一种羊奶奶叶药材的质量检测方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及中药分析技术领域,特别是一种羊奶奶叶药材的质量检测方法。
背景技术
羊奶奶叶(胡颓子叶):根据《贵州省中药材、民族药材质量标准》命名。
【基原】本品为胡颓子科植物胡颓子Elaeagnus pungens Thunb.或宜昌胡颓子Elaeagnus henryi Warb.的干燥叶。夏季采收,晒干。
关于羊奶奶叶的基原,现有标准中,《中国药典》(1977)、《上海市中药材标准》、《广东省中药材标准》、《湖北省中药材标准》、《湖南省中药材标准》、《贵州省中药材、民族药材质量标准》、《云南省中药材标准(彝药)》、《福建省中药材标准》等收载的有关胡颓子属植物叶药用的有“羊奶奶叶/胡颓子叶”、“白绿叶”、“沙枣叶”3个药材品种,涉及6种基原植物(胡颓子E.pungens、宜昌胡颓子E.henryi、蔓胡颓子E.glabra、福建胡颓子E.oldhami、绿叶胡颓子E.viridis、沙枣E.angustifolia),以胡颓子E.pungens使用最为广泛。据实地调查和文献记载,尚有牛奶子E.umbellataThunb.等多种在贵州(苗族)、湖北(土家族)同作“羊奶奶叶”药用。
经对共7种胡颓子属植物的成分分析(成分组成与含量)比较,结果表明:福建胡颓子分布区域较小且成分与胡颓子叶相差较大;胡颓子、宜昌胡颓子2种的成分组成较为相似;其他5种胡颓子属植物的成分组成与胡颓子、宜昌胡颓子的成分组成均有较大差异。经查阅文献及实地调查,胡颓子、宜昌胡颓子分布较为广泛,资源丰富,且有标准收载;福建胡颓子仅分布于福建、广东,主要在福建民间使用;绿叶胡颓子(分布于陕西、湖北)为彝族习用药材;沙枣(分布于西部及华北等)为维吾尔族习用药材;其他种类除蔓胡颓子外尚无标准收载;蔓胡颓子虽分布也较为广泛,也有标准收载,但其成分组成与胡颓子、宜昌胡颓子有较大差异;巴东胡颓子(分布较为广泛)、牛奶子(分布广泛)、长裂胡颓子(仅分布于贵州)尚无标准收载。参考已有标准收载情况,综合考虑,确定“羊奶奶叶/胡颓子叶”的基原为胡颓子E.pungens Thunb.和宜昌胡颓子E.henryi Warb.。
根据《贵州省中药材、民族药材质量标准》确定采收期为“夏季采收”。
【性状】《中国药典》(1977)及湖南、上海、贵州地方标准中有性状规定。据对样品观察,胡颓子和宜昌胡颓子的药材(叶)有一定差异,故对原标准进行了修订,分别规定了2种的性状。
羊奶奶叶主要含有黄酮类、有机酸类及皂苷类:其中黄酮类成分又主要为山柰酚、槲皮素、异鼠李素三种黄酮苷元的衍生物。主要包括:山柰酚、槲皮素、异鼠李素、银锻苷、芦丁、白麻苷等。
《贵州省中药材、民族药材质量标准》、《广东省中药材质量标准》、《湖南省中药材质量标准》规定以对照药材作为薄层鉴别的对照物质,而《湖北省中药材质量标准》规定以齐墩果酸作为薄层鉴别的对照物质。原湖北省标规定的以齐墩果酸为对照的薄层鉴别,鉴于该成分为较多药材的共有成分,专属性较差。
胡颓子属(Elaeagnus)多种植物的叶在汉族、苗族、土家族、彝族、维吾尔族等均药用,也已有处方中使用该类药材的成药上市。但关于其药材质量标准,上述有关标准中仅有【鉴别】(性状、粉末、对照药材薄层)、【检查】等项的规定,标准尚不完善,也制约了其成方制剂的质量控制。因此,亟需研发一种羊奶奶叶药材的质量检测方法来有效控制羊奶奶叶药材的整体质量、保证羊奶奶叶药材临床用药的安全有效性。
发明内容
本发明的目的在于,提供一种羊奶奶叶药材的质量检测方法。本发明具有能够有效控制羊奶奶叶药材的整体质量、保证羊奶奶叶药材临床用药的安全有效性的特点。
本发明的技术方案:一种羊奶奶叶药材的质量检测方法,包括鉴别和/或含量的测定项目;其中对羊奶奶叶药材的鉴别是采用薄层色谱鉴别方法;含量测定是用高效液相色谱法测定羊奶奶叶药材银锻苷的含量。
前述的羊奶奶叶药材的质量检测方法中,薄层色谱鉴别方法包括以下步骤:
(1)称取本品粉末2g,加10倍量水煎煮30分钟,滤过,滤液置水浴上蒸干,残渣加无水乙醇0.5ml使溶解,作为供试品溶液;另取羊奶奶叶对照药材2g,同法制成对照药材溶液;
(2)照薄层色谱法试验
吸取上述两种溶液各5μl,分别于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以醋酸乙酯:水:甲酸=57:10:8.2的混合液作为展开剂,展开,取出,晾干,置365nm的紫外灯下检视,得到供试品色谱。
前述的羊奶奶叶药材的质量检测方法中,供试品色谱中,在与对照药材色谱相应的位置上,应显相同颜色的荧光斑点。
