补体C3裂解抑制剂在制备治疗糖尿病心肌病的药物中的应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明涉及生物医药技术领域,尤其涉及补体C3裂解抑制剂在制备治疗糖尿病心肌病的药物中的应用。
背景技术
糖尿病(Diabetes mellitus,DM)是以高血糖为特征的严重危害人类健康的慢性疾病,且其患病率和发病率均呈逐年增加的趋势。糖尿病心肌病(Diabeticcardiomyopathy,DCM)是糖尿病患者死亡的主要原因之一。DCM发病机制涉及代谢紊乱、炎症、氧化应激和纤维化等诸多方面,然而其具体的机制仍未阐明。
补体系统(Complement system,CS)由血浆蛋白、膜蛋白以及细胞内蛋白组成,参与构成免疫系统,是机体抵御入侵病原体的防线之一。补体在免疫系统中发挥着重要作用,在多种疾病损伤中都能检测到补体水平的升高,而补体的活化往往发生在炎症和氧化应激状态——有研究发现晚期糖基化终末产物(Advanced glycation endproducts,AGEs)、高血糖诱导的活性氧ROS和免疫复合物沉积都能激活补体系统,而DCM的发生发展与AGEs沉积和氧化应激相关,这提示DCM中也存在补体系统的激活。
近年来,大量研究也表明例如补体系统激活与糖尿病的发生发展密切相关,补体C3作为补体激活的关键分子。已有研究证明补体C3水平升高与胰岛素抵抗及糖尿病发病相关。此外,还有研究发现C3裂解产物C3a在糖尿病肾病中起致病作用,且尿C3a水平与肾小球病变严重程度相关,而C3a受体(C3aR)拮抗剂C3aRA能通过抑制TGFβ/Smad3通路减少肾脏纤维化,减轻足细胞线粒体功能障碍,改善糖尿病肾病。其他有关糖尿病并发症的研究中也发现高水平的C3与糖尿病视网膜病变、肾病和神经病变的风险增加相关。但是,关于补体C3及其裂解产物C3a在DCM中的作用研究甚少。
发明内容
本发明的目的在于提供补体C3裂解抑制剂以及补体C3裂解产物C3a的受体拮抗剂在制备治疗糖尿病心肌病的药物中的应用,通过本发明可以看出补体C3裂解抑制剂CP40-KK与C3a的受体拮抗剂C3aRA均能通过抑制C3a-C3aR通路改善糖尿病小鼠的心肌功能,为糖尿病心肌病提供潜在的治疗靶点。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了补体C3裂解抑制剂在制备治疗糖尿病心肌病的药物中的应用。
作为优选,所述补体C3裂解抑制剂包括CP40-KK。
本发明提供了补体C3裂解产物C3a的受体拮抗剂在制备治疗糖尿病心肌病的药物中的应用。
作为优选,所述C3a的受体拮抗剂包括C3aRA。
作为优选,所述C3aRA抑制C3aR下游ERK的磷酸化。
作为优选,所述C3aRA通过抑制NLRP3-Caspase-1-IL-1β/IL-18通路相关分子表达发挥作用。
通过采用上述技术方案,本发明具有如下有益效果:
1.本发明所述的CP40-KK能与人和小鼠的补体C3结合,并抑制C3裂解,并通过C3a-C3aR途径在糖尿病心肌病的发生发展中起作用。
2.本发明发现C3a是其主要的效应分子,CP40-KK与C3a受体拮抗剂均能通过抑制C3a-C3aR通路改善糖尿病小鼠的心功能,减轻心肌肥大程度、降低心脏纤维化程度,对糖尿病心肌病起保护作用。
3.通过本发明可以得到,高糖和C3a诱导的NLRP3活化和氧化应激互相促进,共同参与糖尿病心肌病的发展。C3a受体拮抗剂能抑制NLRP3-Caspase-1-IL-1β/IL-18通路改善糖尿病心肌病;还能降低NADPH途径及线粒体途径的氧化应激水平,改善线粒体功能,减少细胞凋亡,对糖尿病心肌病起保护作用。
附图说明
图1为糖尿病心肌病小鼠造模示意图;
图2为小鼠随机血糖、体重及心重胫骨长度比(图中的A为对照与糖尿病小鼠随机血糖对比,B为对照与糖尿病小鼠体重对比,C为对照与糖尿病小鼠心重胫骨长度比(n=6,**P<0.01));
图3为对照组与糖尿病小鼠心脏超声结果(图中的A为对照组与糖尿病组小鼠左室射血分数(LVEF)对比,B为对照组与糖尿病组小鼠左室缩短分数(LVFS)对比,C为对照组与糖尿病组小鼠E/A比值(n=6,**P<0.01),D为对照组与糖尿病组小鼠M型超声心动图,E为对照组与糖尿病组小鼠典型的E峰和A峰);
图4为小鼠左心室血流动力学检测结果(图中的A为对照组与糖尿病组小鼠dP/dt
图5为组织脱水步骤示意图;
