一种硅基负极复合材料及其制备方法
文献发布时间:2024-04-18 19:44:28
技术领域
本发明涉及电池负极材料技术领域,更具体地说,涉及一种硅基负极复合材料及其制备方法。
背景技术
负极材料是指电池中充当负极的材料,也被称为负极活性材料。在充电时,电池负极材料接受正极材料释放出来的离子,这些离子会通过电解液移动到负极材料中,使其发生化学反应并存储能量。目前广泛应用于锂离子电池的负极材料主要有石墨、硅等。它们具有较高的能量密度和循环寿命,是现代电子设备和电动汽车等领域中常见的能源储存方式之一。而硅(Si)因为理论比容量高达4200mAh g
然而,在锂嵌入/脱出过程中,严重的体积变化(超过300%)将导致硅的粉碎和固体电解质界面(SEI)的连续形成。由于电接触的丧失和与电解质的副反应导致结构完整性的降低和容量的快速衰减。
目前,现有的方法中,通过将材料特征尺寸减小到纳米尺度,各种Si纳米结构材料可以避免机械断裂并改善循环稳定性。另外纳米化、将Si颗粒包裹在弹性和导电的外壳或基质,如金属和氧化物中,增强电子导电性以及稳定性也是改善体积效应的一种途径。
但现有的方法中,纳米化会降低振实密度、成本非常昂贵并且不可避免地在循环期间出现巨大的不可逆容量损失;Si与金属和氧化物复合降低容量发挥,未体现Si负极高容量优势。
因此,应用纳米化的硅材料,或者核-壳结构硅碳材料,依然不能解决问题,存在技术困难,成本高昂且不环保。
发明内容
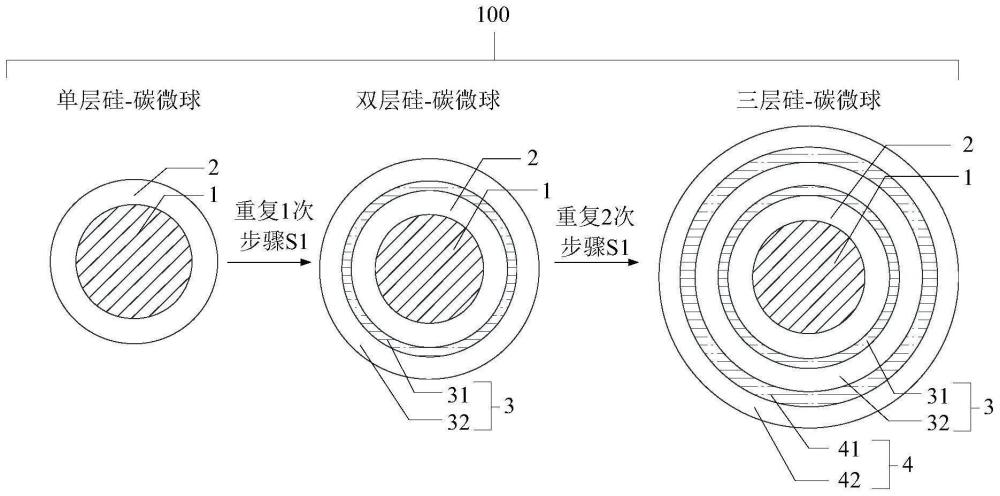
为解决上述缺陷,本发明提供一种硅基负极复合材料,所述硅基负极复合材料的每个颗粒中均为硅-碳微球;
其中,所述硅-碳微球包括设于微球的核心处的硅核,以及包覆在所述硅核外的第一碳层。
优选地,所述硅核的直径范围为258nm-344nm;
所述第一碳层的厚度范围为22nm-65nm。
优选地,所述硅-碳微球还包括包覆在所述第一碳层外有至少一层的复合层;
所述复合层包括由内至外依次包覆的硅底层和第二碳层。
优选地,在所述复合层为单层的复合层时,所述硅核的直径范围为225nm-310nm,所述第一碳层和所述第二碳层的厚度范围为23nm-58nm,所述硅底层的厚度范围为22nm-40nm;
优选地,在所述复合层为两层以上的复合层时,所述硅核的直径范围为180nm-358nm,所述第一碳层和所述第二碳层的厚度范围为14nm-46nm,所述硅底层的厚度范围为17nm-35nm。
此外,为解决上述问题,本发明还提供一种如上述所述硅基负极复合材料的制备方法,所述硅基负极复合材料的颗粒,是在碱性体系溶液的作用下,形成在所述硅核外包覆有所述第一碳层的所述硅-碳微球;
或者,在所述第一碳层外还包覆有至少一层的复合层的所述硅-碳微球。
优选地,所述在碱性体系溶液的作用下,形成在所述硅核外包覆有所述第一碳层的所述硅-碳微球;或者,在所述第一碳层外还包覆有至少一层的所述复合层的所述硅-碳微球,包括:
所述硅基负极复合材料的颗粒,是在碱性体系溶液的作用下基于碳化处理后,经过热还原反应得到的在所述硅核外包覆有所述第一碳层的所述硅-碳微球;或者,在所述第一碳层外还包覆有至少一层的所述复合层的所述硅-碳微球。
优选地,所述硅基负极复合材料的制备方法包括:
S1,利用碳源和硅源制备获得硅碳核;
S2,在所述硅核基础上,通过碳化处理,形成微球粗品;
S3,将所述微球粗品进行热还原反应,利用酸性溶液进行处理后,用水洗涤并干燥,得到终产品;
其中,所述S1,利用碳源和硅源制备获得硅碳核,包括:
S11,基于所述碳源和所述硅源,在碱性溶液体系的条件下,得到所述硅碳核。
优选地,所述S11,基于所述碳源和所述硅源,在碱性溶液体系的条件下,得到所述硅碳核,包括:
S111,通过进行至少一次的包覆处理得到混合产物,并以所述混合产物作为所述核心混合物料;
S112,将所述核心混合物料于室温下搅拌混合30小时,得到乳状混合物;
S112,将所述乳状混合物在180℃条件下水热处理24小时,真空过滤后得到固体产物;
S113,将所述固体产物60℃下干燥12小时,即得到所述硅碳核;
其中,所述包覆处理,包括:
S1111,将水、甲醇或乙醇、氨水和甲醛进行混合,得到碱性体系溶液;
S1112,在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物;
其中,在进行到第二次或第二次以上次数的所述包覆处理时,在步骤S1112中,同时加入前一次包覆处理所得到的混合产物;
优选地,所述步骤S1112,在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物中,混合方式为搅拌;混合时间为第一预设时间;
