吗啡喃衍生物
文献发布时间:2023-06-19 12:21:13
本申请是申请号为201680016083.0、申请日为2016年3月17日、发明名称为“吗啡喃衍生物”的发明申请的分案申请。
技术领域
本发明涉及具有阿片δ受体激动剂作用的吗啡喃衍生物。
背景技术
阿片结合于阿片受体而发挥效果,阿片受体存在μ、δ、κ这3个亚型。已知μ、δ、κ这3个亚型中的任一者的激动剂均具有镇痛作用。
但是,作为对阿片μ受体亲和性高的激动剂的吗啡具有强效的镇痛作用,另一方面,具有依赖性、药物滥用、耐性、呼吸抑制、消化道运动抑制所致的便秘、恶心呕吐、血压下降、心动过慢、咳嗽反射抑制、瞌睡等副作用。
作为阿片κ受体的选择性激动剂的依他佐辛具有强镇痛作用,虽然依赖性、耐性、嗜睡、便秘、呼吸抑制为轻度,但确认了发汗、恶心呕吐、口渴。
另一方面,已知若将阿片δ受体活化,则带来镇痛、抗抑郁和抗焦虑效果。例如,已知作为阿片δ受体的内源性配体的脑啡肽具有镇痛作用。另外,已知在受体缺损小鼠中发生焦虑样和抑郁样行为的增加(非专利文献1)、脑啡肽-δ受体系统的亢进与情绪调节有关(非专利文献2)。进而,在大鼠、小鼠的各种焦虑和抑郁模型中,由于各种δ受体激动剂的抗抑郁·抗焦虑样作用被δ受体拮抗剂拮抗,因此,显示了作为δ受体选择性激动剂的抗抑郁·抗焦虑药的有用性(非专利文献3~7、专利文献1、专利文献2)。可以期待选择性活化阿片δ受体的激动剂不具有或几乎不具有通过阿片μ受体、阿片κ受体的活化而出现的副作用。
此外,暗示了δ受体的活化对帕金森氏病、阿尔茨海默病等神经退行性疾病、缺血、中风、排尿障碍、HIV感染、酒精依赖症、糖尿病等的改善效果(非专利文献8)。迄今为止,作为阿片δ激动剂,报告了各种化合物,其镇痛作用、抗抑郁作用和抗焦虑作用得到证实(专利文献1~6、非专利文献9)。应予说明,报告了SNC80、BW373U86等一部分阿片δ激动剂诱发痉挛(非专利文献5、6和10)。
作为抗抑郁药,除传统的三环系抗抑郁药和单胺氧化酶抑制剂以外,还开发了四环系抗抑郁药、三唑并吡啶系抗抑郁药,近年来,频繁使用选择性血清素再摄取抑制剂(Selective Serotonin Reuptake Inhibitors:SSRI)、血清素·去甲肾上腺素再摄取抑制剂(serotonin-noradrenaline reuptake inhibitor:SNRI)、去甲肾上腺素激动性·特异性血清素激动性抗抑郁药(Noradrenergic and Specific SerotonergicAntidepressant:NaSSA)。然而,对于任一抗抑郁药,在以缓解率观察时有效性均并不高。另外,服用开始后早期的攻击性的亢进、年轻患者的自杀意念、自杀企图的风险增加等,其有用性受到限制。
作为抗焦虑药,广泛使用苯并二氮杂
因此,期望开发出以与目前使用的药剂不同的作用机制发挥效果且副作用和安全性更进一步得到改善的抗焦虑药、抗抑郁药。
现有技术文献
专利文献
专利文献1:日本特表2006-522775
专利文献2:WO2001/046192
专利文献3:WO2008/001859
专利文献4:WO2013/035833
专利文献5:WO2014/021273
专利文献6:WO2014/136305
非专利文献
非专利文献1:Nature Genetics 2000,25,195
非专利文献2:Neuroscience 2005,135,305
非专利文献3:J.Pharmacol.Exp.Ther.2011,338,195
非专利文献4:Trends in Neurosciences 2013,36,195
非专利文献5:Behavioural Brain Research 2011,223,271
非专利文献6:Neuropharmacology,2013,67,485
非专利文献7:Current Neuropharmacology,2012,10,231
非专利文献8:Psychopharmacology(Berl)2013,228,1
非专利文献9:Tetrahedron,2011,67,6682
非专利文献10:The International Narcotics Research Conference2014,July13,2014
发明内容
本发明的课题在于提供有效性高且依赖性、耐性、呼吸抑制、便秘、恶心呕吐、血压降低、心动过慢、咳嗽反射抑制、催眠作用、肌肉松弛、镇静、认知功能降低、发汗、口渴等有害作用少、安全的抗焦虑药、抗抑郁药、镇痛药、帕金森氏病治疗药和尿频·尿失禁治疗药。本发明的课题在于提供还能够同时发挥抗抑郁作用、抗焦虑作用、镇痛作用的安全的药剂,对受困于抑郁、焦虑、疼痛的患者提供福音。而且,本发明的课题在于提供能够以一种药剂同时治疗抑郁、焦虑、疼痛并能够安全地通过口服或注射进行给药(例如皮下注射)的药剂。
(1)本发明涉及以下的通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物。
(式中,R
R
在此,R
R
R
R
R
X表示O或CH
而且,Y表示C(=O)。
其中,R
卤素;羟基;C
而且,R
C
R
进而,R
而且此外,R
另外,本发明涉及一种医药品,由上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物构成。
另外,本发明涉及一种医药品组合物,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种镇痛剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种抗抑郁剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种抗焦虑剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种缓和、预防或治疗抑郁的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及一种缓和、预防或治疗焦虑的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及一种缓和、预防或治疗疼痛的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在缓和、预防或治疗疼痛、抑郁、焦虑中的应用。
