液分离,除去上清液后,用无水乙醇洗涤5次,得到CeO
步骤1-3:在双蒸水中制备分散的CeO
步骤1-4:将40mg十六烷基三甲基溴化铵与660μL超纯水、300μL无水乙醇混合,制得表面活性剂溶液,其中,十六烷基三甲基溴化铵为纯度≥99%。
步骤1-5:将所述预备液滴加到所述表面活性液中,制得混合物,并对混合物进行充分超声分散,将80μL正硅酸乙酯作为前驱体,滴加到混合物中,将上述混合物在室温下持续搅拌18h,然后在18℃,1200r/min离心15min,以去除产品中未反应的化学物质,然后用萃取液20%硝酸铵乙醇溶液洗涤2次,除去多余的十六烷基三甲基溴化铵,得到作为表面活性剂模板的二氧化硅层,再用无水乙醇洗涤1次,得到二氧化铈嵌套介孔硅纳米粒作为空白载药纳米粒(multifunctional envelope-type nanodevice,MEND),其中,所述正硅酸乙酯的纯度≥99%。
步骤1-6:将2mg亲和素加入2mg所述空白载药纳米粒中反应3h,10000r/min离心,采用双蒸水洗涤3次,去除上清液后,采用甘氨酸-氢氧化钠Good's缓冲液重悬,得到空白亲和素化载药纳米粒。
步骤1-7:将1mL 30mg/mL DMSO加入至20%SBE-β-Cd(sulfobutyl ether-β-cycloSPtrin,磺基丁基醚β-环糊精)溶液中,混合均匀,在超声振荡下,加入10mg磺胺吡啶(sulfapyridine,SP)至完全溶解,得到磺胺吡啶前体溶液。其中,20%SBE-β-Cd溶液的制备方法如下:先将2g SBE-β-CD加入5mL 1×PBS(phosphate buffered solution,磷酸盐缓冲盐溶液)中,然后固定体积为10mL 1×PBS,超声处理后,振荡至溶液清澈透明,得到20%的SBE-β-Cd溶液。
步骤1-8:将10mg AEAPS(N-氨乙基-γ-氨丙基三甲氧基硅烷,N-(2-aminoethyl)-3-aminopropyltrimethoxysilane)修饰的空白亲和素化载药纳米粒加入10mL所述磺胺吡啶前体溶液中,在65℃超声振荡分散上述载药纳米粒,在磁力搅拌器下持续高速搅拌24h,将所述载药后的亲和素化载药纳米粒依次采用无水乙醇离心洗涤2次、双蒸水洗涤1次后,去除上清液,烘干沉淀物,得到亲和素化载药纳米粒。
具体的,所述AEAPS修饰的空白亲和素化载药纳米粒的制备方法包括:
步骤1-8-1:将30mg三乙氧基硅烷和50mg三乙胺溶解在10mL无水乙醇中,放入2mg所述空白亲和素化载药纳米粒,在室温下进行受控水解和缩合反应16h,离心采用无水乙醇洗涤2次除去上清液后,制得修饰后的空白亲和素化载药纳米粒。
步骤1-8-2:采用甘氨酸-氢氧化钠Good's缓冲液重悬所述修饰后的空白载药纳米粒,加入2mgAEAPS,高速搅拌2h进行AEAPS修饰,采用双蒸水离心洗涤3次去除上清液后,制得AEAPS修饰的空白亲和素化载药纳米粒。
介孔硅纳米粒是由纳米级别的多介孔的二氧化硅颗粒构成,因而可以很简单的将硅烷基引入到分子中取代活性氢,从而进行硅烷化偶联。活性氢被硅烷基取代后降低了化合物的极性,减少了氢键束缚。在本发明实施例中选用N-氨乙基-γ-氨丙基三甲氧基硅烷作为硅烷修饰剂,旨在对二氧化铈嵌套的介孔硅纳米粒进行硅烷化修饰。
步骤2:制备生物素化水凝胶微球;
步骤2-1:称取20g明胶(Gelatin,GEL),分散于200mLPBS(Phosphate BufferedSolution,磷酸缓冲盐溶液)(0.01M)中,水浴使GEL温度达到60℃,搅拌至溶解澄清,用注射器抽取16mL甲基丙烯酸酐,采用微量以0.25mL/分钟的速度缓慢将甲基丙烯酸酐加入明胶中,加入甲基丙烯酸酐2h后,加入200mLPBS终止上述反应,得到前体甲基丙烯酸酯明胶;
步骤2-2,反应终止15min后,将前体甲基丙烯酸酯明胶放入MWCO 3500透析袋中,38℃下透析过夜,然后在4℃下10000r/min下离心15min,除去不溶物,取上清液,继续透析2-3天,获得成品甲基丙烯酸酯明胶(Methyl methacrylate Gelatin,GelMA)。
步骤2-3,将20g GelMA与0.2mg EDC(1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐,1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride)在高速搅拌下混合,再放入MWCO 3500透析袋中,40℃下透析12h,获得碳二亚胺化反应水凝胶(Carbodiimide Gelatin,Gel-E);
步骤2-4,将Gel-E溶液用MES(2-Morpholinoethanesulphonic acid,2-吗啉乙基磺酸缓冲液)调整为pH值为8的溶液,并加入0.3mgNHS-Biotin(N-hydroxysuccinimidobiotin,生物素-N-琥珀酷亚胺基酯),高速搅拌24h并透析12h,获得最终前体材料生物素化水凝胶(Gel-B);