前述的羊奶奶叶药材的质量检测方法中,所述的羊奶奶叶药材中银锻苷的含量测定方法,照高效液相色谱法测定,包括以下步骤:
1)液相色谱仪的色谱条件
色谱柱为inertsustain C18,以十八烷基硅烷键合硅胶为填充剂;以乙腈和0.4%甲酸水为流动相,洗脱时间为0.01~25min,以25%~29%的乙腈进行等度洗脱,柱温30℃,流速1.0ml/min,检测波长为314nm,测定银锻苷;
2)含量测定对照品溶液的制备:精密称取银锻苷对照品适量,加甲醇溶解制成每ml含
0.22392mg的含量测定对照品溶液;
3)含量测定供试品溶液的制备:精密称取本品粉末1.0g,置具塞锥形瓶中,加入75%甲醇30ml,称重,水浴回流1.5小时,迅速冷却,称重后用75%甲醇补足失重,摇匀,过滤后取续滤液,即得含量测定供试品溶液;
4)测定法:精密吸取以上含量对照品和含量供试品溶液各10μl,注入液相色谱仪,测定,即得羊奶奶叶药材中银锻苷的含量。
前述的羊奶奶叶药材的质量检测方法中,羊奶奶叶药材按干燥品计算,含银锻苷C30H26O13不得少于0.16%。
前述的羊奶奶叶药材的质量检测方法中,步骤1)液相色谱仪的色谱条件中,理论板数按银锻苷计算应均不低于7500,按山柰酚、槲皮素、异鼠李素峰计算均不小于5000。
与现有技术相比,本发明的质量检测方法包括羊奶奶叶药材的薄层鉴别方法和羊奶奶叶药材中银锻苷的含量测定方法。具体的,本申请所采用的羊奶奶叶药材的薄层鉴别方法具有灵敏度高、专属性强以及分离效果好等特点,简便可行,重现性强,能够对药材的真伪进行准确的鉴别;本申请所采用的羊奶奶叶药材中银锻苷的含量测定方法能够准确、快速的测定出羊奶奶叶药材的含量,为羊奶奶叶药材提供有效的定性定量分析手段,从而有利于对羊奶奶叶药材的质量进行控制。采用本发明提供的羊奶奶叶药材的质量检测方法能有效的控制羊奶奶叶药材的整体质量、保证羊奶奶叶药材临床用药的安全有效性。
本发明方法对于羊奶奶叶药材的进一步质量控制具有重要的意义,可为羊奶奶叶药材质量表混的制定提供科学依据。
附图说明
图1是供试品制备方法考察薄层色谱图,(图1中,1、银锻苷对照品;2胡颓子叶超声提取;3、胡颓子叶乙酸乙酯水回流提取;4、胡颓子叶正丁醇水回流提取;5、宜昌胡颓子叶超声提取;6、宜昌胡颓子叶乙酸乙酯水回流提取;7、宜昌胡颓子叶正丁醇水回流提取;8、胡颓子叶超声提取;9、胡颓子叶乙酸乙酯水回流提取);
图2是点样量考察薄层色谱图,(图中,1、银锻苷对照品;2、胡颓子叶点样5μl;3、胡颓子叶点样10μl;4、胡颓子叶点样15μl;5、宜昌胡颓子叶点样5μl;6、宜昌胡颓子叶点样10μl;7、宜昌胡颓子叶点样15μl;8、胡颓子叶点样5μl;9、胡颓子叶点样10μl;10、胡颓子叶点样15μl;11、胡颓子点样叶10μl;12、胡颓子叶点样10μl)。
图3是乙酸乙酯-2-丁酮-甲酸(8:3:0.25)展开体系的薄层色谱图(图中,1、银锻苷;2、胡颓子叶;3、宜昌胡颓子叶;4、胡颓子叶;5、胡颓子叶;6、胡颓子叶);
图4是三氯甲烷-丙酮-甲酸(5:5:0.125)展开体系的薄层色谱图(图中,1、银锻苷;2、胡颓子叶;3、宜昌胡颓子叶;4、胡颓子叶;5、胡颓子叶;6、胡颓子叶);
图5是三氯甲烷-甲醇-甲酸(36:4:0.75)展开体系的薄层色谱图(图中,1、银锻苷;2、胡颓子叶;3、宜昌胡颓子叶;4、胡颓子叶);
图6是30%相对湿度的薄层色谱图;
图7是72%相对湿度的薄层色谱图;
图8是室温6℃时的薄层色谱图;
图9是空调25℃时的薄层色谱图;
图10是S1-S10批次羊奶奶叶样品的薄层色谱图(图中,1代表银锻苷,2-11代表S1-S10批次羊奶奶叶样品);
图11是S11-S18批次羊奶奶叶样品的薄层色谱图(图中,1代表银锻苷,2-9代表S11-S18批次羊奶奶叶样品);
图12是银锻苷对照品UV扫描色谱图;
图13是不同梯度条件下样品色谱图(图中,A梯度1(乙腈-0.4%甲酸水等度洗脱);B梯度2(乙腈-0.4%甲酸水等度洗脱);C梯度3(甲醇-0.4%甲酸水);D梯度4(乙腈-0.4%甲酸水梯度洗脱);E梯度5(乙腈-0.4%甲酸水梯度洗脱);1、银锻苷));
图14是梯度5的对照品与供试品色谱图(图中,A供试品;B对照品;1银锻苷);
图15是银锻苷线性回归曲线;
图16是山柰酚UV谱图;
图17是槲皮素UV谱图;
图18是异鼠李素UV谱图;
图19是各流动相梯度色谱图;
图20是样品色谱图及混合对照品HPLC色谱图(图中,A供试品;B混合对照品;1槲皮素;2山柰酚;3异鼠李素);
图21是槲皮素标准曲线图;
图22是山柰酚线性曲线图;
图23是异鼠李素线性曲线图;