另外,本发明涉及一种缓和、预防或治疗人的疼痛、抑郁、焦虑的方法,所述方法包括对人给予有效量的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物。
另外,本发明涉及一种帕金森氏病的治疗剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种缓和、预防或治疗帕金森氏病的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在缓和、预防或治疗帕金森氏病中的应用。
另外,本发明涉及一种缓和、预防或治疗人的帕金森氏病的方法,所述方法包括对人给予有效量的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物。
另外,本发明涉及一种尿频或尿失禁的治疗剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种缓和、预防或治疗尿频或尿失禁的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在缓和、预防或治疗尿频或尿失禁中的应用。
另外,本发明涉及一种缓和、预防或治疗人的尿频或尿失禁的方法,所述方法包括对人给予有效量的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物。
另外,本发明涉及一种青光眼的治疗剂,含有上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物作为有效成分。
另外,本发明涉及一种缓和、预防或治疗青光眼的方法,包括给予上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的有效量。
另外,本发明涉及上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在缓和、预防或治疗青光眼中的应用。
另外,本发明涉及一种缓和、预防或治疗人的青光眼的方法,所述方法包括对人给予有效量的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物。
本发明提供的化合物即通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物对阿片δ受体具有强效的激动剂活性且对μ和κ受体不显示活化或者仅显示极弱的活化,因此,具有基于阿片δ受体活化的优异的抗抑郁作用、抗焦虑作用、镇痛作用、帕金森氏病的治疗作用、尿频·尿失禁的治疗作用。本发明的化合物对阿片μ受体和κ受体不显示活化或者仅显示极弱的活化,因此,不具有依赖性、药物滥用、耐性、呼吸抑制、消化道的运动抑制所致的便秘、恶心呕吐、血压降低、心动过慢、咳嗽反射抑制、嗜睡、发汗、口渴等副作用,或者非常弱。另外,本发明的化合物在所研究的限度内,对其它受体、通道、酶没有作用或者仅显示极轻微的作用。因此,可以期待本发明的化合物完全没有痉挛、肌肉松弛、镇静、认知功能降低等有害作用或者非常弱。
本发明的化合物通过利用口服或注射进行给药(例如皮下注射)而显示高血中浓度和脑内移行性,因此,可以通过口服给药或注射给药来使用。
本发明的化合物由于在来自肝细胞的微粒体中难以被代谢,因此,从药物代谢的观点考虑是优异的。代谢产物所致的副作用的顾虑也少。
本发明的化合物由于完全不具有对承担心肌活动电位的再极化的锂离子通道即Kv11.1(或hERG;human Ether-a-go-go Related Gene)的抑制活性或者弱到能够忽略的程度,因此,从担心QT间隔延长而导致猝死这样的方面考虑,是安全的药剂。
本发明的化合物是有效性高且安全的药剂。
本发明的化合物能够以一种药剂同时消除抑郁、焦虑、疼痛全部。
附图说明
图1是表示关于化合物1的小鼠高架式十字迷宫试验的结果的图。
图2是表示关于化合物7的小鼠高架式十字迷宫试验的结果的图。
图3是表示关于化合物3的小鼠高架式十字迷宫试验的结果的图。
图4是表示关于化合物9的小鼠高架式十字迷宫试验的结果的图。
图5是表示关于化合物10的小鼠高架式十字迷宫试验的结果的图。
图6是表示关于化合物3、7、10的大鼠高架式十字迷宫试验的结果的图。
具体实施方式
接着,对本发明进一步详细地进行说明。
上述(1)的通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物中,优选可以举出以下的物质。
(2)根据(1)的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(3)根据上述(1)或(2)所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(4)根据(1)的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(5)根据(1)的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(6)根据上述(1)~(5)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(7)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(8)根据上述(1)~(7)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