步骤2-5,在冻干前,将所述生物素化水凝胶储存在冰箱中,备用储存在-80℃的冰箱中,使用时水浴38℃加入2mg光固化剂(Photoinitiator-2959),进行冻干脱水;
步骤2-6,以10%w/v的GEL-B溶于ETOH为分散相,IM(isopropyl myristate,肉豆蔻酸异丙酯)中加入5%
其中,微流控技术是制备微球的主要技术之一。微流控技术制备微球的方法是通过微通道生产液滴。利用体积(如注射器泵)或压力(如压力容器)的驱动力使不相溶的连续相和分散相分别在各自的通道流动,气体或液体在通道的交汇处相交遇,利用连续相对分散相进行挤压或剪切作用,促使界面不稳定从而断裂,生成分散的液滴。微流控芯片的通道基本结构有3种类型:T型微通道芯片、流动聚焦型、共聚焦型。本发明采用了T型微通道芯片。微通道可以制备的微球尺寸在10μm到300μm之间,制作精良的微通道内部结构规整,能够保证液滴在每一条微通道内的微环境的完全相同,在扩大生产规模时,无需再调整微通道的尺寸,只需要并行增加微通道数量或微反应器单元;因此,所有的工艺控制参数均具有延续性。并且通过将黏度、界面张力等参数输入到预先设计的数学模型中,就可以实现对微球粒径的预测,进而优化释放。微流控技术是制备均匀颗粒物的一种新途径,可采用简单的一步法,以低成本的方式控制微球的大小和粒径分布。
步骤3:将所述亲和素化载药纳米粒和所述生物素化水凝胶微球混合后,制得所述抗炎载药缓释制剂;
步骤3-1,将亲和素化载药纳米粒加入至7mL MES buffer(2-(N-Morpholino)ethanesulfonic acidhydratete,2-吗啉乙基磺酸缓冲液)和1mLPEG溶液(poly(ethyleneglycol)2000,聚乙二醇溶液),再加入2mg EDC(O-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride,1-(3-二甲氨基丙基)-3-2-乙基碳二亚胺盐酸盐)和2mgMSR-Biotin反应30h至反应结束后,得到抗炎载药缓释微球;
步骤3-2,依次用乙醇洗涤1次,双蒸水(ddH
参阅图1,图1为本发明制备抗炎载药缓释制剂的合成路线图。图1中(a)部分为制备得到的载药纳米粒,图1中(b)部分为亲和素化载SP纳米粒,显示出泡沫样表面结构,图1中(c)部分为生物素化水凝胶缓释微球,图1中(d)部分为通过亲和素-生物素系统结合的SP@MSR-Granule,载药纳米粒在SP@MSR-Granule内形成纤维骨架结构,图1中(e)部分为SP@MSR-Granule在模拟关节液内15天后崩解。
参阅图2,图2为本发明通过微流控制备的SP@MSR-Granule。图2中(a)部分为在T型微通道芯片中微球制作过程,图2中(b)部分为扫描电镜下缓释微球的表征,图2中(c)部分为冻干的生物素化水凝胶样品,图2中(d)部分为加双蒸水复胀的生物素化水凝胶,呈现半透明,图2中(e)部分为结合载药纳米粒后的复胀后的生物素化水凝胶,呈现漫反射样非完全透明。
冻干样本呈现“塑料泡沫”样形态,加入光固化剂在40℃下水浴可进行液化。液化后的Gel-B可以迅速被交联成“果冻样”的形态,与GelMA交联的形态,水凝胶形态的Gel-B颜色更偏白,并且机械强度更有增强。T型芯片微流控的收集池中可见,如图2中(a)部分所示,在剪切力的作用下,收集池以约1500粒/分钟的速度被固化为水凝胶微球(multifunctional-slow-release-Granule,MSR-Granule)。如图2中(b)部分扫描电镜结果所示,微流控制作的MSR-Granule被表征为一个150-160μm的球形颗粒。粒径分析表示通过此方法进行的制作的MSR-Granule粒径分布均匀,并且可以通过调整微流控速度对粒径进行调整。
本发明通过制备亲和素化载药纳米粒以及生物素化水凝胶冻干微球,并通过生物素-亲和素系统使两者结合,形成多功能嵌套的抗炎载药缓释制剂用于关节腔内注射治疗关节炎。在此基础上,通过活化羧基策略修饰亲和素可长期有效的滞留在关节腔内。此外,由于抗炎载药缓释制剂具有理想的关节内靶向和缓释性能,采用第一代磺酰胺类抗生素之一的磺胺吡啶作为载药。抗炎载药缓释制剂抗炎效果理想,还具有显著的抗滑膜炎性生发中心作用,可避免炎症的复发。
本发明制备抗炎载药缓释制剂的方法简单易行、成本低、效率高,低污染且重复性好、性质稳定,可以进行大规模应用,且具有多孔结构、仅有微米尺寸,不仅能够实现可调节的降解率,还能发挥水凝胶的生物相容性;通过增加目标功能基团,来发挥其特定作用;通过自体溶胀特性来负载药物等作用物。
实施例2:一种抗炎载药缓释制剂的表征