图6为对照组与糖尿病组小鼠心脏组织切片HE和WGA染色结果(图中的A为心肌组织苏木素-伊红染色(标尺=50μm)图像,B为苏木素-伊红染色心肌细胞横截面积大小统计,C为心肌组织WGA染色(标尺=20μm)图像,D为WGA染色心肌细胞横截面积大小统计(n=6,**P<0.01));
图7为对照与糖尿病小鼠心脏组织切片天狼猩红及Masson染色结果(图中的A为心肌组织天狼猩红染色(上,红色为胶原纤维)及Masson染色(下,蓝色为胶原纤维)典型图片(标尺=100μm),B为天狼猩红及Masson染色阳性区域面积统计(n=6,**P<0.01));
图8为对照与糖尿病小鼠血浆C3浓度;
图9为对照与糖尿病小鼠心脏组织C3蛋白表达水平;
图10为对照与糖尿病小鼠心脏组织C3蛋白免疫印迹条带灰度值定量分析(Mean±SD,n=6,**P<0.01));
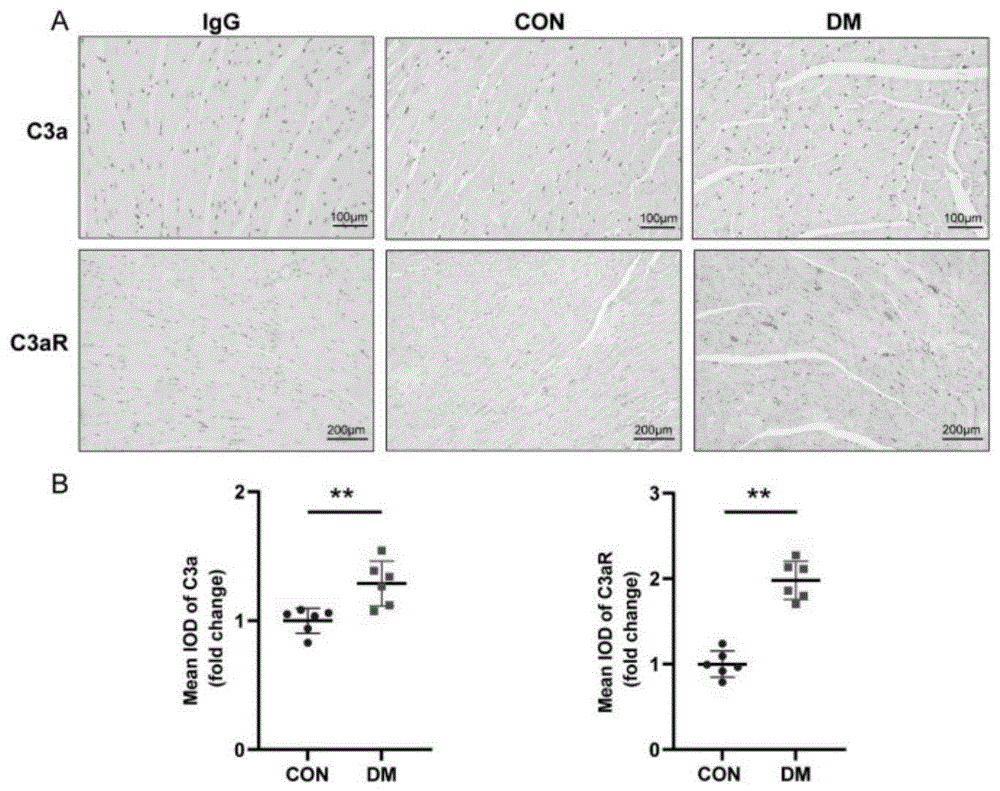
图11为对照与糖尿病小鼠心脏组织C3及C3a表达水平(图中的A为心脏组织C3a(标尺=100μm)及C3aR(标尺=200μm)免疫组织化学染色,B为C3a和C3aR免疫组织化学染色阳性区域比较(Mean±SD,n=6,**P<0.01));
图12为高糖(HG,30mmol/L)干预对H9c2细胞C3、C3a及C3aR表达量影响(图中的A为H9c2细胞总蛋白Westernblot条带,B为C3 Westernblot条带灰度定量分析,C为C3aWesternblot条带灰度定量分析,D为C3aRWesternblot条带灰度定量分析(Mean±SD,n=6,**P<0.01,*P<0.05));
图13为高糖干预对H9c2细胞C3aR核浆定位的影响(图中的A为高糖干预后H9c2细胞核浆蛋白Westernblot结果(NG:正常糖,HG:高糖),B为高糖干预后H9c2细胞C3aR免疫荧光染色结果(标尺=50μm));
图14为糖尿病心肌病小鼠造模及干预示意图;
图15为小鼠血糖、体重及心重胫骨长度比(图中的A为小鼠随机血糖浓度,B为小鼠体重,C为小鼠心重胫骨长度比(n=6,*P<0.05,ns,non-significant));
图16为小鼠左心室血流动力学结果(图中的A为dP/dt
图17为小鼠超声心动图结果(图中的A为M型超声心动图,B为小鼠左室射血分数比较,C为小鼠左室短轴缩短率比较,D为小鼠二尖瓣血流E峰和A峰图像,E为小鼠二尖瓣血流E/A统计图(n=6,**P<0.01,ns,non-significant));
图18为心脏损伤指标ANP、BNP、β-MHC的mRNA表达水平(n=6,**P<0.01,*P<0.05,ns,non-significant);
图19为小鼠心脏组织HE及WGA染色(图中的A为心脏组织HE染色(标尺50μm)及心肌细胞横截面积统计结果,B为心脏组织WGA染色(标尺20μm)及心肌细胞横截面积统计结果(n=6,**P<0.01,ns,non-significant));
图20小鼠心脏组织切片天狼猩红及Masson染色(图中的A为心肌组织天狼猩红染色(红色为胶原纤维,标尺=100μm)及染色阳性区域面积统计,B为Masson染色(蓝色为胶原纤维,标尺=100μm)及染色阳性区域面积统计(n=6,**P<0.01,ns,non-significant));
图21为人和小鼠C3与CP40-KK结合的SPR感测图(图中的A为人补体C3与CP40-KK结合SPR感测图,B为小鼠补体C3与CP40-KK结合SPR感测图));
图22为小鼠血浆过敏毒素C3a浓度(n=6,**P<0.01,*P<0.05,ns,non-significant);
图23为小鼠心肌组织C3片段蛋白表达水平(图中的A为小鼠心脏组织C3裂解片段Western blot条带,B为C3裂解片段Westernblot灰度定量分析(n=6,**P<0.01));
图24为小鼠心脏组织ERK磷酸化水平(图中的A为小鼠心脏组织磷酸化ERK1/2及总ERK1/2Westernblot条带,B为磷酸化ERK1/2及总ERK1/2条带灰度值之比(n=6,**P<0.01,ns,non-significant));
图25小鼠心脏组织炎症指标水平(图中的A为小鼠心脏组织Western blot条带,B为NLRP3的Westernblot灰度值定量分析,C为Caspase-1的Westernblot灰度值定量分析,D为IL-1β的Westernblot灰度值定量分析,E为IL-18的Westernblot灰度值定量分析(n=6,**P<0.01,ns,non-significant));