图24是29个样品聚类分析图;
图25是银锻苷含量条形图;
图26是29批次药材三种苷元含量统计图。
具体实施方式
下面结合附图和实施例对本发明作进一步的说明,但并不作为对本发明限制的依据。
实施例。一种羊奶奶叶药材的质量检测方法,
仪器:
岛津LC-20AT型液相色谱仪,含自动进样器,DAD检测器。labsolution色谱系统;色谱柱:inertsustain C18(4.6mm×250mm,5μm);AL204型电子分析天平(Mettler Toledo仪器(上海)有限公司);VORTEX3旋涡混匀器(德国IKA公司);SHHW21型恒温水浴锅(北京科伟永兴仪器有限公司);BS224S型电子天平:万分之一,Sartorius;KQ-5200DB型超声清洗机(昆山市超声波仪器公司);DHG-9140A电热恒温鼓风干燥箱:巩义市予华仪器有限责任公司;8-10型箱式电阻炉:中国沈阳市节能电炉厂制造;三用紫外线分析仪:上海顾村电光仪器厂。
试剂与试药
1.对照品
银锻苷(Z30J7S16999)、山柰酚(RO3F6C1)、槲皮素(C2016Y1722)、异鼠李素(PO8J7F15965)对照品均购置于上海源叶生物科技有限公司,纯度≥98%。
2.试剂
乙腈为色谱纯(德国默克公司),甲酸为色谱纯(天津大茂化学试剂),浓盐酸(西陇化工股份有限公司),三氯甲烷、丙酮、甲酸、乙酸乙酯、2-丁酮、甲醇均为分析纯(国药集团化学试剂有限公司);硅胶GF254(薄层色谱用)。超纯水(Millipore-Simplicity超纯水处理系统,默克密理博,德国)。
药材
胡颓子属植物药用的种类较多,现有标准中收载的以叶入药的“羊奶奶叶/胡颓子叶/沙枣叶”的基原共有6种,本研究所用29批次样品(包括叶、根、茎、果实)均为自采或委托采集、购买,经江西中医药大学民族药研究中心慕泽泾讲师鉴定,共7种,其中有标准收载的4种,有文献记载药用的3种。样品具体来源信息见表1。
表1样品信息表
9【药材名称】羊奶奶叶。228
羊奶奶叶(胡颓子叶):根据《贵州省中药材、民族药材质量标准》命名。
本研究中,对原贵州省标规定的对照药材薄层鉴别进行了重复符合实验,结果表明方法可行。另,原湖北省标规定的以齐墩果酸为对照的薄层鉴别,鉴于该成分为较多药材的共有成分,专属性较差。考虑到黄酮类成分为土大黄的主要成分类型,故研究中建立了以银椴苷为对照的薄层间鉴别方法,结果表明方法可行。综合考虑,银椴苷在土大黄中含量较高,最终确定将该成分作为含测指标性成分,故本修订标准仍采用了对照药材的薄层鉴别,将银椴苷作为含测指标成分。
薄层鉴别:取本品粉末2g,加10倍量水煎煮30分钟,滤过,滤液置水浴上蒸干,残渣加无水乙醇0.5ml使溶解,作为供试品溶液。另取羊奶奶叶对照药材2g,同法制成对照药材溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述两种溶液各5μl,分别于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以醋酸乙酯-水-甲酸(57:10:8.2)为展开剂,展开,取出,晾干,置紫外灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。
1)制备方法考察
称取羊奶奶叶样品粉末1.0g,分别考察30ml水+30ml乙酸乙酯、30ml水+30ml正丁醇回流提取1h和60ml 75%甲醇超声提取对点样效果的影响,结果见图1。
薄层板:硅胶G板;点样量:5μl;展开剂:三氯甲烷-丙酮-甲酸(5:5:0.25);展开方式与展距离:上行展开7cm;显色:喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;检视:365nm紫外灯下检视。
由图1可知,直接超声提取得到的斑点颜色较淡,而正丁醇水、乙酸乙酯水溶液回流得到的斑点清晰且差异不大。考虑到正丁醇毒大,且沸点较高,选择乙酸乙酯水作为供试品制备提取溶媒。因此,供试品制备方法确定为:取羊奶奶叶粉末1.0g,精密称定,置于具塞锥形瓶中,精密加入30ml水和30ml乙酸乙酯溶液,摇匀,水浴回流提取60min,迅速过滤,续滤液置于分液漏斗中,静置分层,取上清液蒸干,取甲醇溶解定容于5ml容量瓶中,即得。
2)点样量考察
选择乙酸乙酯加水回流提取基础上,对点样量进行了考察图2,结果发现,点样量5-15μl均可,选择点样10μl。
薄层板:硅胶G板;展开剂:三氯甲烷-丙酮-甲酸(5:5:0.125);展开方式与展距离:上行展开8cm;显色:喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;检视:366nm紫外灯下检视。