在本发明实施例中以亲和素化载药纳米粒和生物素化水凝胶微球制备的抗炎载药缓释制剂(SP@MSR-Granule)为例进行说明,具体的表征操作如下:
A:模拟关节液中,SP@MSR-Granule中药物SP的释放实验及电镜表征
模拟关节液的制备:取100ml载药纳米粒,100ml透明质酸,50ml乳酸林格氏液,5mlTGF-β3和2.5ml的骨形态发生蛋白(bone morphogenetic protein,BMP)7,2mg的卵磷脂,充分溶解,依次混合均匀得到模拟关节液。
精密称取SP@MSR-Granule 1mg,加入上述模拟关节液中,并放入37℃温箱,分别在第1天、第6天、第9天、第12天、第15天、第21天,使用倒置相差显微镜进行记录,观察所述SP@MSR-Granule的崩解过程。在第15天根据光镜观测结果,在SEM电镜下观察15天后破碎微球的形态和纳米粒负载情况。
参阅图3,图3中(a)部分为MSR-Granule的光镜下表征,拍摄于第1天、第6天、第9天、第12天、第15天、第21天,图3中(b)部分为第15天的扫描电镜图像,显示MSR-Granule的崩解,图3中(c)部分为同一图像(图3中(b)部分)的局部高分辨率放大图像,显示内部纤维骨架结构,图3中(d)部分为同一图像(图3中(b)部分)的局部高分辨率放大图像,显示载药纳米粒组成其内部纤维骨架,图3中(e)部分为同一图像(图3中(b)部分)的局部高分辨率放大图像,显示其作为骨架的载药纳米粒细部。
所制备的SP@MSR-Granule具有在15天的时间内稳定破裂释放载药米粒治疗单元,在21天时完全破碎释放,这归功于其显微的网络纤维结构。如图3所示为一个破碎的SP@MSR-Granule的扫描电镜结构,放大后可以看到,SP@MSR-Granule对于载药纳米粒的载荷量。由于载药纳米粒与MSR的结合是通过亲和素-生物素系统,所以与单纯依靠复胀吸附于表面的载荷不同,BMP是吸附与MSR的内部纤维结构上的,因此所储蓄的SP量也随之增加。
参阅图4,图4中(a)部分为SP溶液的药物标准曲线,图4中(b)部分为空白载药纳米粒的载药曲线。根据药物释放实验,不同的药物浓度比例,负载的SP的量与空白载药纳米粒的质量比呈正比,可以很容易地通过混合不同药物的组分并在水性溶剂中孵育混合物过夜来获得。获得载药曲线,先需要计算SP溶液的药物标准曲线如图所示,如图4中(a)部分所示,SP溶液浓度与OD值的线性关系为y=0.0014x-0.0103(R
B:将GelMA、Gel-E和Gel-B的冻干样品进行氢核磁质谱分析
本材料的缓释单元MSR-Granule,是由Gel-B在T型芯片在微流控下进行光固化制造的,对GelMA、Gel-E和Gel-B的冻干样品进行氢核磁质谱分析,参阅图5,图5中(a)部分为甲基丙烯酸酐化水凝胶Gel-MA的合成氢谱表征,图5中(b)部分为碳二亚胺功能化水凝胶Gel-E的合成氢谱表征,图5中(c)部分为生物素化水凝胶Gel-B的合成氢谱表征,图5中(d)部分为氢谱磁共振表征。
如图5所示,前体材料Gel-B的氢谱表征数据提示,化学位移在2.5ppm-0ppm附近处为长链上的氢信号,5.0ppm~3.0ppm附近处为与N,O等吸电子基团相连接的氢信号,是由于受到N,O等吸电子基团的影响使其周围电子云密度降低,去屏蔽作用增强,信号移动像低场,化学位移相对升高在3.9ppm附近处,为结构当中的=CH
在对缓释单元MSR-Granule进行可靠性验证:首先,通过在制备过程中对T型芯片的微流控设备进行拍摄,以确保制备流程正确,经过扫描电镜可见,微球结构清晰,大小符合设计。第二,通过水凝胶合成氢谱表征可以证实碳二亚胺化及生物素化水凝胶制备成功。第三,载药后的治疗单元载药纳米粒和缓释单元MSR-Granule结合后,通过电镜表征可见内部纤维骨架结构,并在15天内缓慢释放,释放时间符合预算设计。由此,可以验证本实施例成功制备了一种可有效载药、粒径符合细胞内作用、可在关节腔内缓慢释放的长效制剂。
实施例3:SP@MSR-Granule的体外试验
由于SP具有肾脏损伤和胃肠道反应等不良作用,将SP嵌套至载药纳米粒内,探讨其在细胞内产生的功效和安全性。
A:C-28/I2、MH7A、HUVEC细胞的培养
C-28/I2、MH7A、HUVEC细胞分别为人正常软骨C-28/I2细胞系、关节炎成纤维细胞MH7A、人脐静脉内皮细胞HUVEC。
从液氮罐中取出细胞迅速放入36℃~37℃水浴,扣上盖并不时摇动,尽快解冻。用吸管吸出细胞悬液,装入离心管中,再补加10ml培养液,吹打使细胞悬浮。低速离心(500~1000r/min)5分钟,取上清后再重复用培养液漂洗、离心。加入培养液适当稀释后,再移装入培养瓶中,置温箱培养,次日更换一次培养液后再继续传代培养。
B:SP@MSR-Granule对于C-28/I2细胞的细胞毒性实验