(9)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(10)根据上述(1)~(6)或(9)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(11)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(12)根据上述(1)~(6)或(11)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(13)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(14)根据上述(1)~(6)或(13)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(15)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(16)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(17)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(18)根据上述(1)~(6)或(17)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(19)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(20)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(21)根据上述(1)~(6)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(22)根据上述(1)~(6)或(21)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(23)根据上述(1)~(22)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,X为CH
(24)根据上述(1)~(23)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(25)根据上述(1)~(23)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(26)根据上述(1)~(23)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(27)根据上述(1)~(23)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(28)根据上述(1)~(23)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(29)根据上述(1)~(28)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(30)根据(1)的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
R
R
在此,R
R
X为CH
而且,Y为C(=O),
其中,R
卤素;羟基;C
而且,R
C
R
进而,R
(31)根据(1)或(30)所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(32)根据(1)、(30)或(31)所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(33)根据(1)或(30)所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(34)根据(1)或(30)所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(35)根据(1)或(30)~(34)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(36)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(37)根据(1)或(30)~(36)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(38)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(39)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(40)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(41)根据(1)、(30)~(35)或(40)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(42)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(43)根据(1)、(30)~(35)或(42)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(44)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(45)根据(1)、(30)~(35)或(44)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(46)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(47)根据(1)、(30)~(35)或(46)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(48)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(49)根据(1)、(30)~(35)或(48)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(50)根据(1)或(30)~(35)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(51)根据(1)、(30)~(35)或(50)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(52)根据(1)或(30)~(51)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(53)根据(1)或(30)~(51)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(54)根据(1)或(30)~(51)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(55)根据(1)或(30)~(51)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(56)根据(1)、(30)~(51)中任一项所述的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,其中,R
(57)一种化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,该化合物选自:
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物、
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-6-甲基吡啶-2(1H)-酮、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮、
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-2,4(1H,3H)-二酮、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-4(1H)-酮、
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-4(1H)-酮、
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)哒嗪-3(2H)-酮、
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)喹啉-2(1H)-酮、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-2H-吡喃-2-酮、
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-4H-吡喃-4-酮、
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-4(1H)-酮、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡嗪-2(1H)-酮、
2-((1S,3aR,5aS,6R,11bR,11cS)-10-乙酰氧基-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡嗪-2(1H)-酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-2,4(1H,3H)-二酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-乙基吡啶-2(1H)-酮、
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-4(3H)-酮以及
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-乙基吡啶-2(1H)-酮。
(58)一种化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物,该化合物选自:
6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基-1,2-二氢-3H-吡唑-3-酮、
5-氯-3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮、
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1,3-二甲基嘧啶-2,4(1H,3H)-二酮以及
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-甲氧基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮。
在本说明书中,
作为C
作为C
作为被1~3个卤素取代的C
作为C
作为环烷基部分的碳原子数为3~6、亚烷基部分的碳原子数为1~5的环烷基烷基,可以举出被环丙基、环丁基、环戊基或环己基等C
作为芳基部分的碳原子数为6~10、亚烷基部分的碳原子数为1~5的芳烷基,可以举出苄基或苯乙基。
作为C
作为C
作为含有选自N、O和S中的1~4个杂原子作为环构成原子的杂芳基,可以举出吡啶基、呋喃基、咪唑基、吡唑基、嘧啶基、吡嗪基、哒嗪基或噻唑基等。