优选地,所述第一预设时间为25小时-35小时;
优选地,所述碳源为可溶性有机物,包括烃类化合物、酮类化合物、醇类化合物、醛类化合物和酚类化合物中的一种或多种;
优选地,所述硅源包括三甲基氯硅烷、四甲基硅烷、二甲基二氯硅烷、四氯化硅、乙烯基硅烷、氨基硅烷、四乙氧基硅烷和甲基丙烯酰氧基硅烷中的一种或多种;
优选地,所述碱性体系溶液的pH为7.1-9.0。
优选地,满足以下条件中的至少一个:
A、所述水热处理的温度为100℃-250℃;
B、所述水热处理的时间为10小时-30小时;
C、所述碳化处理的温度为800℃-1200℃;
D、所述碳化处理的时间为2小时-10小时。
优选地,所述S3,将所述微球粗品进行热还原反应,利用酸性溶液进行处理后,用水洗涤并干燥,得到终产品,包括:
S31,将所述微球粗品与还原剂进行共同研磨,得到研磨混合物;
S32,将所述研磨混合物加热第二预设时间,得到加热粉末;
S33,将所述加热粉末用所述酸性溶液处理,再用水洗涤后,得到处理粉末;
S34,将所述处理粉末干燥第三预设时间,得到所述终产品;
优选地,所述还原剂为镁;
优选地,所述第二预设时间为5小时-10小时;
优选地,所述第三预设时间为10小时-15小时。
本发明提供一种硅基负极复合材料及其制备方法,其中,所述硅基负极复合材料的每个颗粒中均为硅-碳微球;其中,所述硅-碳微球包括设于微球的核心处的硅核,以及包覆在所述硅核外的第一碳层。本发明所提供的硅基负极复合材料,与现有的核-壳结构相比,硅-碳微球具有其核和壳具有多孔的优点,这使得其更容易渗透电解质,从而缩短Li
附图说明
图1为本发明硅基负极复合材料的结构示意图;
图2为本发明硅基负极复合材料检测实验中颗粒1电镜图;
图3为本发明硅基负极复合材料检测实验中颗粒5电镜图;
图4为本发明硅基负极复合材料检测实验中表2的参数对比图;
图5为本发明硅基负极复合材料检测实验中颗粒6电镜图;
图6为本发明硅基负极复合材料检测实验中颗粒10电镜图;
图7为本发明硅基负极复合材料检测实验中表3的参数对比图;
图8为本发明硅基负极复合材料检测实验中颗粒11电镜图;
图9为本发明硅基负极复合材料检测实验中颗粒15电镜图;
图10为本发明硅基负极复合材料检测实验中表4的参数对比图;
图11为本发明硅基负极复合材料检测实验中颗粒16电镜图;
图12为本发明硅基负极复合材料检测实验中颗粒20电镜图;
图13为本发明硅基负极复合材料检测实验中表5的参数对比图;
图14为本发明硅基负极复合材料检测实验中颗粒21电镜图;
图15为本发明硅基负极复合材料检测实验中颗粒25电镜图;
图16为本发明硅基负极复合材料检测实验中表6的参数对比图;
图17为本发明硅基负极复合材料检测实验中颗粒26电镜图;
图18为本发明硅基负极复合材料检测实验中颗粒27电镜图;
图19为本发明硅基负极复合材料检测实验中表7的参数对比图;
图20为本发明电化学性能测试的3A/20A循环放电容量曲线图;
图21为本发明电化学性能测试的3A/20A循环放电容量保持率曲线图。
附图标记:
100,硅基负极复合材料的硅-碳微球颗粒;1,硅核;2,第一碳层;3,第1层的复合层;31,第1层的硅底层;32,第1层的第二碳层;4,第2层的复合层;41,第2层的硅底层;42,第2层的第二碳层。
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
下面详细描述本申请的实施例,实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本申请,而不能理解为对本申请的限制。
在本申请的描述中,需要理解的是,术语“中心”、“纵向”、“横向”、“长度”、“宽度”、“厚度”、“上”、“下”、“前”、“后”、“左”、“右”、“竖直”、“水平”、“顶”、“底”、“内”、“外”、“顺时针”、“逆时针”、“轴向”、“径向”、“周向”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本申请和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本申请的限制。
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者更多个该特征。在本申请的描述中,“多个”的含义是两个或两个以上,除非另有明确具体的限定。