C-28/I2细胞传代到第四代,将细胞接种在96孔板上,细胞数量至少为1000个/孔(100μL培养基)与用0、0.01、0.1、0.5、1、5、10μg/mL的SP与等量浓度的SP@MSR-Granule(经载药曲线换算浓度为0、0.1、1、5、10、50、100μg/mL)、SP@MEND(经载药曲线换算浓度为0、0.013、0.13、0.65、1.3、6.5、13μg/mL)共孵育24小时,然后更换补充含有10%的CCK-8溶液的培养基。2小时后,使用酶标仪测量450nm波长处的吸光度,对照组为(SP、SP-载药纳米粒),参考波长为570nm。以浓度为X轴,细胞活性百分比值为Y轴制柱状图。
将上述同批次的C-28/I2细胞,将细胞接种在96孔板上细胞数量至少为1000个/孔(100μL培养基),用0、1、5、10、100mg/ml浓度的空白MEND,共孵育24小时,然后更换补充含有10%的CCK-8溶液的培养基。2小时后,使用酶标仪测量450nm波长处的吸光度,以浓度为X轴,CCK-8细胞OD值为Y轴制柱状图。
参阅图6,图6为SP@MSR-Granule对C28-I2细胞的共培养CCK-8细胞毒性结果。图6中(a)部分为空白MEND在不同浓度下的CCK-8细胞毒性实验直方图,纵坐标为CCK-8细胞OD值,横坐标为MEND浓度,图6中(b)部分:SP(磺胺吡啶)、SP@MEND(载SP纳米粒)、SP@MSR-Granule(抗炎载药缓释制剂),在不同磺胺吡啶预制浓度下细胞活性直方图。纵坐标为细胞活性百分比,横坐标为浓度。注:每个*代表P值小于0.001。
对SP@MSR-Granule在软骨细胞中CCK-8的细胞毒性测试。当浓度升高到100mg/ml的MEND时,仍旧没有观测到该MEND对软骨细胞有明显的细胞毒性,本发明实施例1制备的MEND生物相容性极佳,如图5中(a)部分所示,共培养CCK-8组间对照结果显示,当浓度上升到0.01μg/ml的SP载量时,SP组和SP@MSR-Granule开始出现显著性差异,当浓度上升到1μg/ml的SP载量时,可以显著的观察到SP组和SP@MEND组都出现显著的细胞活性降低,而SP@MSR-Granule组则有效的保护了软骨细胞的活性,如图6中(b)部分所示,使用MSR-Granule进行SP载药可以有效的减少对于软骨细胞的细胞毒性,而MEND本身没有细胞毒性。
C:C-28/I2细胞对SP@MEND的摄取实验
C-28/I2细胞传代到第四代,将细胞接种道6孔板上,细胞数量至少为15000个/孔(600μL培养基)。然后用6μM的SP@MEND用以浸涂法将过量的浓度为5μL/mL的异硫氰酸荧光素(FITC)和相同量的同批次未修饰的MEND同样以浸涂法进行5μL/mL的AF594染色,并洗涤3次。共孵育5分钟、10分钟、15分钟、30分钟后,用DAPI(4',6-二脒基-2苯基吲哚)染色C-28/I2细胞并以倒置荧光显微镜检验其摄取能力。
参阅图7,图7中(a)部分为在荧光倒置显微镜下,C28-I2细胞对SP@MEND的摄取结果。左向右图列依次为明场、DAPI、AF594(空白载药纳米粒)、FITC(亲和素化载药纳米粒)、合成图像。从上到下图行依次为在5分钟、10分钟、15分钟、30分钟拍摄。图7中(b)部分为图7中(a)部分的差异性分析直方图,横坐标为时间,纵坐标为C28-I2的细胞摄取百分比。注:每个*代表P值小于0.001。
通过观察AF594(红色荧光)处理的空白载药纳米粒和FITC(绿色荧光)处理的亲和素化载药纳米粒的荧光共定位判断载空白药纳米粒均为细胞核优先摄取,如图7所示,细胞核摄取之后MEND均匀分布于整个细胞。与AF594处理的空白载药纳米粒(红色荧光)相比,FITC(绿色荧光)处理的亲和素化载药纳米粒在30分钟的共培养过程中始终表现出更高的向C-28/I2细胞的核摄取效率。在相同浓度的空白载药纳米粒与亲和素化载药纳米粒(5μg/ml)以时间依赖的方式进入C-28/I2细胞。当共培养达仅仅达到5分钟和10分钟时,亲和素化载药纳米粒对C-28/I2细胞的摄取率即达到24.42%和58.31%,而空白载药纳米粒基本未观测到明显的细胞摄入,但10分钟开始观测到细胞聚集的倾向。15分钟后,亲和素化载药纳米粒的摄取率高达82.54%并均匀分散于细胞中,空白载药纳米粒摄取率达61.09%但仍以核聚集摄取为主。30分钟之后,可以观察到无论是空白载药纳米粒还是亲和素化载药纳米粒均对C-28/I2拥有高摄取率。由此判断,亲和素修饰的多功能嵌套纳米器相比于未修饰的空白纳米器,更高程度的提高了软骨细胞的摄取效率。该结果提示亲和素化载药纳米粒在滑液代谢快速的动态关节腔代谢的IAIs治疗中有者更大优势和潜力。
D:SP-Avdin@MEND干预HUVEC/MH7ATranswell共培养成血管实验
取3个Transwell培养皿,将1×10