3)展开剂考察
以银锻苷为对照品,分别考察三氯甲烷-丙酮-甲酸(5:5:0.125),三氯甲烷-甲醇-甲酸(36:4:0.75),乙酸乙酯-2-丁酮-甲酸(8:3:0.25)三种展开体系。根据图3-5结果,三种体系分离效果均较好,但三氯甲烷-丙酮-甲酸(5:5:0.125)条件斑点集中,明显。
薄层板:硅胶G板;展开方式与展距离:上行展开7cm;显色:喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;检视:365nm紫外灯下检视。
4)环境条件的耐用性考察
在选定展开系统为三氯甲烷-丙酮-甲酸(5:5:0.125)的条件下,开展环境条件的耐用性实验考察。分别开展了30%和72%相对湿度,以及室温6℃和空调25℃的温度考察,均取得良好地分离效果,见图6~9。
薄层板:硅胶G板;展开剂:三氯甲烷-丙酮-甲酸(5:5:0.125);展开方式与展距离:上行展开7cm;显色:喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰;检视:365nm紫外灯下检视。
5)18批药材的薄层色谱图
从药材中选择S1-S18批次羊奶奶叶样品粉末,按条件制备供试品溶液,以三氯甲烷-丙酮-甲酸(5:5:0.125)为展开系统,同时吸取供试品和对照品溶液10μl点于硅胶G板上。供试品和对照品在薄层版相同距离上,呈现相同的荧光斑点。见图10及11。
【含量测定】原标准中无含量测定规定。
止咳平喘是羊奶奶叶/胡颓子叶的主要功效之一。据文献报道,胡颓子属植物中含有挥发油、黄酮类、皂苷、萜类和甾体、脂肪酸、生物碱、酚类、鞣质、木脂素、有机酸、糖苷等成分;有关胡颓子E.pungens的药理学研究表明其叶的正丁醇萃取部位有较好的镇咳、祛痰作用,明显抑制豚鼠气管平滑肌收缩,与胡颓子叶的药用功效一致,为其有效部位之一,但对胡颓子E.pungens的成分研究较少。鉴于此,本修订中采用UPLC/Q-TOF-MS/MS,对胡颓子E.pungens的正丁醇萃取部位进行了初步成分分析,结果表明,该部位主要含有黄酮苷类、酚酸类及皂苷类成分,推测出了其中的49个成分,并结合对照品对照和结构解析,确认了其中的6个黄酮类成分,即槲皮素、山柰酚、异鼠李素、白麻苷(槲皮素-3-O-槐糖苷)、异鼠李素-3-O-槐糖苷、(异鼠李素苷)、银锻苷(山奈酚苷)。
根据上述研究结果,暂定选择黄酮类成分作为含量测定的指标成分,同时考虑到需与胡颓子属其他种类之间的比较鉴别、提高含测指标成分的专属性的需要,故对银椴苷、山柰酚、槲皮素、异鼠李素等4个成分的含量进行了考察。由于“羊奶奶叶”中所含黄酮苷类成分多达数十种,而各单体成分的含量较低,且多为黄酮氧苷,可能受植物生长环境等诸多因素的影响,在药材中的含量并不稳定,故将样品经盐酸水解后测定山柰酚、槲皮素、异鼠李素等苷元。
供试品溶液:称取羊奶奶叶粉末约1.0g,精密称定,置于50ml锥形瓶中,加75%甲醇30ml,称重,水浴回流1.5h,迅速冷却,用75%甲醇补足失重,摇匀,过滤后续滤液用0.45μm微孔滤膜滤过,即得。
对照品溶液:精密称取银锻苷对照品适量,加甲醇溶解制成每ml含0.22392mg的对照品溶液。
精密称取槲皮素、山柰酚、异鼠李素对照品适量,加甲醇溶解制成每ml含0.0888mg山柰酚、0.1208mg槲皮素、0.0414mg异鼠李素的混合对照品溶液。
色谱条件以十八烷基硅烷键合硅胶为填充剂;以乙腈和0.4%甲酸水为流动相,柱温30℃,流速1ml/min,检测波长为314nm。理论板数按银锻苷计算应均不低于7500,按山柰酚、槲皮素、异鼠李素峰计算均不小于5000。以乙腈(A)和0.4%甲酸水(B)为流动相(0.01~25min,25%~29%A等度洗脱),柱温30℃,流速1.0ml/min,检测波长为314nm,测定银锻苷。
1、银锻苷的含量测定
1.1检测波长的选择及流动相的考察
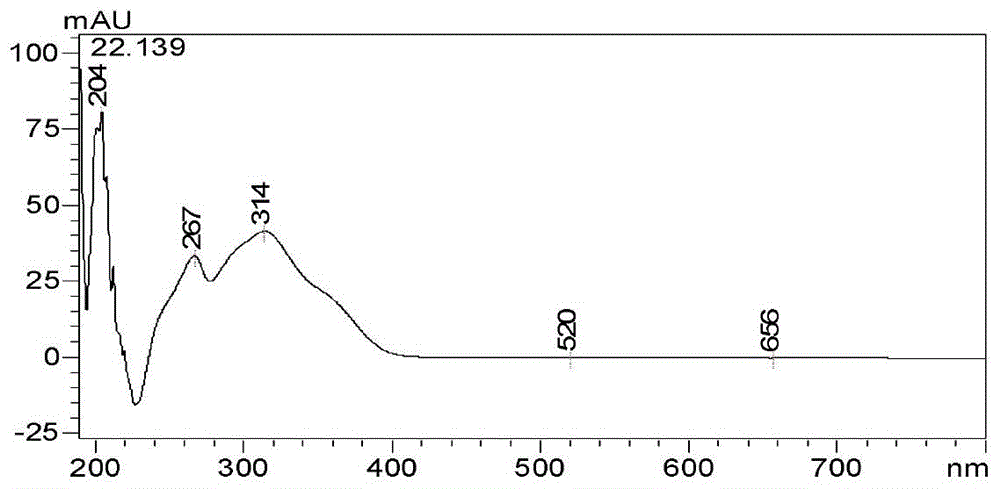
根据银锻苷UV扫描结果(见图12),银锻苷在314nm有最大吸收,由此选择为检测波长。考虑到黄酮苷类成分因带有较多羟基,加入一定的酸能有效改善峰形。以1.0ml·min-1为洗脱流速分别考察了乙腈-0.4%甲酸水等度洗脱、乙腈-0.4%甲酸水梯度洗脱、甲醇-0.4%甲酸水梯度洗脱等流动相洗脱比例。色谱梯度见表7,对应液相色谱图见图13。
表7流动相梯度设置