图26为C3a及C3aRA对心肌细胞C3aR的作用验证(图中的A为心肌细胞AC16总蛋白Western blot结果,B为AC16细胞磷酸化ERK、总ERK Western blot条带灰度定量分析(n=3,**P<0.01,*P<0.05,ns,non-significant));
图27为C3aRA抗炎效应验证(图中的A为心肌细胞AC16总蛋白Westernblot结果,B为NLRP3的Western blot条带灰度定量分析,C为Caspase-1的Westernblot灰度值定量分析,D为IL-1β的Western blot灰度值定量分析,E为IL-18的Western blot灰度值定量分析(n=3,**P<0.01,*P<0.05,ns,non-significant));
图28为鼠心脏组织NADPH氧化酶催化亚基表达水平(图中的A为小鼠心脏组织NADPH氧化酶催化亚基Western blot结果,B为小鼠心脏组织切片p47-phox免疫组织化学染色为,C为gp91-phox的Western blot条带灰度扫描定量分析,D为p67-phox的Western blot条带灰度扫描定量分析,E为p47-phox的Westernblot条带灰度扫描定量分析(n=6,**P<0.01,*P<0.05,ns,non-significant));
图29为心肌细胞AC16 NADPH氧化酶亚基及SOD2表达水平(图中的A为心肌细胞NADPH氧化酶亚基及SOD2 Western blot结果,B为gp91-phox的Western blot条带灰度扫描定量分析,C为p67-phox的Westernblot条带灰度扫描定量分析,D为p47-phox的Westernblot条带灰度扫描定量分析,E为SOD2的Western blot条带灰度扫描定量分析(n=3,**P<0.01,*P<0.05,ns,non-significant));
图30为心肌细胞MitoSOX染色(标尺=200μm,红色为MitoSOX,反映线粒体内ROS水平,蓝色为细胞核);
图31为心肌细胞JC-1染色(标尺=50μm,绿色为JC-1单体,表示线粒体膜电位降低,红色为JC-1多聚体,表示线粒体膜电位正常);
图32为心肌细胞DHE染色(标尺=100μm,红色为DHE,反映细胞内ROS水平,蓝色为Hochest,代表细胞核);
图33为小鼠心脏组织TUNEL染色,白色箭头所指为凋亡细胞(标尺=100μm,绿色荧光为TUNEL染色,表示凋亡细胞核,蓝色荧光表示细胞核);
图34为C3aRA减少AC16心肌细胞凋亡(图中的A为心肌细胞AC16总蛋白凋亡指标Westernblot条带,B为Bax/Bcl-2灰度值定量分析(n=3,**P<0.01,*P<0.05,ns,non-significant),C为剪切的Caspase-3条带灰度值定量分析(n=3,**P<0.01,ns,non-significant),D为AC16心肌细胞TUNEL染色(标尺=200μm));
图35为MCC950降低ROS表达水平(图中的A为AC16心肌细胞DHE(染色标尺=50μm),B为DHE染色阳性细胞数统计图(n=3,**P<0.01,ns,non-significant));
图36为NAC影响NLRP3的表达(图中的A为心肌细胞AC16 DHE染色(标尺=50μm),B为NLRP3Western blot条带,C为AC16 DHE染色阳性细胞数统计图(n=3,**P<0.01,ns,non-significant),D为NLRP3蛋白Western条带灰度扫描统计图(n=3,**P<0.01,*P<0.05,ns,non-significant))。
具体实施方式
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1糖尿病心肌病小鼠模型建立
(一)糖尿病心肌病小鼠造模
1.实验动物
实验所用小鼠为7周龄雄性C57BL/6J小鼠,购自北京维通利华公司,饲养于华中科技大学同济医学院附属同济医院实验动物中心SPF级动物房,饲养环境温度设置为20-26℃,每日光照12小时,黑暗12小时,以此循环,并提供充足的水和食物。动物实验已通过华中科技大学同济医学院实验动物中心伦理委员会批准。
2.实验方法