参阅图8,图8中(a)部分为空白对照组成管实验结果,图8中(b)部分为亲和素化载药纳米粒组成管实验结果,图8中(c)部分为亲和素化载SP纳米粒组成管实验结果,图8中(d)部分为体外血管生成的连接数直方图,图8中(e)部分为体外血管生成的成结数直方图。注:直方图中纵轴为连接数/成结数,Avidin@MEND为亲和素化载药纳米粒组,SP-Avdin@MEND为亲和素化载SP纳米粒组,每个*代表P值小于0.001。
通过NIHImageJ软件分析了人脐静脉内皮细胞HUVEC成管效果的三个主要参数:体外血管生成的连接数(红点)、成结数(蓝色圆圈)和总长度(黄线),如图8中(a)、(b)和(c)所示,这三个指标可以反映血管生成的程度。连接和成结越多,总长度越长,血管生长得越旺盛。因此,从图8可以看出,相比于空白对照组和亲和素化载药纳米粒组,亲和素化载SP纳米粒组的连接点的数量减少了十倍,这证实了SP对于血管成型的抑制和亲和素化载药纳米粒提高SP的生物利用度。此外,成结数和总长度结果表明,亲和素化载SP纳米粒组比空白对照组和亲和素化载药纳米粒组具有更少的成结和短10%的长度,这再次证明了SP血管干扰效应的协同治疗。
本发明中SP@MSR-Granule局部给药,细胞通过内吞作用摄取载药MEND,在提高药效的同时减少了毒副作用且输送系统能够有效地穿透整个软骨层。以SP作为一种抗炎药物,试图在“一次注射”中满足所有的需求。充分利用所涉及的所有材料的全部特性,并合理组合,本发明实施例制备的SP@MSR-Granule可能为未来开发理想的关节腔内注射用抗炎产品提供一定的启示。
实施例4:SP@MSR-Granule的动物治疗效果实验
本发明采用SpragueDawley远交群大鼠(SpragueDawleyoutbredrats,SD大鼠)来进行CIA(胶原诱导关节炎动物模型,Collageninducedarthritis)造模。
本发明选用24只8周龄SPF级SpragueDawley远交群大鼠,体重为(230±20)g,全部为雌性。所有动物均在SPF级别下照料,在相同环境下进行试验。所有本课题试验动物的饲养、管理和试验均遵循《北京大学医学部伦理委员会》的相关规定。
A:对SD大鼠的局部不完全弗氏佐剂和脂多糖加强的CIA造模和对于SP@MSR-Granule的IAIs(Intra-articular injections,关节腔内注射)治疗
适应性喂养1周后,用1-24随机数生成法,将24只SD大鼠分为4组,并进行标记,分别为:空白对照组(blank组)、CIA模型对照组(CIA组)、CIA模型的SP灌胃(灌胃组)对照组和CIA模型的SP@MSR-Granule治疗组(SP@MSR-Granule组),并进行分笼。
将牛II型胶原与等体积的不完全弗氏佐剂在0℃(冰水混合物中)下进行充分混匀和乳剂化,配置为1g/mL。该乳剂在水中滴落一滴后可形成不分散液滴。空白对照组大鼠继续进行常规喂养。将CIA组、灌胃组与SP@MSR-Granule大鼠进行后爪关节的腔内注射,在后爪两个关节部位进行免疫诱导,每次注射80μL。在初次诱导后的第7天,再注射乳剂100μL和LPS50μL的免疫加强。CIA组、灌胃组与SP@MSR-Granule组进行CIA造模成功后,将200mL浓度为100mg/L的SP对灌胃组大鼠进行分次灌胃。经过载药率部分结果换算相同剂量SP的SP@MSR-Granule为0.38mg,称取0.38mg SP@MSR-Granule并均匀分散于200μLPBS中,对SP@MSR-Granule组大鼠的后爪趾关节进行IAIs。对空白组、灌胃组和CIA模型组的大鼠后抓趾关节进行200μLPBS注射。四组大鼠继续喂养10天后,拍摄大鼠后爪大体相。建模成功10天后对SD大鼠的CIA造模进行关节炎指数(arthritisindexindex,AI)评分:采用关节炎指数积分评价其关节炎症程度。趾间是否发炎对每一足爪分别进行打分,以0~4分记录,累积得分即为每只大鼠的关节炎指数。0分:无红斑和肿胀的证据;1分:红斑和轻度肿胀局限于足中段或踝关节;2分:红斑和轻度肿胀从踝关节蔓延至足中段;3分:红斑和轻度肿胀从踝关节蔓延至关节;4分:红斑和重度肿胀包括了踝、足和趾。对血清,趾关节软骨和滑膜进行取材。血清采集方法:处死前麻醉后尾静脉采集,离心方式为3000转/分,3分钟。关节滑液取材方法:将大鼠下爪趾关节组织整体取下,移到EP离心管中用蚊头尖镊小心的挑开关节囊,与预冷的0.9%NaCl 0.5mL稀释,室温血液自然凝固10-20分钟,离心20分钟左右(2000-3000转/分),取上清。采集软骨和滑膜的组织块,用0.1mL的PBS按照1:10的重量混匀后和进行匀浆,再放入组织破碎机中15000转/分钟研磨40次,每次10秒。收集匀浆后6000转/分离心10分钟取上清。
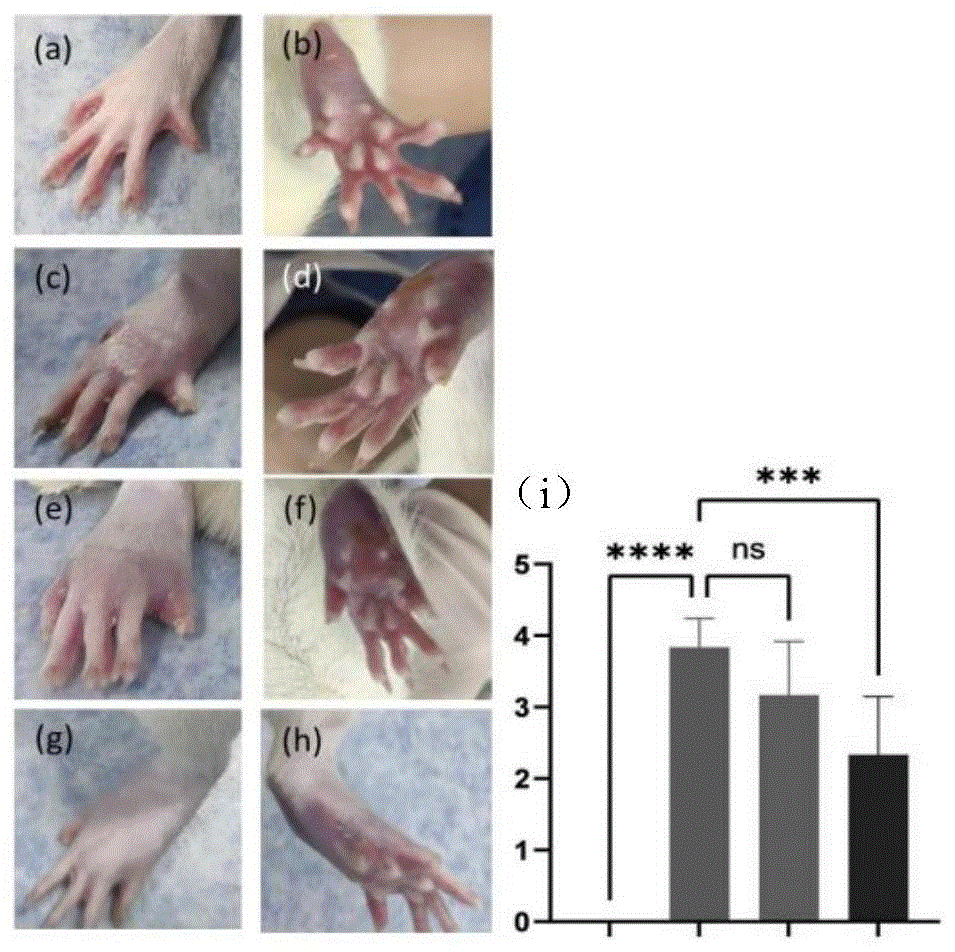