由上图的结果可知,梯度1和2均为乙腈-0.4%甲酸水等度洗脱,两条件下银锻苷色谱峰分离度均为1.0左右,达不到较好的分离效果,继续延长时间可能对分离度的提高效果不大。梯度3为以甲醇-0.4%甲酸水作为梯度洗脱流动相,银锻苷色谱峰分离度在1.5以上,但色谱峰较宽,且时间相对较长;梯度4和梯度5均为乙腈-0.4%甲酸水梯度洗脱,其中,梯度4银锻苷色谱峰分离度小于1.5,梯度5银锻苷色谱峰分离度在1.5以上,且出峰时间适中,峰形尖锐对称。故选择梯度5作为羊奶奶叶银锻苷含量测定的流动相梯度。
1.2、不同流速及柱温的考察
以梯度5作为流动相洗脱梯度,分别对流速0.8ml/min、1.0ml/min、1.2ml/min三个不同的流速和20℃、30℃、40℃三个不同温度对银锻苷色谱峰分离的影响进行了考察,结果表明,不同流速和不同温度对银锻苷色谱峰分离效果影响不大。综合考虑效率和能源等因素,选择1.0ml/min和30℃作为羊奶奶叶银锻苷含量测定流动相流速和柱温箱温度。
1.3、色谱条件的确定
根据上述试验结果,确定羊奶奶叶银锻苷含量测定的色谱条件为:色谱柱为inertsustain C18(4.6mm×250mm,5μm);流动相为乙腈(A)-0.4%甲酸水(B);梯度洗脱(0.01~25min,25%~29%A);流速:1.0ml·min-1;柱温:30℃;检测波长314nm;进样量10μl;检测时间25min。在以上梯度下,对照品和供试品色谱图见图14.
1.4提取方法考察
1.4.1提取方式考察
称取羊奶奶叶S14及S17号粉末各2份,每份约1.0g,精密称定,加入40ml甲醇,称重,75℃水浴回流1.5h,放冷,用甲醇补足失重,摇匀,过滤,取续滤液用0.45μm微孔滤膜滤过,即得供试品液。另取S14号及S17号样品羊奶奶叶粉末各2份,每份约1.0g,精密称定,加入40ml甲醇,称重,超声1h,放冷,用甲醇补足失重,摇匀,过滤,取续滤液用0.45μm微孔滤膜滤过,即得供试品液。按1.3色谱条件进样测定,结果见表8。
表8提取方式的考察
由上表中结果可知,两份样品回流提取时银锻苷提取效率均比超声提高30%以上,据此选择回流提取作为供试品溶液制备的提取方法。
1.4.2提取溶媒的考察
称取同批次羊奶奶叶粉末共6份,每份1.0g,精密称定,放入具塞锥形瓶中,分别加入50%甲醇、75%甲醇、甲醇、50%乙醇、75%乙醇、乙醇溶液40ml,称重,水浴回流提取1.5h,放冷,用对应的溶媒补足失重,摇匀,过滤,取续滤液用0.45μm微孔滤膜滤过,即得。按1.3色谱条件进样测定,测定结果见表9。
表9提取溶媒的考察
综合表3结果可知,甲醇提取效率要比乙醇稍高,且75%甲醇提取效率最好。据此选择以75%甲醇作为供试品溶液制备的提取溶媒。
1.4.3溶媒用量的考察
称取同批次样品羊奶奶叶粉末共3份,每份1.0g,精密称定,分别加入30ml、40ml、50ml 75%甲醇,称重,水浴回流提取1.5h,放冷,用75%甲醇补足失重,摇匀,过滤,取续滤液用0.45μm微孔滤膜滤过,即得。按1.3色谱条件进样测定,结果见表10。
表10提取溶媒用量的考察
综合上表结果可知,当溶媒用量为50ml时,银锻苷的提取效率最好,其次为30ml,但两者相差不多。考虑到节省溶剂等原因,选择30ml作为供试品溶液制备的提取溶媒用量。
1.4.4提取时间考察
称取同批次羊奶奶叶样品共3份,每份1.0g,精密称定,加入75%甲醇30ml,称重,分别水浴回流提取1h、1.5h、2h,用75%甲醇补足失重,摇匀,过滤,取续滤液用0.45μm微孔滤膜滤过,既得。按1.3色谱条件进样,结果见表11。
表11提取时间的考察
综合上表结果可知,随着提取时间增加,银锻苷的提取效率缓慢增加。综合考虑提取时间和提取效率,选择1.5h作为供试品溶液制备的提取时间。
根据以上考察结果,最终确定供试品溶液制备方法为:称取羊奶奶叶粉末约1.0g,精密称定,置于50ml锥形瓶中,加75%甲醇30ml,称重,水浴回流1.5h,迅速冷却,用75%甲醇补足失重,摇匀,过滤后续滤液用0.45μm微孔滤膜滤过,即得。
1.5方法学验证
1.5.1线性关系考察
精密称取11.196mg干燥后的银锻苷对照品,置于50ml容量瓶中,加入甲醇溶解,定容,摇匀,制成每ml含0.22392mg的对照品储备溶液。储备液依次稀释2、4、8、16、32、64倍,取储备液和各稀释液分别按1.3项下进样10μl,进样三次,测定并计算平均峰面积,以对照品浓度对峰面积进行线性回归,绘制标准曲线及回归方程。银锻苷线性考察结果见表12,回归曲线见图15。
表12银锻苷线性关系考察结果
由以上图表可知,银锻苷浓度在3.499μg·ml
1.5.2精密度试验
精密吸取稀释4倍的储备液,注入液相色谱仪,按1.3项下条件进行测定,共六次,记录峰面积。结果见表13。