购买7周龄野生型雄性C57BL/6J小鼠,到达动物房并适应饲养环境1周后给予60mg/kg剂量的链脲佐菌素(STZ,美国Sigma-Aldrich)连续5天腹腔注射,对照组注射等体积溶剂(0.05mol/L柠檬酸钠缓冲液,pH 4.5),造模流程见图1。最后一次注射完成2周后取尾静脉血测随机血糖,当小鼠随机血糖高于16.7mmol/L时纳入糖尿病模型组。继续饲养小鼠至26周龄,期间对于血糖>27.7mmol/L的鼠进行每周2次,每次0.5U长效胰岛素皮下注射以维持其存活,饲养至34周龄。
3.结果
在34周龄时称量小鼠体重,检测随机血糖。比较发现,DCM组小鼠随机血糖显著升高(图2中的A),体重明显下降(图2中的B),糖尿病造模成功。处死后称量心脏重量,测量胫骨长度,并计算心重胫骨长度比(HW/TL),结果显示糖尿病组小鼠心重胫骨长度比显著增加,提示可能存在心肌肥厚(图2中的C)。
(二)糖尿病心肌病小鼠心功能检测
1.小鼠超声心动图检查
在处死小鼠前对其进行超声心动图检查。提前用棉签蘸取脱毛膏去除小鼠心前区毛发,将小鼠用胶带呈仰卧位固定于测试台上,在四肢涂少量超声耦合剂,保证其与测试台的电极接触良好。固定好后,借助呼吸机辅助用异氟烷进行持续气体麻醉,调节麻醉剂剂量,使小鼠心率稳定500-600bpm,在心前区涂抹适量超声耦合剂,记录长轴和短轴的二维超声心动图及M型超声心动图。移动探头位置至心尖部,同时调整麻醉剂量,控制小鼠心率在500bpm,记录二尖瓣血流E峰、A峰。结果显示见图3,反映了小鼠心功能的各项指标如左室射血分数(LVEF),左室缩短分数(LVFS),E/A等在糖尿病小鼠中均显著下降,提示糖尿病组小鼠心脏收缩及舒张功能受损。
2.左室血流动力学检查
小鼠用1%戊巴比妥钠腹腔注射麻醉(4μL/g),仰卧位固定,在小鼠颈部正中做纵行切口,切口范围尽可能大,以方便后续操作。用镊子钝性分离肌肉,暴露并分离右颈总动脉。接着用4-0的手术缝线结扎远心端,向外拉并固定线头,待动脉充盈后,用动脉夹夹闭动脉的近心端。在体式显微镜直视下用显微剪在靠近结扎处剪一小口,并用胰岛素针辅助将Millar导管送入动脉,松开动脉夹,将导管插入左心室,采集血流动力学信号。
测量完成后计算左室压力最大下降速率-dP/dt
3.组织石蜡包埋切片
(1)取材:处死小鼠后取新鲜心脏组织,在心尖部往上2-3mm的地方做横切,切取宽度2mm的心室环,放入4%多聚甲醛固定24小时以上。
(2)脱水:从固定液中取出组织,置于脱水盒内并放入脱水机,按图5步骤依次梯度酒精脱水。
(3)包埋:使用包埋机进行包埋,在包埋框内加入适量融化的蜡,在蜡凝固之前从脱水盒内取出组织,心脏横切面朝上放入包埋框,等待其凝固。
(4)切片:用石蜡切片机做组织切片,厚度4μm,在温水中展平后用载玻片捞起,放入60℃烘箱内烤片。
4.WGA染色和HE染色
对得到的切片进行苏木素-伊红(HE)和小麦凝集素(WGA)染色,观察心肌细胞形态变化。通过两组形态对比可以看出,糖尿病组小鼠心肌细胞横截面积显著大于对照组(见图6)。
5.天狼猩红染色和Masson染色
对小鼠心脏组织进行天狼猩红和Masso染色来检测心脏纤维化程度。通过两种染色实验得到,所述糖尿病组小鼠心脏血管周围纤维化程度明显高于对照组(见图7)。
实施例2糖尿病心肌病小鼠体内C3、C3a及C3aR表达情况
(一)糖尿病小鼠血浆及心脏组织C3表达水平
1.血清补体C3浓度测定
血清补体C3浓度测定使用南京建成生物工程研究所的补体C3检测试剂盒(货号E032-2),测定原理为免疫比浊法。
测定过程:配制反应体系,见表1;轻轻振荡孔板,37℃孵育5分钟,340nm处读取吸光度A1;向每个孔内加入50μL R2;轻轻振荡孔板,37℃孵育5分钟,340nm处读取吸光度A2;计算ΔA=A2-A1,以标准品不同浓度与其对应的ΔA作标准曲线,代入测定孔ΔA求浓度。结果见图8。
表1补体C3浓度测定反应体系
2.心脏组织总蛋白提取
(1)从-80℃超低温冰箱取出冻存的组织,并迅速转移至盛有适量液氮的研钵中,将组织研磨成粉末状,取1/2放入在液氮中预冷过的EP管内用于心脏组织总蛋白提取,1/3用于心脏组织核蛋白及浆蛋白提取,剩余1/6用于组织RNA提取。(2)向装有组织的管子内加入500μL RIPA裂解液(裂解液内已提前加入蛋白酶及磷酸酶抑制剂),置于冰上裂解15分钟以上。(3)裂解完成,在提前遇冷至4℃的离心机内以12000g的转速离心15分钟,得到的上清液即为总蛋白。(4)测定总蛋白浓度,因组织蛋白浓度较高,将蛋白稀释20倍(用PBS进行稀释)后再进行浓度测定。测浓度使用武汉博士德生物的BCA蛋白浓度测定试剂盒,每次均需设置标准曲线,采用倍比稀释法配制标准曲线,见表2。(5)配制BCA工作液,试剂A:试剂B=50:1,现配现用,充分混匀。(6)每孔加95μL BCA工作液及5μL待测样品,振荡30秒,混匀,置于37℃温箱孵育30分钟。每个样品设置两个复孔。(7)取出板子,冷却至室温,用酶标仪检测吸光度,波长设置562nm。(8)根据标准品浓度及对应的吸光度绘制标准曲线,计算样品蛋白浓度,对于浓度较高的样品,加入适量RIP裂解液,调整所有样品浓度为1μg/μL。取800μL蛋白,加入200μL 5×还原型蛋白上样缓冲液,沸水煮10分钟,煮完后分装于多个螺旋尖底管内,冻于-80℃,避免反复冻融。