参阅图9,图9中(a)部分为未处理组,爪背面照片,图9中(b)部分为未处理组,同爪的掌面照片,图9中(c)部分为CIA模型组,爪背面照片,图9中(d)部分为CIA模型组,同爪的掌面照片,图9中(e)部分为SP灌胃组,爪背面照片,图9中(f)部分为SP灌胃组,同爪的掌面照片,图9中(g)部分为抗炎颗粒SP@MSR-Granule注射组,爪背面照片,图9中(h)部分为抗炎颗粒SP@MSR-Granule注射组,同爪的掌面照片,图9中(i)部分为各组的关节炎大体评分直方图,纵坐标为关节炎大体评分,横坐标左到右依次为未处理组、CIA模型组、SP灌胃组、SP@MSR-Granule注射组。
CIA建模成功后,进行对应干预10天。通过四组(空白对照、CIA、SP治疗CIA和SP@MSR-Granule)的AI评分发现,SP灌胃组和SP@MSR-Granule组对于CIA模型的均有显著变化,而SP@MSR-Granule组对于CIA模型的治疗效果优于相同剂量的SP灌胃组(p<0.05)。此外,SP@MSR-Granule对于CIA的全身性症状改善也比较明显,干预后的大鼠的活动度比SP灌胃组要灵活。而SP组的关节肿胀度和皮肤破溃程度虽然与CIA相比有明显改善,但是大鼠仍旧不喜运动,触碰诱导炎症的关节部位时有明显抗拒反应,这与CIA组大鼠症状相似。
B:SD大鼠CIA模型对于SP@MSR-Granule的IAIs治疗在炎性分子指标的组间对照试验
将上述的空白组、CIA组、灌胃组与SP@MSR-Granule组大鼠分别进行血清、软骨、滑膜的取材并进行分别测定以下指标。
Rat-IL-1β、IL-6、IL-10和TNF-α(IL-1β为细胞因子,IL-6、IL-10和TNF-α为关节液及外周血的炎性因子)的ELISA-kit(酶联免疫吸附法试剂盒)测定:在酶标包被板上设标准品孔10孔(稀释后各孔加样量都为50μl,浓度分别为60ng/L,40ng/L,20ng/L,10ng/L,5ng/L)。分别设空白孔(空白对照孔不加样品及酶标试剂,其余各步操作相同)、待测样品孔。在酶标包被板上待测样品孔中先加样品稀释液40μl,然后再加待测样品10μl(样品最终稀释度为5倍)。加样将样品加于酶标板孔底部,尽量不触及孔壁,轻轻晃动混匀。随后,每孔加入酶标试剂50μl,空白孔除外,再用封板膜封板后置37℃温育30分钟。将30倍浓缩洗涤液用蒸馏水30倍稀释后备用。小心揭掉封板膜,弃去液体,甩干,每孔加满洗涤液,静置30秒后弃去,如此重复5次,拍干。每孔先加入显色剂100μl轻轻震荡混匀,37℃避光显色10分钟。以空白孔调零,450nm波长依序测量各孔的吸光度(OD值)。测定应在加终止液后15分钟以内进行。
MMP-3、P65和β-actin(内参)的Westernblot测定:采用SDS/PAGE技术(sodiumdodecyl sulfate-polyacrylamide gel electrophoresis,聚丙烯酰胺凝胶电泳)对相应蛋白进行分离,将样品壁上缘用小刀去除,然后再凝胶胶板右上角进行标记方便定位,再小心的放入转移缓冲液中待用打开转移盒后,转移缓冲液浸润的湿海绵铺在筛孔板上,在海绵垫上放置3MM色谱层析纸,注意将排空气泡。将PVDF(聚偏氟乙烯)膜放置在凝胶板上并再以3MM色谱层析纸覆盖。关闭转移盒,将转移盒正确的方向放入转移槽中,转移盒的黑色筛孔板靠近转移槽的黑色一端。最后,连接电源,在4℃条件下维持恒压100V。1小时后,断开电源,将转移盒移出,取出PVDF膜,用TBS缓冲液(Tris缓冲盐水)进行冲洗,从膜上剪下宽约5mm的片段,在染色液中浸泡30分钟。然后,倒掉TBS缓冲液,加入10mL 0.5%封闭缓冲液及一抗,用TTBS缓冲液(TTBS Washing Bufferd Solution forWestern Blots)清洗2次,每次约10分钟。加入5mL 0.5%封闭缓冲液和二抗,用TTBS缓冲液清洗2次后,加入显影剂。看到清晰的显带后,用蒸馏水清洗来终止显色反应。
参阅图10,图10中(a)部分为在不同样本中各实验组TNF-α的ELISA实验结果,图10中(b)部分为在不同样本中各实验组IL-6的ELISA实验结果,图10中(c)部分为在不同样本样本中IL-10各实验组的ELISA实验结果,图10中(d)部分为在各样本中各实验组IL-1β的ELISA实验结果。图10中(a)、(b)、(c)、(d)部分对应的纵坐标均为浓度(ng/L)。注①:各结果图中样本从左至右依次为血清、软骨、滑膜。注②:直方图中从左至右依次为未处理组、CIA模型组、SP灌胃组、SP@MSR-Granule注射组。注③:注②:每个*代表P值小于0.001。