表13精密度考察
由上表结果可知,6次测定银锻苷峰面积RSD低于1%,表明仪器精密度良好。
1.5.3重复性试验
按上述确定的供试品溶液制备方法,平行制备6份羊奶奶叶药材供试品溶液进行测定,计算其RSD值,结果见表14。
表14重复性试验结果
结果表明,平行制备6份羊奶奶叶药材供试品溶液进行测定,银锻苷RSD值为1.032%,说明该法重复性良好。
1.5.4重复性实验
取同一批次羊奶奶叶样品共6份,每份1.0g,精密称定,按1.4项下方法制备供试品溶液,按1.3项下条件进样测定,记录峰面积。结果见表15。
表15重复性考察
由上表结果可知,6份羊奶奶叶样品中含量测定值的RSD低于1%,表明试验方法重复性良好。
1.5.5溶液稳定性试验
取羊奶奶叶供试品按1.4项下方法制备供试品溶液,在0,1,2,4,8,12,24h后分别按1.3项下条件进样测定,记录峰面积。结果见表16。
表16稳定性考察
由上表结果可知,供试品在24小时内测定得的峰面积RSD为0.47%小于1%,表明供试品溶液在24h内稳定性良好。
1.5.6加样回收率
取已知含量的同一批羊奶奶叶样品9份,每份约1.0g,精密称定,分成三组,分别加入药材中银锻苷含量的约80%、100%、120%的对照品,按2.2.5项下制备供试品溶液,按2.1.3项下条件进样测定,计算加样回收率。结果见表17。
表17银锻苷加样回收率实验结果
/>
由上表结果可知,银锻苷在三个试验水平下平均回收率分别为98.24%、98.80%和102.69%,RSD均小于2%,表明试验方法的加样回收率良好。
1.5.7样品含量测定
取10批次羊奶奶叶药材各0.5g,精密称定,按供试品溶液的制备方法制备,各取10μl进样,记录峰面积,以回归方程计算各成分的质量分数,结果见表12。
分别取29批次样品粉末各1.0g,按1.4项下制备供试品溶液,按1.3项下条件进样测定,结果见表18。
表18 29批次样品银锻苷测定结果
结果表明,7种胡颓子属植物中以胡颓子E.pungens、宜昌胡颓子E.henryi的叶中含量较高,其他种类及部位中的含量明显低于叶。巴东胡颓子叶含量极低,不在线性范围内。
2槲皮素、山柰酚、异鼠李素的含量测定
2.1流动相的考察
由山柰酚、槲皮素、异鼠李素的UV扫描结果(见图16~图18)可知,3个对照品均在370nm左右有最大吸收,据此确定检测波长为370nm。以乙腈-0.4%甲酸水溶液为流动相,考察不同梯度流动相等度洗脱和梯度洗脱对色谱分离度的影响。梯度设置见表19,梯度色谱图见图19。
表19流动相各梯度设置
由上述图谱可知,梯度1和梯度2为梯度洗脱,山柰酚和异鼠李素达不到理想分离状态。梯度3-5均达到1.5以上。鉴于梯度5各目标峰的分离度均在2以上,而且出峰时间适中,峰形对称,据此选择梯度5作为羊奶奶叶含量测定梯度。
2.2色谱条件的确定
以梯度5作为流动相洗脱梯度,经对不同流速、柱温对各成分分离效果的考察,最终确定色谱条件为:色谱柱inertsustain C18(4.6mm×250mm,5μm)。等度洗脱:乙腈(29%A)-0.4%甲酸水(71%B);流速:1.0ml·min-1;柱温:30℃;检测波长370nm。进样量10μl。采样时间35min。在上述色谱条件下,样品和混合对照品色谱图见图20。
2.3供试品制备方法的考察
2.3.1提取方式的考察
称取样品14号粉末约1.0g,精密称定,加入40ml75%甲醇,称重,超声60min,放冷,用对应溶剂补足失重,摇匀,过滤,取20ml续滤液加入3ml浓盐酸,称重,置80℃水浴回流1h,迅速冷却,补足失重,摇匀,过滤后续滤液用0.45μm微孔滤膜滤过,即得。另取14号样品羊奶奶叶粉末约1.0g,精密称定,加入40ml75%甲醇,称重,水浴回流1.5h,放冷,用对应溶剂补足失重,摇匀,过滤,取20ml续滤液加入3ml浓盐酸,称重,置80℃水浴回流1h,迅速冷却,补足失重,摇匀,过滤后续滤液用0.45μm微孔滤膜滤过,即得。取以上制备的供试品,按2.2项下方法进样分析,结果见表20。
表20提取方式考察
上表结果表明,回流提取对3个化合物提取效率均比超声提取提升30%以上,可能是由于黄酮苷类成分在热溶剂中的溶解度较高导致。因此,选择回流提取作为供试品溶液制备的提取方法。
2.3.2提取溶媒的考察
称取同一批次样品粉末(过60目筛)共6份,每份1.0g,精密称定,放入具塞锥形瓶中,分别加入50%甲醇、75%甲醇、甲醇、50%乙醇、75%乙醇、乙醇溶液40ml,称重,水浴回流提取1.5h,放冷,用对应的溶媒补足失重,其余按3.3.1项下的操作,按2.2项下色谱条件进样测定,测定结果见表21。