表2蛋白浓度测定标准曲线配制表
此外,对提取得到的小鼠心脏组织总蛋白进行蛋白免疫印迹(Western Blot)检测,结果发现,小鼠心脏组织内也有补体C3的表达,且糖尿病小鼠心脏组织补体C3水平显著高于对照组(见图9和图10)。
(二)C3a及C3aR在糖尿病小鼠心脏组织表达水平
进一步的对心脏组织石蜡切片进行了C3a及C3a受体(C3aR)的免疫组织化学染色(Immunohistochemistry,IHC)。心脏组织切片染色结果(见图11中的A)表示,糖尿病组小鼠心脏组织C3a和C3aR的蛋白表达水平明显升高,且染色阳性区域集中在细胞核周围。使用Image J图片分析软件对染色阳性区域进行定量分析,发现糖尿病组染色阳性区域面积显著高于对照组(见图11中的B),且两组之间有统计学差异。即说明了,在糖尿病状态下,C3的产生和裂解均增加,导致心脏组织局部C3a水平升高。增加的C3a进一步诱导心脏组织局部C3aR产生增多。C3a通过结合心脏局部的C3aR,调控下游分子,在DCM中发挥作用。
实施例3高糖刺激影响H9c2细胞C3、C3a及C3aR的表达
(一)高糖干预促进心肌细胞C3、C3a及C3aR表达
在大鼠心肌细胞系H9c2(购自American Type Culture Collection(ATCC))上也进行了验证。使用含10%FBS的低糖DMEM培养基(葡萄糖浓度5mmol/L)培养大鼠心肌细胞系H9c2,FBS在使用前进行补体灭活(56℃,30分钟)。在细胞密度约50%的时候换无血清培养基饥饿处理12小时,处理完后换回完全培养基,加高糖(终浓度为30mmol/L)干预48小时后提取细胞总蛋白,进行Westernblot检测心肌细胞内C3、C3a和C3aR表达量。
根据图12可以看出,心肌细胞H9c2能表达C3、C3a及C3aR,且高糖干预能刺激其表达增加。
(二)高糖干预促进心肌细胞C3aR由胞核向胞浆转移
心脏组织免疫组织化学染色显示C3aR染色集中在心肌细胞核附近,且H9c2细胞蛋白Westernblot结果显示心肌细胞内有C3aR的表达。为了探究高糖刺激对心肌细胞C3aR的影响,给予H9c2细胞高糖刺激48小时,干预完成后提取核浆蛋白,并进行细胞免疫荧光染色,明确C3aR的表达量及位置。
根据图13可以看出,高糖干预之后,H9c2细胞浆内C3aR表达量明显增加,而细胞核内表达量下降,即说明高糖刺激会使细胞内的C3aR由细胞核向胞浆转移。
实施例4C3通过C3a-C3aR途径参与糖尿病心肌病的发生
Compstatin家族衍生物是针对补体C3的靶向抑制剂,它们通过空间阻碍C3与其转化酶相结合,进而阻止C裂解。CP40便是其改良产物之一,由D-酪氨酸取代Compstatin的N-末端乙酰基而形成。本研究所选用的多肽CP40-KK是在CP40的基础上添加两个亲水的赖氨酸残基形成的,相较于CP40,它的溶解性更高,与C3的结合力更强。C3aR拮抗剂C3aRA(SB290157)是选择性C3a受体拮抗剂,能阻断C3a受体下游ERK等信号分子的激活。
(一)C3aR拮抗剂及CP40-KK干预能改善糖尿病小鼠的心功能
对糖尿病心肌病小鼠模型分别给予CP40-KK和C3aRA干预,通过糖尿病组、抑制C裂解与只抑制C3a-C3aR通路三组小鼠之间的对比,明确C3在糖尿病心肌病中的作用及其主要作用途径。
1.在小鼠26周龄时将糖尿病小鼠随机分为3组:DCM组,DCM+CP40-KK组,DCM+C3aRA组。DCM组每天给予等体积的生理盐水注射,后两组每天分别给予CP40-KK(2mg/kg/d,溶于生理盐水)及C3aRA(30mg/kg/d,溶于10%乙醇/蒸馏水中)腹腔注射给药,连续给药8周。在小鼠34周龄时进行超声心动图及左室血流动力学检测,处死,留取血液及组织(见图14)。
CP40-KK多肽序列:
DTyr-Ile-[Cys-Val-Trp(Me)-Gln-Asn-Trp-Sar-Ala-His-Arg-Cys]-mIle-Lys-Lys-NH