参阅图11,图11中(a)部分为在滑膜样本中各实验组的P65蛋白表达结果,图11中(b)部分为在滑样本中各实验组的MMP-3蛋白表达结果,图11中(c)部分为在滑膜样本各实验组的WB条带,上到下依次为P65、MMP-3、内参β-actin,图11中(d)部分为在软骨样本中各组的P65蛋白表达结果,图11中(e)部分为在软骨样本中各组的MMP-3蛋白表达结果,图11中(f)部分为在软骨样本各实验组的WB条带,上到下依次为P65、MMP-3、内参β-actin。注①:直方图中从左至右依次为未处理组,CIA模型组、SP灌药组、SP@MSR-Granule组。注②:每个*代表P值小于0.001。
由Elisa结果得知,白介素10(IL-10)作为炎症与免疫抑制因子在SP灌胃组和SP@MSR-Granule相比于CIA模型组获得了显著的提升,证明了SP对于CIA模型的炎性抑制作用。其余三个炎性标志物,TNF-α、IL-6、IL-1β作为关节炎最常用的三个代表,在SP灌胃组和SP@MSR-Granule组与CIA模型组的比较均存在明显降低,值得注意的是白介素6,作为炎症发生时最早升高的标志物,在SP@MSR-Granule组滑膜和软骨中相比较灌胃组,明显的浓度更低(p<0.00001)。这可能表明,SP@MSR-Granule可能存在更长效的抗炎效果。由Western-blot结果得知,滑液和软骨组织的MMP-3对于SP@MSR-Granule组来说已经与空白对照组无显著性差异且与灌胃组相比含量更少。但对于P65来说,SP@MSR-Granule组的下降与SP灌胃组无显著性差异。
C:SD大鼠CIA模型对于SP@MSR-Granule的IAIs治疗在免疫组化的组间对照试验
将上述的空白组、CIA组、灌胃组与SP@MSR-Granule组大鼠分别进行软骨取材。对取材的软骨组织进行石蜡切片后,将APES(3-氨丙基-3-乙氧基硅烷)胶片进行烤片(65℃下烤90分钟),直至蜡融化组织贴在玻片上。依次放入二甲苯(I)、二甲苯(II),二甲苯(III)各10分钟,直至蜡消失。然后,使用(100%、90%、80%)的酒精进行梯度水化,直至玻片没有油腻感,干净透亮。用3%H
参阅图12,图12中(a)、(b)、(c)和(d)部分为各组小鼠软骨切片的番红-固绿染色结果。图12中(a)部分为未处理组,图12中(b)部分为CIA模型组,图12中(c)部分为SP灌胃组,图12中(d)部分为SP@MSR-Granule注射组,图12中(e)部分为Mankin评分结果,横坐标从左至右依次为未处理组、CIA组、SP组、SP@MSR-Granule注射组,纵坐标为Mankin评分。
对大鼠行关节软骨的组织形态学观察及Mankin评分后,形态学上SP@MSR-Granule治疗组的软骨表面光整,软骨四层结构清晰可辨,软骨细胞排列有序,基质染色均匀。SP灌胃组关节软骨层变薄,软骨表面破损,部分软骨细胞核固缩,潮线紊乱。Mankin评分显示了CIA造模的成果,差异具有统计学意义。灌胃组和SP@MSR-Granule,都对CIA造成的软骨破坏具有保护和缓解作用。SP@MSR-Granule组的关节表明番红层更加明显,说明胶原特性更好,可以使CIA造模的中重度软骨损伤提供理想保护。
D:SP@MSR-Granule对SD大鼠负重关节区域的滞留效果——在体表征试验
利用Cy7-NHSester(Cyanine 7monosuccinimidyl ester,Cy7-NHS酯)上琥珀酰亚胺基与信号肽末端以及侧链氨基的反应,将荧光分子Cy7-NHSester连接到SP@MSR-Granule的BAS多肽上,形成Cy7-信号肽的荧光多肽SP@MSR-Granule(探针)。将1mg Cy7-NHSester溶解于200μL PBS,取2mg SP@MSR-Granule,溶解于0.1MNaHCO
参阅图13,图13中(a)部分为小动物活体荧光成像图,从上至下分别为1小时、24小时、5天、15天的小动物活体荧光成像图。图中红色箭头处代表SP@MSR-Granule的注射位置,对侧下肢为对照。绿色箭头处示意膀胱代谢。图13中(b)部分为左侧小动物活体荧光范围折线图(内圈、外圈),图13中(c)部分为右侧小动物活体荧光范围折线图(内圈、外圈),图13中(d)部分为左侧小动物活体荧光范围折线图(高度、宽度),图13中(e)部分为左侧小动物活体荧光范围折线图(高度、宽度),图13中(f)部分为小动物活体荧光浓度折线图,红色箭头处显示第二峰。
本实施例中,采取同体双下肢対照实验。每只大鼠右趾关节为SP@MSR-Granule装载的Cy7探针注射,左趾关节为Cy7的林格氏液对照组。通过荧光范围结果(长度和宽度)可以得知,相比于对照组,SP@MSR-Granule的荧光扩散更加平缓,且存在维持的(平台释放期)。通过荧光浓度f的结果可以得知,SP@MSR-Granule具有CRDDS(ControlledReleaseDrug Delivery Systems,是指药物以受控形式恒速从制剂中释放至靶部位而发挥疗效的给药系统)的“双峰释放”特性,第二释放峰值在5天后,浓度甚至超过首次注射后1小时。
E:SD大鼠CIA模型对于SP@MSR-Granule的IAIs治疗的抗氧化应激指标的组间对照试验