表21提取溶剂考察
由上表结果可知,随着甲醇浓度的增高,3个化合物的提取效率逐渐提高。随着乙醇浓度的上升,3个化合物的提取效率先上升后下降,其中75%乙醇对三个化合物的提取效率最好。据此选择75%乙醇作为提取溶媒。
2.3.3溶媒用量的考察
称取同一批次样品粉末(过60目筛)共3份,每份1.0g,精密称定,分别加入25ml、40ml、55ml 75%乙醇,称重,水浴回流提取1.5h,放冷,用对应的溶媒补足失重,摇匀,过滤,其余按2.3.1项下的操作,按2.2项下色谱条件进样测定,测定结果见表22。
表22溶媒用量的考察
由上表中结果可知,随着提取溶媒用量的增大,3个化合物的提取效率先升后降。据此选择40ml作为溶媒提取用量。
2.3.4盐酸用量的考察
称取同一批次羊奶奶叶粉末(过60目筛)共3份,约1.0g,精密称定,置于100ml锥形瓶中,加入75%乙醇40ml,称重,水浴回流1h,迅速冷却,补足失重,摇匀,过滤,取20ml续滤液置于具塞锥形瓶中,另加入3、4、5ml浓盐酸,其余按2.3.1项下的操作制备,按2.2色谱条件进样测定,测定结果见表23。
表23盐酸用量的考察
由上表中结果可知,随着盐酸用量的增加,3个化合物的水解效率逐渐上升,但考虑到过酸性的供试品可能对色谱柱柱效产生影响,选择5ml盐酸作为供试品制备用量。
2.3.5提取时间的考察
在以上条件下,考察水浴回流60min、90min、120min对三个成分提取的影响,结果见表24。
表24回流时间考察
结果表明,不同提取时间中以120min提取效率最高,但各提取时间对提取率的影响并不显著,且变化趋势不一致,可能与不同成分的水解过程有关。综合考虑,确定以60min作为水浴回流提取时间。
2.3.6供试品制备方法的确定
综合以上考察项结果,确定供试品制备方法为:取供试品粉末约1.0g,精密称定,置于具塞锥形瓶中,加入75%乙醇40ml,称重,水浴回流1h,迅速冷却,补足失重,摇匀,过滤,取20ml续滤液置于具塞锥形瓶中,另加入5ml盐酸,称重,80℃水浴回流1h,迅速冷却,补足失重,摇匀,过滤后续滤液用0.45μm微孔滤膜滤过,即得供试品溶液。
2.4线性范围的考察
分别精密称取山柰酚对照品4.44mg、槲皮素对照品6.04mg、异鼠李素对照品2.07mg,用甲醇定容至50ml,超声助溶,过0.45μm微孔滤膜后制成每ml含0.0888mg山柰酚、0.1208mg槲皮素、0.0414mg异鼠李素的混合对照品储备液。储备液依次稀释2、4、8、16、32、64、128倍,取储备液和各稀释液分别按2.2项下条件进样10μl,各进样3次,测定并计算平均峰面积,以对照品浓度对峰面积进行线性回归,绘制标准曲线及回归方程。各化合物线性考察结果见表25~27,回归曲线见图21~23。
表25槲皮素线性关系考察结果
/>
表26山柰酚线性关系考察表
表27异鼠李素线性关系考察表
结果表明,槲皮素在0.94~120.8μg·ml
2.5精密度实验
精密吸取对照品溶液,注入液相色谱仪,按2.2项下条件进样分析,共6次,记录峰面积。结果见表28。
表28精密度考察
综合上表结果,各化合物6次测定峰面积RSD均低于1%,表明仪器的精密度良好。
2.6稳定性实验
取供试品按2.3.6项下方法制备供试品溶液,在0,1,2,4,8,12,24h后分别按2.2项下条件进样测定,记录峰面积。结果见表29。
表29稳定性考察
以上结果表明,3个待测成分在24小时内多次测定的峰面积RSD低于2%,表明供试品溶液在24小时内稳定性良好。
2.7重复性考察
取同一批次羊奶奶叶样品(过60目筛)共6份,每份1.0g,精密称定,按2.3.6项下方法制备供试品溶液,按2.2项下条件进样测定,记录峰面积。结果见表30。
表30重复性考察
由上表结果可知,3个待测成分6次重复试验测定的含量的RSD均小于3%,表明方法的重复性良好。
2.8加样回收实验
取已知含量的同一批羊奶奶叶样品6份,每份约0.5g,精密称定,分别加入药材中3种苷元含量的约100%的对照品,按2.3.6项下制备供试品溶液,按2.2项下条件进样测定,计算加样回收率。结果见表31。
表31加样回收率
结果表明,3个化合物的加样回收率良好。
2.9含量测定
分别取29批次样品粉末各1.0g,精密称定,按2.3.6项下制备供试品溶液,按2.2项下条件进样测定。结果见表32。
表32 29批次样品含量测定
有上表可知,3种黄酮苷元的含量以叶中较高,茎中含量较低,根中不含有(或衡量)该3种苷元。3种苷元的含量及其组成比例以及总量在不同种植物的叶中的含量均存在较大差异。
3不同胡颓子属植物成分的比较
3.1聚类分析