在本申请中,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”、“固定”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或成一体;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通或两个元件的相互作用关系。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本申请中的具体含义。
在本申请中,除非另有明确的规定和限定,第一特征在第二特征“上”或“下”可以是第一和第二特征直接接触,或第一和第二特征通过中间媒介间接接触。而且,第一特征在第二特征“之上”、“上方”和“上面”可是第一特征在第二特征正上方或斜上方,或仅仅表示第一特征水平高度高于第二特征。第一特征在第二特征“之下”、“下方”和“下面”可以是第一特征在第二特征正下方或斜下方,或仅仅表示第一特征水平高度小于第二特征。
实施例:
参考图1,本实施例提供一种硅基负极复合材料,所述硅基负极复合材料的每个颗粒中均为硅-碳微球;其中,所述硅-碳微球包括设于微球的核心处的硅核,以及包覆在所述硅核外的第一碳层。
上述,硅基负极复合材料,具有多层的复合结构。硅基负极复合材料为颗粒,每个颗粒中,均为硅-碳微球。
上述,硅-碳微球中,由内至外,依次为硅核和第一碳层。
其中,硅核,设在微球核心处;第一碳层,包覆在核心外。
本实施例所提供的硅基负极复合材料,与现有的核-壳结构相比,硅-碳微球具有其核和壳具有多孔的优点,这使得其更容易渗透电解质,从而缩短Li
进一步的,所述硅核的直径范围为258nm-344nm;例如,可以为260nm、270nm、280nm、290nm、300nm、310nm、320nm、330nm、340nm、344nm等。
所述第一碳层的厚度范围为22nm-65nm;例如,可以为22nm、25nm、30nm、35nm、40nm、45nm、50nm、55nm、60nm、65nm等等。
进一步的,所述硅-碳微球还包括包覆在所述第一碳层外有至少一层的复合层;
所述复合层包括由内至外依次包覆的硅底层和第二碳层。
上述,硅基负极复合材料,可以为多层的复合结构。
在第一碳层外还包覆有复合层。因此,在硅基负极复合材料中,颗粒中的复合层的数量可以为0、1、2、3等。其复合结构可以参考如下表中的结构,表中从上至下,为复合结构中由内至外排列的不同层的部分。
表1、包覆不同层数复合层的硅-碳微球结构(复合结构由内至外排列)
上述,本实施例中,提供的硅基负极复合材料中的硅-碳微球,为使用第一碳层包覆硅核以及多层复合层包覆在第一碳层外,从而构成多层的复合结构,能够有效通过多层碳层的弹性,吸收充放电过程中硅的体积效应带来的应力,保护结构的完整性。
上述,在多层复合结构中,包含有的多层硅层的设计,还可以加大粒径,有利于提高压实密度从而发挥更高容量。
进一步的,在所述复合层为单层的复合层时,所述硅核的直径范围为225nm-310nm;例如,可以为225nm、230nm、235nm、240nm、245nm、250nm、255nm、260nm、270nm、280nm、290nm、300nm、310nm等等。
所述第一碳层和所述第二碳层的厚度范围为23nm-58nm;例如,可以为23nm、25nm、30nm、35nm、40nm、45nm、50nm、55nm、58nm等等。
所述硅底层的厚度范围为22nm-40nm;例如,可以为22nm、25nm、27nm、30nm、32nm、35nm、37nm、40nm等等。
进一步的,在所述复合层为两层以上的复合层时,所述硅核的直径范围为180nm-358nm;例如,可以为180nm、190nm、200nm、220nm、240nm、260nm、280nm、300nm、320nm、340nm、358nm等等。
所述第一碳层和所述第二碳层的厚度范围为14nm-46nm;例如,可以为14、16、18、20、25、30、35、40、46等等。
所述硅底层的厚度范围为17nm-35nm;例如,可以为17nm、19nm、20nm、22nm、25nm、27nm、29nm、30nm、31nm、33nm、35nm等等。
上述,所述第一碳层和所述第二碳层的厚度可以相同,也可以不同,但均在上述限定的范围之内。
此外,本实施例中还提供一种如上述所述硅基负极复合材料的制备方法,所述硅基负极复合材料的颗粒,是在碱性体系溶液的作用下,形成在所述硅核外包覆有所述第一碳层的所述硅-碳微球;
或者,在所述第一碳层外还包覆有至少一层的复合层的所述硅-碳微球。
在上述方法中,采用碱性体系的溶液是为了形成固定的微球表面,从而形成稳定的复合材料微球。并且,采用碱性条件还可以促进乳液的凝胶化,快速形成结构稳定的小球体,在后续的热处理过程中,可以避免产物在高温下的分解和变形等问题。因此,采用碱性体系的溶液可以有效地改善复合材料的形貌和性能,并且提高制备效率。