参照实验A中的方法重新建模,用随机数生成法,将大鼠分为5组,并进行标记,分别为:空白对照组(blank组)、CIA模型对照组(CIA组)、CIA模型的SP灌胃(灌胃组)对照组、CIA模型的SP@MSR-Granule治疗组(SP@MSR-Granule组)和未载SP的空白MSR-Granule组,并进行分笼。按照实验A中的方法,取每组关节滑液针对MDA(Malondialdehyde,丙二醛),SOD(superoxide dismutase,超氧化物歧化酶),CAT(catalase,过氧化氢酶),GPx(Glutathione Peroxidase,谷胱甘肽过氧化物酶)指标进行ELISA分析,进行差异性统计和制图。
参阅图14,图14中(a)部分为各实验组MDA的ELISA实验结果,纵坐标为MDA浓度(nmol/ml),图14中(b)部分为各实验组SOD的ELISA实验结果,纵坐标为SOD浓度(U/ml),图14中(c)部分为各实验组CAT的ELISA实验结果,纵坐标为CAT浓度(U/ml),图14中(d)部分为各实验组GPx的ELISA实验结果,纵坐标为GPx浓度(U/ml)。横坐标从左往右依次均为空白对照组、CIA组、SP组、SP@MSR-Granule组、MSR-Granule组。注①每个*代表P值小于0.001。
由图14可见,在MDA指标上,CIA-SD大鼠组较SP@MSR-Granule组以及未负载SP的空白MSR-Granule组有显著的下降,而SP@MSR-Granule组以及未负载SP的空白MSR-Granule组之间没有显著的统计学差异。这表明影响MDA指标的能力是可能由核心二氧化铈纳米粒起抗氧化作用,而非SP。在SOD、CAT、GPx指标上CIA-SD大鼠组较SP@MSR-Granule组以及未载磺胺吡啶的空白MSR-Granule组有显著的上升,而SP@MSR-Granule组以及未载磺胺吡啶的空白MSR-Granule组之间没有显著的统计学差异。这可能也是意味着该系统的抗氧化能力是由核心二氧化铈纳米粒起抗氧化的作用。
在大鼠的关节炎评分中,治疗组的关节炎评分有显著的下降。在关节炎的炎性指标中,如MMP-3、P64、TNF-α、IL-6、IL-10等表达水平上,无论是滑膜还是软骨标本,SP@MSR-Granule组相对于SP灌胃对照组都体现了更好的抗炎能力。且SP@MSR-Granule组在软骨免疫组化染色结果提示了软骨保护能力。此外,考虑到关节炎中存在氧化应激相关的病理生理机制,本实施例测定了抗氧化应激的相关指标如MDA、SOD、GPx等,结果显示主要是作为核心的载药纳米粒中的二氧化铈纳米颗粒发挥了抗氧化应激作用,这对载药系统本身选用材料的思路具有重大意义。在众多炎性指标中,SP@MSR-Granule组相比于CIA组IL-6的下调最为显著。IL-6对关节炎的疾病发展过程中具有广泛的影响,并且在免疫稳态破坏过程易于动态监测。急性感染或炎症可诱导IL-6的快速释放,并通过促进急性期炎性蛋白释放来激活免疫反应。同时,IL-6的大量释放和失控的IL-6受体信号传导对关节炎发病机制至关重要,现在被认为是临床干预的重要靶点。此外,IL-6被认为是炎性活动中反应最快的标志物,那么可以认为本材料相比于其他组出现了IL-6的显著下调,标志着更好的抗炎作用。通过抗氧化应激结果表明,载SP与未载SP的缓释载药制剂的抗氧化作用基本一致,说明铈离子作为强效催化剂可以有效的清除活性氧以减轻细胞中氧化应激损伤。进一步证明本发明实施例制备的抗炎载药缓释制剂的核心抗氧化单元(铈离子)起到了关键的抗炎收尾作用。炎症条件下的软骨细胞被NF-κB激活,使细胞代谢向糖酵解途径转换,并在缺氧环境下细胞内乳酸脱氢酶A与烟酰胺腺嘌呤二核苷酸结合,这个结合促进了活性氧的产生,而活性氧可以将IκB-ζ稳定(软骨细胞中的关键促炎介质),从而导致分解代谢和炎症基因(如MMPs和白细胞介素)的表达显著增加,同时降低了软骨细胞合成代谢因子(如聚集蛋白聚糖)的表达。因此,炎症和新陈代谢可以相互调节来促进软骨降解。这表明炎症介导的代谢转移可能是关节炎中关节软骨降解的病理基础,因而通过调节软骨细胞中的活性氧来降低炎症反应和减少分解代谢蛋白的产生,是一个很有潜力的治疗方式。
以上对本发明所提供的一种抗炎载药缓释制剂、制备方法及其应用,进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
- 一种载药止血海绵的制备方法及其制备的载药止血海绵
- 一种具有pH响应性的席夫碱共聚物包覆的介孔二氧化硅载药纳米粒子及其制备方法和应用
- 一种二硫化钼纳米载药复合物及其制备方法和应用
- 一种基于阿魏酸聚合物的载药纳米粒及其制备方法和应用
- 一种用于抗炎的载药纳米纤维膜制备方法
- 一种载带si-RNA的纳米递药体系及其制备方法和在抗耐曲妥珠单抗乳腺癌中的应用