如表18及表29所示,各基原物种及不同药用部位间银锻苷和三种黄酮苷元的含量均存在显著差异。为探讨此种差异是否可用来区分各基原物种,采用SPSS18.0软件,以银锻苷及3种苷元成分的含量为指标变量,采用ward法和欧式距离聚类方法,对所有样品进行了系统聚类分析。结果见图24。
聚类分析图表明,所测定成分能较好地区分不同基原物种。16批次胡颓子叶中的15批次聚为1大类,仅有样品S14与巴东胡颓子叶聚为一类。S14样品为20kg大量商品药材与其他15批次1kg小样不同,可能是由于药材采集量大,采集区域较广,掺杂了其他同属植物的叶,导致化学成分的差异,尤其是异鼠李素苷元含量明显高于其他批次样品。宜昌胡颓子与胡颓子聚为一大类,表明宜昌胡颓子叶与胡颓子叶在两类成分含量上更为相近。三批次蔓胡颓子聚为一小类,两批次牛奶子聚为一小类,其余长裂胡颓子叶、福建胡颓子叶等各自分别聚为1小类,表明不同基原物种间的化学成分的差异显著。以上结果显示,银椴苷及3种苷元成分的含量及其组成能较好地区分胡颓子属的7种植物。而药材的茎、根、果和叶不聚为一类,表明其与叶的成分差异较大,表明根、叶、果实在分别药用时各有其不同的临床对症有一定的科学依据。
3.2不同种间银椴苷和3种苷元成分的比较
胡颓子属7种植物及其不同部位中的银椴苷、槲皮素、山柰酚、异鼠李素含量比较结果见图25、26。
银椴苷宜昌胡颓子叶中银锻苷的含量最高,达3.1704mg·g-1,其次为胡颓子叶,其余种含量均较低,表明种间差异较大,是否可以相互混用仍需结合市场及临床使用调查、品种与基原整理、活性和质量评价等进行深入研究。茎和果中含量不及叶中十分之一,而根中不含银锻苷,提示不同部位应分别作为不同的药材使用。
槲皮素、山柰酚、异鼠李素可能由于植物体内代谢途径不同,各不同种间三个化学成分的相应比值有一定规律。胡颓子叶中含量山柰酚>槲皮素>异鼠李素,且当以槲皮素含量为1时,16批次胡颓子叶除S14以外,槲皮素与山柰酚和异鼠李素的比值稳定在1∶1.31~2.38∶0.16~0.64之间,宜昌胡颓子叶比值与此接近。蔓胡颓子叶中异鼠李素含量要明显高于山柰酚和槲皮素。牛奶子叶中山柰酚与槲皮素比值大于胡颓子叶,且异鼠李素含量极低,比值为1∶2.94~3.89∶0.05。福建胡颓子叶槲皮素含量最高,且槲皮素>山柰酚>异鼠李素。巴东胡颓子叶中异鼠李素与山柰酚比值接近1∶1。三种苷元总量以福建胡颓子叶最高,其次为蔓胡颓子叶、胡颓子叶和宜昌胡颓子叶及长裂胡颓子叶。牛奶子叶和巴东胡颓子叶最低。茎、叶、果中含量均比叶低,根中不含。
3.3基原物种的取舍
胡颓子叶中两类成分的含量普遍较高,且分布广泛,用药历史悠久,因此作为主要基原物种与其他物种比较。贵州省标(2003版)曾收载羊奶奶叶(胡颓子叶)基原植物为胡颓子E.pungens Thunb.、宜昌胡颓子E.henryi Warb.、蔓胡颓子E.glabra Thunb.。福建省标(2006版)将当地分布较多的福建胡颓子E oldhami Maxim也作胡颓子叶药用。由聚类分析图及两类成分的含量可知,宜昌胡颓子与胡颓子成分较为相似,故保留为基原物种。蔓胡颓子及福建胡颓子中成分与胡颓子叶差异较大。贵州及其他地方同做胡颓子叶药用的牛奶子、巴东胡颓子、长裂胡颓子等亦与胡颓子叶差异较大。综上所述,羊奶奶叶(胡颓子叶)基原植物以胡颓子E.pungens Thunb.、宜昌胡颓子E.henryi Warb.为宜,而蔓胡颓子E.glabra Thunb.同用是否合理还有待研究,暂不作为基原植物。
4含量测定项指标成分的选择及其限量
黄酮是胡颓子属植物中的主要成分,也是其有效部位之一。前述研究表明,胡颓子叶中含有数十种黄酮类成分,这些成分均为以山柰酚、槲皮素及异鼠李素为苷元与多种糖或香豆素、阿魏酸或芥子酸形成的多种黄酮苷。对银椴苷及山柰酚、槲皮素及异鼠李素3种苷元的含量分析表明,总体上,银椴苷在胡颓子和宜昌胡颓子中含量较高且较为一致,而3种苷元及其组成比例在各种中均存在较大差异,文献报道银椴苷也是具有生物活性的成分之一。综合考虑,选定银椴苷作为胡颓子叶药材的含量测定指标成分。
表17中,胡颓子E.pungens和宜昌胡颓子E.henryi的叶共17个样品中,第14号样品为异常值(0.05%),取其他16个样品计算,银椴苷的含量在0.1471~0.31704%之间,平均含量为0.1961%,取均值下浮20%为0.16%,17批样品中14批合格,合格率为82%。据此确定胡颓子叶中银椴苷的含量不得少于0.16%。
- 一种吴茱萸药材特征图谱的构建方法和吴茱萸药材质量检测方法
- 一种山楂叶药材、山楂叶提取物或其制成品的质量检测方法
- 一种山楂叶药材、山楂叶提取物或其制成品的质量检测方法