上述制备方法中,可以根据需要进行制备如下两大类型的硅基负极复合材料:
(1)只有一层碳层的硅-碳微球:在所述硅核外包覆有所述第一碳层的所述硅-碳微球。其复合层的层数为0。
(2)具有复合层的硅-碳微球:在所述硅核外包覆有所述第一碳层后,在所述第一碳层外还包覆有至少一层的所述复合层的所述硅-碳微球。其复合层的数量为N,N≥1。
进一步的,所述在碱性体系溶液的作用下,形成在所述硅核外包覆有所述第一碳层的所述硅-碳微球;或者,在所述第一碳层外还包覆有至少一层的所述复合层的所述硅-碳微球,包括:
所述硅基负极复合材料的颗粒,是在碱性体系溶液的作用下基于碳化处理后,经过热还原反应得到的在所述硅核外包覆有所述第一碳层的所述硅-碳微球;或者,在所述第一碳层外还包覆有至少一层的所述复合层的所述硅-碳微球。
上述,热还原反应是指在高温条件下,将一种物质还原成另一种物质的化学反应。在这种反应中,还原剂通常会被加热至高温,以产生足够的能量来提供电子给被还原物种,从而实现还原。
热还原反应常用于金属和非金属之间的反应,其中非金属被还原为金属或金属化合物。在这些反应中,通常需要使用还原剂,例如碳、氢气、氧化铝、镁等,来提供电子以实现还原。
在本实施例中,在制备多层硅/碳微球的过程中采用了热还原反应技术,热还原反应被用于将二氧化硅和碳材料还原为硅和碳,其目的在于:
(1)硅和碳具有更高的稳定性:硅和碳比起对应的氧化物和碳氢化合物,具有更高的稳定性。在高温条件下,将氧化物和碳还原为硅和碳可以提高材料的稳定性。
(2)避免产生大量的气体:在常温下使用还原剂将氧化物还原为金属会产生大量气体,而高温条件下可以使产生的气体尽可能地被排出,从而避免对反应造成干扰。
(3)提高反应速率:高温条件下分子的运动速度增加,反应速率也随之增加,这有利于加快反应速率和提高产物纯度。
因此,在制备多层硅/碳微球的过程中,采用了热还原反应来还原氧化物和碳材料,以得到稳定性更高的硅和碳材料。
进一步的,所述制备方法包括:
S1,利用碳源和硅源制备获得硅核;
其中,步骤S1中的工艺可以包括如下特征:
(1)所述碳源为可溶性有机物,包括烃类化合物、酮类化合物、醇类化合物、醛类化合物和酚类化合物中的一种或多种。
(2)所述硅源包括三甲基氯硅烷、四甲基硅烷、二甲基二氯硅烷、四氯化硅、乙烯基硅烷、氨基硅烷、四乙氧基硅烷和甲基丙烯酰氧基硅烷中的一种或多种。
(3)所述碱性体系溶液可以为水、乙醇、氨水和甲醛等组成的混合体系;
(4)所述碱性体系溶液的pH为7.1-9.0。例如,可以为7.1、7.5、7.9、8.0、8.2、8.5、8.7、8.9、9.0等等。
S2,在所述硅核基础上,通过碳化处理,形成微球粗品;
S3,将所述微球粗品进行热还原反应,利用酸性溶液进行处理后,用水洗涤并干燥,得到终产品;
其中,所述S1,利用碳源和硅源制备获得硅碳核,包括:
S11,基于所述碳源和所述硅源,在碱性溶液体系的条件下,得到所述硅碳核。
上述,在步骤S1中,碱性体系溶液可以为水、乙醇、氨水的混合溶液,其混合体系中,水和乙醇的比例可以为1:7、1:10等。
上述,在步骤S1中,可以发生化学反应。当碳源为醇类化合物时,它们可以与硅源发生缩合反应,生成硅氧烷链和醇类化合物的缩合产物,同时放出水分子。这种反应被称为水解缩合反应。
例如,当乙二醇作为碳源,三甲基氯硅烷作为硅源时,可能的反应式如下:
2HOCH
在此反应中,乙二醇与三甲基氯硅烷缩合形成[(HOCH
当硅源为TEOS和碳源为间苯二酚时,其反应的化学方程式可以为:
Si(OC
该反应同样是一种缩合反应,也称为凝胶化反应。在此反应中,TEOS分子与间苯二酚分子通过羟基(-OH)团进行缩合,在生成的产物中,硅原子与苯环上的羟基形成了Si-O-C键,产生了四面体结构的Si(C
这个过程通常用于制备有机-无机复合材料或者纳米材料,可以得到高度透明、高质量、高硬度的薄膜或粉末。
上述,在S1中,具体可以为:
S11,基于所述碳源和所述硅源,在碱性溶液体系的条件下,得到所述硅碳核。
所述S11,基于所述碳源和所述硅源,在碱性溶液体系的条件下,得到所述硅碳核,包括:
S111,通过进行至少一次的包覆处理得到混合产物,并以所述混合产物作为所述核心混合物料;
S112,将所述核心混合物料于室温下搅拌混合30小时,得到乳状混合物;
S112,将所述乳状混合物在180℃条件下水热处理24小时,真空过滤后得到固体产物;
S113,将所述固体产物60℃下干燥12小时,即得到所述硅碳核;
其中,所述包覆处理,包括:
S1111,将水、甲醇或乙醇、氨水和甲醛进行混合,得到碱性体系溶液;
S1112,在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物;
其中,在进行到第二次或第二次以上次数的所述包覆处理时,在步骤S1112中,同时加入前一次包覆处理所得到的混合产物;
进一步的,所述步骤S1112,在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物中,混合方式为搅拌;混合时间为第一预设时间;
进一步的,所述第一预设时间为25小时-35小时;
上述,碱性体系溶液为水、乙醇和氨水的混合体系,或者水、乙醇、氨水和甲醛的混合体系,其pH为7.1-9.0,其配置方法例如可以为如下方法:
将去离子水和乙醇以1:7的比例混合成均匀溶液400mL,再依次加入1.5mL的氨水溶液(28wt%)、2.8mL的甲醛(RF)(37wt%),将上述均相溶液搅拌均匀,即得到所述碱性体系溶液。
上述,混合方式为搅拌时,其混合的时间可以为25小时-35小时,例如为25小时、26小时、27小时、28小时、29小时、30小时、32小时、34小时、35小时等等。
上述,在步骤S1中,采用碱性体系的溶液是为了将碳源(例如间苯二酚)作为硅源的前驱体固定在微球表面。由于碳源(间苯二酚)分子本身不与水或乙醇发生反应,因此需要使用一些添加剂来促进其在乳液中的分散和稳定。
碱性体系溶液中,氨水的加入可以使体系呈现碱性,进而引起甲醛缩合反应,生成DMTP有机化合物。这也有助于提高间苯二酚在乳液中的溶解度,增加与硅源(例如TEOS)的反应时间和交联反应,从而形成稳定的复合材料微球。
此外,采用碱性条件还可以促进乳液的凝胶化,快速形成结构稳定的小球体,在后续的热处理过程中,可以避免产物在高温下的分解和变形等问题。因此,采用碱性体系的溶液可以有效地改善复合材料的形貌和性能,并且提高制备效率。
需要说明的是,本实施例中,通过所述硅基负极复合材料的制备方法,制备得到符合要求的硅基负极复合材料颗粒。
通过步骤S111中的包覆处理的次数,控制终产品,即硅基负极复合材料颗粒的具体形态。
例如,针对于单层的硅-碳微球、包覆1层复合层的硅-碳微球和包覆2层复合层的硅-碳微球,分别可以为如下情况和步骤:
1、制备如表1中的单层的硅-碳微球(单层硅-碳微球),则进行1次包覆处理,具体的:
(1)将水、甲醇或乙醇、氨水和甲醛进行混合,得到碱性体系溶液;
(2)在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物,以所述混合产物作为所述核心混合物料;
(3)将所述核心混合物料于室温下搅拌混合30小时,得到乳状混合物;
(4)将所述乳状混合物在180℃条件下水热处理24小时,真空过滤后得到固体产物;
(5)将所述固体产物60℃下干燥12小时,即得到所述硅碳核;
2、制备如表1中的包覆1层复合层的硅-碳微球(双层硅-碳微球),则进行2次包覆处理,具体的:
(1)将水、甲醇或乙醇、氨水和甲醛进行混合,得到碱性体系溶液;
(2)在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物A;
(3)再次配置碱性体系溶液,或采用前述的配置好的碱性体系溶液;
(4)在所述碱性溶液体系中加入所述碳源、所述硅源,以及步骤(1)中得到的所述混合产物A进行混合溶解,得到新的混合产物B,以所述混合产物B作为所述核心混合物料;
(5)将所述核心混合物料于室温下搅拌混合30小时,得到乳状混合物;
(6)将所述乳状混合物在180℃条件下水热处理24小时,真空过滤后得到固体产物;
(7)将所述固体产物60℃下干燥12小时,即得到所述硅碳核;
3、制备如表1中的包覆2层复合层的硅-碳微球(三层硅-碳微球),则进行3次包覆处理,具体的:
(1)将水、甲醇或乙醇、氨水和甲醛进行混合,得到碱性体系溶液;
(2)在所述碱性溶液体系中加入所述碳源和所述硅源进行混合溶解,得到混合产物A;
(3)再次配置碱性体系溶液,或采用前述的配置好的碱性体系溶液;
(4)在所述碱性溶液体系中加入所述碳源、所述硅源,以及步骤(1)中得到的所述混合产物A进行混合溶解,得到新的混合产物B;
(5)再次配置碱性体系溶液,或采用前述的配置好的碱性体系溶液;
(6)在所述碱性溶液体系中加入所述碳源、所述硅源,以及步骤(4)中得到的所述混合产物B进行混合溶解,得到新的混合产物C,以所述混合产物C作为所述核心混合物料;
(7)将所述核心混合物料于室温下搅拌混合30小时,得到乳状混合物;
(8)将所述乳状混合物在180℃条件下水热处理24小时,真空过滤后得到固体产物;