干预完成后,同样对小鼠进行了血糖、体重及胫骨长度的测量,结果显示糖尿病小鼠血糖明显升高,体重明显下降,而C3aRA及CP40-KK干预对糖尿病小鼠的血糖及体重无明显改善。但是,糖尿病组小鼠心重胫骨长度比显著高于对照,而C3aRA及CP40-KK干预均能降低小鼠心重胫骨长度比,改善心脏肥大程度,且小鼠的心重胫骨长度比在两个药物干预组之间无明显差异(见图15)。
对小鼠还进行了左室血流动力学(图16)及超声心动图检查(图17)。糖尿病组小鼠的dP/dt
2.酶联免疫吸附实验。采用小鼠血清C3aELISA试剂盒,购自武汉华美生物有限公司。
3.心脏组织RNA提取
(1)处死小鼠后,取1/6研磨成粉末状的组织,放入用液氮中预冷过的EP管内。(2)向EP管中加入500μL Trizol,振荡混匀后置于室温,静置5分钟。(3)在通风橱中打开EP管,向其中加入100μL氯仿,使用涡旋振荡器振荡15秒,再置于室温静置5分钟。(4)静置时预冷低温离心机至4℃,设置离心力为10000g,4℃离心15分钟。(5)离心完毕后取出E管置于冰盒内,此时管内液体分层,上层为无色水相,RNA主要存在于该层,中间层呈乳白色,下层为粉红色有机相。转移最上层无色水相至新的EP管,注意避免吸到中间层。(6)向水相中加入与其等体积的异丙醇,颠倒混匀,室温孵育10分钟。(7)10000g,4℃离心15分钟,弃去上清。(8)向管内加入500μL 75%乙醇(用DEPC水配制),剧烈振荡。(9)7500g,4℃离心5分钟,弃上清,管底可见极少量白色胶状沉淀。(10)重复步骤9的离心,用10μL微量移液器吸去残余液体,将EP管倒扣于干净的纸上,室温晾干。(11)晾干后观察沉淀量,加入20-40μL无酶水溶解沉淀,并用涡旋振荡器振荡混匀。混匀后使用超微量核酸分析仪测量每个样品的RNA浓度及OD260/280的值。
4.逆转录。使用武汉爱博泰克生物科技有限公司的逆转录试剂盒,所得产物cDNA存放于-80℃超低温冰箱。
5.实时荧光定量PCR,并检测心脏损伤指标ANP、BNP、β-MHC的mRNA表达水平。使用武汉爱博泰克生物科技有限公司SYBR Green Fast qPCR Mix试剂盒。引物序列见表3。
表3小鼠引物序列表
从图18中可以看出,糖尿病组小鼠心肌损伤指标显著升高,而C3aRA及CP40-KK干预组显著降低,提示C3aRA或CP40-KK能改善糖尿病导致的心脏损伤。
(二)C3aR拮抗剂及CP40-KK干预能改善糖尿病小鼠心肌细胞肥大
在小鼠34周龄时处死小鼠,留取心脏组织。切取心尖往上2-3mm的心室部分进行石蜡包埋切片。采用苏木素-伊红(HE)染色及小麦凝集素(WGA)对石蜡切片进行染色,明确心肌细胞的大小及形态变化,结果见图19。
对心肌细胞横截面积进行统计分析,发现糖尿病小鼠心肌细胞明显肥大,排列紊乱,而C3aRA及CP40-KK干预组心肌细胞肥大情况明显改善,与糖尿病组相比有统计学差异,但是,对C3aRA与CP40-KK两个药物干预组小鼠的心肌细胞横截面积大小进行比较,发现两者之间无统计学差异,提示相较于C3b途径而言,C3a-C3aR途径在糖尿病心肌病中发挥着重要作用。
(三)C3aR拮抗剂及CP40-KK干预能改善糖尿病小鼠心脏组织纤维化程度
进一步的对小鼠心脏组织进行天狼猩红及Masson染色来观察心脏组织纤维化程度,结果表示(见图20)糖尿病组小鼠心脏血管周围有较多胶原纤维沉积,纤维化程度显著高于对照组,而C3aRA或CP40-KK干预能改善血管周围纤维化程度,在糖尿病心肌病中发挥保护作用。
实施例5C3抑制剂CP40-KK与C3aR拮抗剂作用验证
(一)CP40-KK与补体C3结合并抑制C3裂解
在给药之前,采用表面等离子共振法(surface plasmon resonance technology,SPR)对合成的CP40-KK与小鼠补体C3结合的动力学亲和力进行了检测。结果表明(见图21),人的补体C3能以1.53μM的亲和常数(KD)与CP40-KK结合,而小鼠补体C3与CP40-KK结合的亲和常数为26.7μM。
表4CP40-KK与人及小鼠补体C3动力学亲和力分析
干预完成后,留取小鼠血浆,检测血浆C3a水平,并提取小鼠心脏组织蛋白,检测心脏组织内C3裂解片段的蛋白表达水平。结果显示,糖尿病小鼠血浆C3a的水平明显高于对照组,而CP40-KK干预组小鼠血浆C3a浓度显著下降,表明CP40-KK能抑制小鼠补体C3裂解(见图22);Western blot结果显示CP40-KK干预组C3裂解片段的蛋白水平显著低于DCM组和C3aRA组,再次证明CP40-KK能抑制补体C3裂解(见图23)。