(9)将所述固体产物60℃下干燥12小时,即得到所述硅碳核;
通过反复进行N次的包覆处理流程,从而得到对应的硅碳核,并通过进一步的水热处理、碳化处理等步骤,得到符合目标要求的终产品,终产品的复合结构,即为与包覆处理的进行次数相对应的复合结构。总之,本实施例所提供的方法,能够更加方便的控制终产品的结构,从而方便的制备得到更加符合进一步的电化学性能的浆料和电池产品,通过制备方法所制备得到的硅基负极复合材料,与现有的核-壳结构相比,硅-碳微球具有其核和壳具有多孔的优点,这使得其更容易渗透电解质,从而缩短Li+的扩散长度。并且,多层硅/碳微球在充电/放电过程中能够表现出优异的机械和电化学性能,既降低了制造成本,又具有绿色环保的特性。
进一步的,所述硅基负极复合材料的制备方法满足以下条件中的至少一个:
A、所述水热处理的温度为100℃-250℃;
B、所述水热处理的时间为10小时-30小时;
C、所述碳化处理的温度为800℃-1200℃;
D、所述碳化处理的时间为2小时-10小时。
上述,水热处理可以在反应釜中进行,例如可以为具有Teflon衬里的高压釜。
上述,水热处理的方式例如可以为:
将混合溶液在Teflon衬里的高压釜中在180℃下水热处理24小时。
上述,在步骤S3,将所述第二混合溶液固液分离后,收集固体产物并干燥,通过碳化处理得到微球粗品中,通过碳化处理得到了微球粗品。
其中,碳化处理的温度为800℃-1200℃,例如可以为1000℃。碳化处理的时间可以为2小时-10小时,例如可以为3小时、4小时。
上述,步骤S3可以为:
通过真空过滤,收集固体产物,并在60℃的烘箱中干燥12小时。
将上述产物在1000℃下在氩气流动管式炉中碳化3小时,加热速率为5℃/min,得到微球粗品。
进一步的,所述S3,将所述微球粗品进行热还原反应,利用酸性溶液进行处理后,用水洗涤并干燥,得到终产品,包括:
S31,将所述微球粗品与还原剂进行共同研磨,得到研磨混合物;
S32,将所述研磨混合物加热第二预设时间,得到加热粉末;
S33,将所述加热粉末用所述酸性溶液处理,再用水洗涤后,得到处理粉末;
S34,将所述处理粉末干燥第三预设时间,得到所述终产品;
进一步的,所述还原剂为镁;
进一步的,所述第二预设时间为5小时-10小时;例如可以为5小时、6小时、7小时、8小时、9小时、10小时等等。
进一步的,所述第三预设时间为10小时-15小时。例如,可以为10小时、11小时、12小时、13小时、14小时、15小时等等。
在上述方法中,使用的还原剂是镁粉。但是,在制备多层硅/碳微球的过程中,也可以考虑使用其他还原剂来进行还原。例如,一些常用的还原剂包括氢气、亚铁离子、钠、锌、铝等物质。还原剂的选择取决于实验条件、所需反应速率以及具体反应类型。
上述,加入镁粉是为了将硅氧化物和碳材料还原为硅和碳。这是一种热还原反应,也被称为焙烧还原。具体的反应可以用以下化学方程式表示:
SiO
在该反应中,镁粉充当还原剂,将SiO
下面通过具体的实施例进一步说明本发明,但是应当理解为,这些实施例仅仅是用于更详细地说明之用,而不应理解为用于以任何形式限制本发明。
实施例1:
(1)将去离子水和乙醇以1:7的比例混合成均匀溶液400mL,再依次加入1.5mL的氨水溶液(28wt%)、2.8mL的甲醛(RF)(37wt%),将上述均相溶液搅拌均匀,得到碱性体系溶液;随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源),将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第一混合溶液;
(2)将第一混合溶液在Teflon衬里的高压釜中在180℃下水热处理24小时,得到第二混合溶液;
(3)通过真空过滤对所述第二混合溶液进行固液分离,收集固体产物,并在60℃的烘箱中干燥12小时。将上述产物在1000℃下在氩气流动管式炉中碳化3小时,加热速率为5℃/min,得到微球粗品;
(4)将3g微球粗品用2.4g镁粉(还原剂)研磨;然后,将研磨混合物放置在管式炉中,在650℃下在氩气氛下以2L/min的流速加热6小时(第二预设时间);将获得的棕色粉末用2M HCl溶液处理以除去MgO和多余的Mg;最后,用去离子水洗涤所得产物,然后在鼓风烘箱中80℃下干燥12小时(第三预设时间),得到终产品(复合结构中无复合层,只有一层第一碳层,简称单层硅-碳微球,)。
实施例2:
(1)将去离子水和乙醇以1:7的比例混合成均匀溶液400mL,再依次加入1.5mL的氨水溶液(28wt%)、2.8mL的甲醛(RF)(37wt%),将上述均相溶液搅拌均匀,得到碱性体系溶液;随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源),将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第一预混液;
将所述第一预混液重复再进行第(1)步1次,即再进行如下步骤:
配置得到碱性体系溶液后,随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源)和所述第一预混液,将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第一混合溶液;
(2)将第一混合溶液在Teflon衬里的高压釜中在180℃下水热处理24小时,得到第二混合溶液;
(3)通过真空过滤对所述第二混合溶液进行固液分离,收集固体产物,并在60℃的烘箱中干燥12小时。将上述产物在1000℃下在氩气流动管式炉中碳化3小时,加热速率为5℃/min,得到微球粗品;
(4)将3g微球粗品用2.4g镁粉(还原剂)研磨;然后,将研磨混合物放置在管式炉中,在650℃下在氩气氛下以2L/min的流速加热6小时(第二预设时间);将获得的棕色粉末用2M HCl溶液处理以除去MgO和多余的Mg;最后,用去离子水洗涤所得产物,然后在鼓风烘箱中80℃下干燥12小时(第三预设时间),得到终产品(在所述第一碳层外还包覆有1层的所述复合层的所述硅-碳微球,简称双层硅-碳微球)。
实施例3:
(1)将去离子水和乙醇以1:7的比例混合成均匀溶液400mL,再依次加入1.5mL的氨水溶液(28wt%)、2.8mL的甲醛(RF)(37wt%),将上述均相溶液搅拌均匀,得到碱性体系溶液;随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源),将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第一预混液;
将所述第一预混液重复再进行第(1)步2次,即再进行如下步骤:
A、配置得到碱性体系溶液后,随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源)和所述第一预混液,将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第二预混液;
B、配置得到碱性体系溶液后,随后加入2g间苯二酚(碳源)、14mL正硅酸乙酯(TEOS,硅源)和所述第二预混液,将混合物在室温下剧烈搅拌30小时(第一预设时间),得到第一混合溶液;
(2)将第一混合溶液在Teflon衬里的高压釜中在180℃下水热处理24小时,得到第二混合溶液;
(3)通过真空过滤对所述第二混合溶液进行固液分离,收集固体产物,并在60℃的烘箱中干燥12小时。将上述产物在1000℃下在氩气流动管式炉中碳化3小时,加热速率为5℃/min,得到微球粗品;
(4)将3g微球粗品用2.4g镁粉(还原剂)研磨;然后,将研磨混合物放置在管式炉中,在650℃下在氩气氛下以2L/min的流速加热6小时(第二预设时间);将获得的棕色粉末用2M HCl溶液处理以除去MgO和多余的Mg;最后,用去离子水洗涤所得产物,然后在鼓风烘箱中80℃下干燥12小时(第三预设时间),得到终产品(在所述第一碳层外还包覆有2层的所述复合层的所述硅-碳微球,简称三层硅-碳微球)。
对比例1:
对比例1中,使用商用研磨法制备的硅碳材料。
硅碳材料的制备方法为:
(1)将单质硅研磨至100nm左右后与石墨混合均匀;
(2)使用软碳进行包覆后烧结制程,得到硅碳材料终产品。
上述方法所得到的硅碳材料终产品的形貌不规则,粒径D50约为10um-15um,比表面积约为3m
对比例2:
对比例2中,使用商用液相法制备的硅碳材料。
硅碳材料制备方法为:
(1)将需要与硅复配的石墨完全浸泡至硅基溶液中;
(2)后将硅在石墨负极中进行还原得到硅碳材料终产品(硅碳石墨负极)。
上述方法制得的硅碳材料终产品中,硅晶尺寸约为150nm,大于研磨法硅碳中硅晶尺寸,材料粒径D50约为10μm-15μm,比表面积约为2m
制成电池后,实施例与对比例负极克容量均调节至400mAh/g-405mAh/g,导电剂1.5%,粘结剂3%,正极使用NCM三元材料。
检测实验:
1、针对于实施例1中所制备的终产品(单层硅-碳微球)中的颗粒进行测量,得到如下数据:
(1)控制实施例1中所制备的终产品的第一碳层厚度下,调整硅核直径,数据如下表:
表2、颗粒1-颗粒5的单层硅-碳微球中各部分参数
参考图2为颗粒1的电镜图,图3为颗粒5的电镜图,图4为表2的参数比对图。