(二)C3aR拮抗剂能抑制小鼠心脏组织的C3aR激活
有研究表明,C3aR被激活之后可以直接促进ERK1/2的激活,因此通过检测其下游磷酸化ERK1/2的水平来反映C3aR的激活水平,结果见图24,糖尿病小鼠心脏ERK1/2磷酸化增加,而C3aRA干预能显著降低糖尿病小鼠心脏组织ERK1/2的磷酸化水平,证明C3aRA能有效发挥拮抗剂的作用,抑制C3aR激活。
实施例6C3aR拮抗剂改善糖尿病心肌病的机制探究
基于上述的研究结果,糖尿病小鼠血浆C3和C3a水平显著增加,且心肌细胞C3、C3a和C3aR表达上调,由此推测,来源于循环或者心脏局部产生的过敏毒素C3a可能通过结合心肌细胞的C3aR发挥作用。C3aR被激活后,通过下游ERK等信号分子增强炎症反应,提高氧化应激水平,进而促进心脏损伤。也进一步的进行体内和体外研究,确定C3a对心肌细胞具体的调控机制。
(一)C3aR拮抗剂通过抑制炎症小体NLRP3-Caspase-1-IL-1β/IL-18通路改善糖尿病心肌病
1.C3aR拮抗剂能减轻糖尿病心肌病小鼠心脏组织炎症水平
为了明确小鼠心脏组织的炎症小体NLRP3下游通路激活水平,我们留取部分心脏组织,提取组织蛋白,用Westernblot检测了NLRP3-Caspase-1-IL-1β/IL-18通路相关蛋白的表达水平,结果见图25,糖尿病小鼠心脏组织的炎症小体NLRP3、活化的Caspase-1以及活化的IL-1β、IL-18水平均显著高于对照组,而在给予C3aRA干预之后,这些炎症指标蛋白表达水平显著下调,提示DCM小鼠心脏组织内存在NLRP3及其下游信号通路的激活,而C3aRA干预能通过阻断NLRP3-Caspase-1-IL-1β/IL-18通路,降低心脏组织炎症水平,改善糖尿病心肌病。
2.C3aR拮抗剂能阻断心肌细胞C3a-C3aR通路,降低C3a诱导的炎症水平
(1)验证C3aRA的抗炎效应,在人心肌细胞系AC16(购自上海淳麦生物科技有限公司)上进行了相关实验。细胞实验分对照、C3aRA、C3a、C3a+C3aRA四组。干预前先换无血清培养基饥饿处理12小时,处理完之后更换为完全培养基,加C3aRA(1mmol/L)预处理半小时,再加C3a(0.5mmol/L)刺激,48时后提取细胞总蛋白。验证C3a对心肌细胞C3aR的激活作用以及C3aRA的拮抗作用,结果表明(图26),外源性C3a干预能激活心肌细胞AC16的C3aR,直接导致下游ERK磷酸化增加,而C3aRA也能作用于AC16,发挥拮抗剂的作用,抑制C3aR的激活。
(2)进一步研究C3a对心肌细胞AC16的炎症诱导效应及C3aRA的保护作用。心肌细胞AC16用高糖培养基(25mmol/L葡萄糖)培养,接种至六孔板,在干预时分对照、C3aRA、C3a、C3a+C3aRA四组。干预前先换无血清培养基饥饿处理12小时,处理完之后更换为完全培养基,加C3aRA(1mmol/L)预处理30分钟,再加C3a(0.5mmol/L)刺激,48小时后提取细胞总蛋白,用Westernblot检测NLRP3、Caspase-1、IL-1β、IL-18的表达情况。细胞实验结果与动物实验一致,高糖培养的细胞NLRP3炎症小体蛋白表达水平在各组之间无差别,而C3a干预使Caspase-1、IL-1β和IL-18的活化显著增加,提示在高糖环境下,C3a能发挥参与NLRP3活化的第二信号,刺激Caspase-1和IL-1β的剪切活化,而C3aRA能拮抗C3a的促炎效应,降低下游效应分子的活化水平,表明C3a-C3aR轴在炎症小体激活过程中发挥着重要作用(见图27)。
(二)C3aR拮抗剂通过降低心肌细胞氧化应激水平改善糖尿病心肌病
1.C3aR拮抗剂能降低DCM小鼠心脏组织NADPH氧化酶亚基表达水平
留取小鼠心脏组织,提取组织蛋白,用Westernblot及免疫组织化学染色检测了NADPH氧化酶亚基的表达水平,结果(图28)得到,糖尿病组小鼠心脏组织的gp91-phox、p67-phox、p47-phox的表达水平显著高于对照,表明NADPH氧化酶在糖尿病小鼠心脏组织内激活,而C3aRA干预能在一定程度上降低其表达水平,从而发挥保护作用。
2.C3aR拮抗剂能抑制心肌细胞NADPH氧化酶活性
为了验证动物实验的结论,在人心肌细胞系AC16上进行了相同的实验。细胞干预同样分为对照、C3aRA、C3a、C3a+C3aRA四组,加C3a(0.5mmol/L)刺激48小时后提取细胞总蛋白,用Westernblot检测NADPH氧化酶亚基表达情况(图29)。C3a干预使心肌细胞NADPH氧化酶亚基gp91-phox、p67-phox、p47-phox表达增加,过氧化物歧化酶SOD2表达下降,表明C3a干预使心肌细胞NADPH氧化酶活性增加,而抗氧化能力下降,C3aRA干预能降低其氧化应激水平。
3.C3aR拮抗剂能减少C3a诱导的线粒体损伤,改善线粒体氧化应激
探究C3a及C3aRA对线粒体氧化应激的影响。对心肌细胞AC16进行了相同的干预,并对细胞进行MitoSOX染色和JC-1染色检测线粒体ROS累积水平及线粒体膜电位的变化。由于MitoSOX能靶向定位到线粒体,结合线粒体中的阴离子超氧化物,氧化后与线粒体DNA结合,发红色荧光,故使用MitoSOX染色来反映线粒体氧化应激水平,结果见图30,C3a干预后细胞内线粒体ROS水平显著增加,而C3aRA干预能抑制其升高。
在线粒体膜电位正常时中,JC-1以多聚体的形式存在,发红色荧光,而在线粒体膜电位下降或消失时JC-1以单体的形式存在于胞浆,发绿色荧光。因此,JC-1染色可以反映线粒体是否受损及其膜电位的变化情况。JC-1染色结果显示(图31),阳性对照CCCP作用于细胞30分钟后即可使线粒体膜电位消失,C3a干预之后线粒体膜电位下降,染色以绿色荧光为主,而C3aRA对线粒体损伤有一定的改善作用。
综上可以得到,C3a能导致线粒体损伤,使线粒体膜电位下降,ROS产生增加,而C3aRA干预能在一定程度上减轻线粒体损伤,降低线粒体氧化应激水平,对心肌细胞起保护作用。
4.C3aR拮抗剂通过降低胞内ROS水平,减少细胞凋亡,在糖尿病心肌病中发挥保护作用
(1)探究细胞内总的ROS水平。对C3a及C3aRA干预的心肌细胞进行超氧化物阴离子荧光探针(Dihydroethidium,DHE)染色,结果表明(图32),C3a干预组在一个视野内的DHE染色阳性细胞数明显多于对照组,而C3aRA干预能在一定程度上阻断C3a对氧化应激的诱导作用。
(2)探究C3aRA对凋亡的影响。对小鼠心脏组织进行了TUNEL染色,结果(图33)表示,糖尿病小鼠心肌细胞凋亡增加,而C3aRA有一定的保护作用。
(3)进一步的在体外培养的心肌细胞AC16上进行了验证。高糖培养的AC16心肌细胞分4组,干预同前,干预完成后提取细胞总蛋白,用Westernblot检测凋亡指标,并进行TUNEL染色,结果(图34)发现,C3a干预组Bax/Bcl-2和剪切的Caspase-3水平显著升高,而C3aRA干预有一定的改善作用。TUNEL染色结果与动物组织切片一致,C3a干预组TUNE染色阳性细胞数显著高于其余三组,提示在高糖环境下,C3a能增加心肌细胞ROS累积,促进细胞凋亡,而该致病作用能被C3aRA所阻断。
(三)高糖及C3a诱导的NLRP3激活与ROS生成互相影响,共同参与糖尿病心肌病的发病
1.NLRP3抑制剂MCC950能降低胞内ROS水平
选择NLRP3抑制剂MCC950干预AC16心肌细胞,并对细胞进行DHE染色,观察干预对胞内ROS水平的影响。结果表明(图35),同时给予高糖和C3a干预的细胞DHE染色阳性细胞数显著高于对照,而使用MCC950抑制NLRP3后,胞内ROS水平显著下降,即NLRP3通路的抑制能减少胞内ROS的积累。
2.ROS抑制剂N-乙酰半胱氨酸能抑制NLRP3表达上调
选择ROS抑制剂N-乙酰半胱氨酸(NAC)干预心肌细胞AC16,并对胞内ROS及NLRP3水平进行了检测。结果如图36,NAC干预组DHE染色阳性细胞数显著低于C3a+HG干预组,证明NAC确实能发挥抑制ROS的作用。此外,Westernblot结果显示NAC干预组NLRP3蛋白表达水平与对照组相当,并显著低于C3a+HG干预组,提示抑制胞内ROS水平可以阻止C3a+HG诱导的NLLRP3表达升高。
由以上实施例可知,C3a是其主要的效应分子,C3aR可以作为糖尿病慢性并发症的一个治疗靶点。C3a通过作用于心肌细胞的C3aR,诱导NLRP3-Caspase-1-IL-1β/IL-18通路激活以及心肌细胞胞内NADPH氧化酶途径和线粒体途径的ROS累积,导致氧化应激水平升高,细胞凋亡增加。C3a诱导的炎症与氧化应激两者之间互为因果,相辅相成,共同参与糖尿病心肌病的发生发展。C3aR拮抗剂通过抑制NLRP3通路的激活、降低氧化应激水平,减少心肌细胞凋亡,进而在糖尿病心肌病中发挥保护作用。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。