(2)控制实施例1中所制备的终产品的硅核直径下,调整第一碳层厚度,数据如下表:
表3、颗粒6-颗粒10的单层硅-碳微球中各部分参数
参考图5为颗粒6的电镜图,图6为颗粒10的电镜图,图7为表3的参数比对图。
2、针对于实施例2中所制备的终产品(双层硅-碳微球)中的颗粒进行测量,得到如下数据:
(1)控制实施例2中所制备的终产品的第一碳层、硅底层和第二碳层厚度下,调整硅核直径,数据如下表:
表4、颗粒11-颗粒15的双层硅-碳微球中各部分参数
参考图8为颗粒11的电镜图,图9为颗粒15的电镜图,图10为表4的参数比对图。
(2)控制实施例2中所制备的终产品的硅核直径下,调整第一碳层、硅底层和第二碳层厚度,数据如下表:
表5、颗粒16-颗粒20的双层硅-碳微球中各部分参数
参考图11为颗粒16的电镜图,图12为颗粒20的电镜图,图13为表5的参数比对图。
3、针对于实施例3中所制备的终产品(三层硅-碳微球)中的颗粒进行测量,得到如下数据:
(1)控制实施例3中所制备的终产品的第一碳层、第1层的硅底层和第1层的第二碳层、第2层的硅底层和第2层的第二碳层厚度下,调整硅核直径,数据如下表:
表6、颗粒21-颗粒25的三层硅-碳微球中各部分参数
参考图14为颗粒21的电镜图,图15为颗粒25的电镜图,图16为表6的参数比对图。
(2)控制实施例2中所制备的终产品的硅核直径下,调整第一碳层、硅底层和第二碳层厚度,数据如下表:
表7、颗粒26-颗粒30的三层硅-碳微球中各部分参数
参考图17为颗粒26的电镜图,图18为颗粒27的电镜图,图19为表7的参数比对图。
通过上述实验结果可以得出,碳层、硅核/硅层的厚度或直径会随着碳源和硅源的用量变化呈正向变化。在合成过程中通过控制碳源和硅源的用量,可以有效调节材料中碳层、硅层/硅核的厚度或直径。并且通过控制碳层、硅层/硅核的厚度或直径可以调整硅碳球的直径。硅核/硅层的直径、厚度以及硅碳球的直径对电池的循环和倍率性能有重要影响,过大的硅核/硅层的直径、厚度会导致更严重的体积膨胀和极片掉粉,会使电池的循环性能急剧衰减,缩短循环寿命,对比大倍率放电同样不利。碳层厚度和层数对材料电化学性能同样重要,通过控制厚度和层数可以吸收部分硅负极充放电过程中的体积膨胀,降低体积效应对极片的不利影响。硅碳球的直径会在加工过程中直接影响浆料分散、压实等性能。
4、电化学性能测试:
分别对实施例1-3和对比例1-2进行了内阻测试和电池性能测试,测试结果如下:
表8、实施例1-2和对比例1-2的内阻数据分布表(内阻值(mΩ))
通过内阻数据分布表,可以了解电池内阻分布的情况,以及电池在生产和使用过程中的质量控制水平。这些信息可以帮助评估电池的性能和寿命,并为电池的设计和优化提供重要的参考。参考表8中的内阻数据分布,其中横向的分别为对比例1和2,以及实施例1-3测试的内阻值(mΩ),通过表8可见,采用本发明所提供方法制备得到的实施例1-3的各测试单体所测得的内阻值均小于对比例1和2。
参考图20为3A/20A循环放电容量曲线图,该曲线图显示了在给定的放电条件下电池的容量随时间的变化情况。通过这个曲线图,可以了解电池在不同的放电条件下的容量表现,以及电池的寿命和性能如何受到放电条件的影响。参考图21为3A/20A循环放电容量保持率曲线图,该曲线图显示了在给定的放电条件下电池容量的保持率随时间的变化情况。通过这个曲线图,可以了解电池在循环放电过程中容量的保持程度,以及电池的衰减速度和寿命如何受到循环放电的影响。
由图20和图21可以得出,采用本发明所提供的硅基负极复合材料的实施例1-3,与对比例1-2相比,初始容量发挥相差不大,随着循环周数增加,对比例1-2循环衰减速度加快,容量保持率降低。循环至600周时,实施例1-3容量保持率大于70%,对比例1与对比例2容量保持率约为51.85%和47.99%,实施例容量保持率优势明显。
因此可以得出结论:在相同条件下,采用实施例1-3的复合材料与对比例1-2相比均可明显改善电池循环容量保持率。
总之,本发明所提供的硅基负极复合材料,与现有的核-壳结构相比,硅-碳微球具有其核和壳具有多孔的优点,这使得其更容易渗透电解质,从而缩短Li
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本申请的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
尽管上面已经示出和描述了本申请的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本申请的限制,本领域的普通技术人员在本申请的范围内可以对上述实施例进行变化、修改、替换和变型。