作为杂芳基含有选自N、O和S中的1~4个杂原子作为环构成原子、亚烷基部分的碳原子数为1~5的杂芳基烷基,可以举出(吡啶-2-基)甲基、(吡啶-3-基)甲基、(吡啶-4-基)甲基、(呋喃-2-基)甲基、(呋喃-3-基)甲基、(咪唑-2-基)甲基、(咪唑-4-基)甲基、(咪唑-5-基)甲基、(噻唑-2-基)甲基、(噻唑-4-基)甲基、(噻唑-5-基)甲基、2-(吡啶-2-基)乙基、2-(吡啶-3-基)乙基、2-(吡唑-1-基)乙基、2-(噻吩-2-基)乙基或2-(噻吩-3-基)乙基等。
作为C
作为C
作为C
作为烷氧基部分的碳原子数为1~6的烷氧基羰基,可以举出甲氧基羰基或乙氧基羰基等。
作为卤素,可以举出氟、氯、溴或碘等。
作为被1~3个卤素取代的C
作为被1~6个卤素取代的C
作为烷基的碳原子数为1~3的苯基烷基,可以举出苄基等。
作为C
作为C
作为酰基部分的碳原子数为2~6的酰基氨基,可以举出乙酰基氨基等。
作为C
作为烷基部分的碳原子数为1~6的烷基氨甲酰基,可以举出乙基氨甲酰基等。
作为烷基部分的碳原子数为1~6的二烷基氨甲酰基,可以举出二乙基氨甲酰基等。
作为烷基部分的碳原子数为1~6的烷基磺酰基,可以举出甲基磺酰基等。
作为烷基部分的碳原子数为1~6的烷基亚磺酰基,可以举出甲基亚磺酰基等。
作为烷基部分的碳原子数为1~6的烷硫基,可以举出甲硫基等。
作为芳基部分的碳原子数为6~10的芳基羰基,可以举出苯甲酰基等。
作为R
作为R
(A)吡啶1-氧化物、2-甲基吡啶1-氧化物等可以被选自被1~3个氟取代的C
(B)吡啶-2(1H)-酮、1-甲基吡啶-2(1H)-酮、1-乙基吡啶-2(1H)-酮、6-甲基吡啶-2(1H)-酮、6-乙基吡啶-2(1H)-酮或6-三氟甲基吡啶-2(1H)-酮等可以被选自被1~3个氟取代的C
(C)吡啶-4(1H)-酮、1-甲基吡啶-4(1H)-酮、1-乙基吡啶-4(1H)-酮或1-(氟乙基)吡啶-4(1H)-酮等可以被选自被1~3个氟取代的C
(D)哒嗪-3(2H)-酮、2-甲基哒嗪-3(2H)-酮等可以被选自被1~3个氟取代的C
(E)吡嗪-2(1H)-酮、1-甲基吡嗪-2(1H)-酮等可以被选自被1~3个氟取代的C
(F)4H-吡喃-4-酮、3-甲基-4H-吡喃-4-酮、2H-吡喃-2-酮、5-甲基-2H-吡喃-2-酮等可以被选自被1~3个氟取代的C
(G)喹啉-2(1H)-酮、6-甲基喹啉-2(1H)-酮、喹啉-1-氧化物、4-甲基喹啉-1-氧化物等可以被选自被1~3个氟取代的C
(H)嘧啶-4(3H)-酮、嘧啶-2,4(1H,3H)-二酮等可以被选自被1~3个氟取代的C
作为上述通式(I)所示的化合物的互变异构体,可以举出上述R
在上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物中,作为药学上允许的盐,优选可以举出酸加成盐,作为酸加成盐,可以举出盐酸盐、硫酸盐、富马酸、草酸盐、甲磺酸盐、樟脑磺酸盐等与有机酸或无机酸的盐。
在上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物中,作为立体异构体,可以举出顺式、反式异构体、外消旋体、光学活性体等。
在上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物中,作为溶剂化物,是本发明的化合物或其盐的医药品上允许的溶剂化物,也包含水合物。
另外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物也可以为以在体内或到达目标部位后转换为药理活性物质而发挥(活化)药理效果的方式进行化学修饰而成的前药。
作为该前药,例如构成前药的基团存在于羟基时,可以举出低级酰基、低级烷氧基羰基等通常的羟基的保护基团,存在于氮原子时,可以举出低级酰基、低级烷氧基羰基等通常的氨基的保护基团或导入到羧酸部位的前药基团、例如新戊酰氧基甲基(tBu-C(O)O-CH2-)基、4-羟甲基-5-甲基-碳酸亚乙烯酯(Medoxomil)基、环己基-1-羟乙基碳酸酯(Cilexetil)基等。
进而此外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物可以被氘等稳定同位素取代。
接着,将上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的制造方法示于以下。
本说明书中所使用的缩写如下所述。
缩写表
Boc:叔丁氧基羰基
CPM:环丙基甲基
DMA:N,N-二甲基乙酰胺
DMAP:N,N-二甲基-4-氨基吡啶
DMF:N,N-二甲基甲酰胺
DMSO:二甲基亚砜
HATU:1-[双(二甲基氨基)亚甲基]-1H-1,2,3-三唑并[4,5-b]吡啶
HOAt:1-羟基-7-氮杂苯并三唑
HOBT:1-羟基苯并三唑
Me:甲基
Ms:甲磺酰基
Ph:苯基
TBS:叔丁基二甲基甲硅烷基
THF:四氢呋喃
TLC:薄层色谱法
Ts:甲苯磺酰基
WSC:1-乙基-3-(3-二甲基氨基丙基)碳二亚胺盐酸盐
(制造方法)
作为本发明提供的化合物的下述化合物(I)例如可以通过从下述的化合物(I-A)向化合物(I)的脱保护反应而得到。
[式中,R
在上述的制造方法中,上述化合物(I)可以通过对上述化合物(I-A),根据需要利用适当公知的一般的脱保护反应将R
另外,上述化合物(I-A)中的R
R
这些R
上述化合物(I-A)可以通过例如以下所示的反应式中的对下述化合物(I-B)的一般的酰基化反应而得到。
[式中,R
在上述的制造方法中,根据需要在HOBT、DMAP等添加剂、三乙基胺、二异丙基乙基胺等碱的存在下使上述化合物(I-B)、羧酸(R
另外,通过使上述化合物(I-B)与羧酰氯(R
R
此外,也可以通过Christian A.G.N.Montalbetti,et al,Tetrahedron,61(46),2005,10827-10852.中解说的缩合反应由上述化合物(I-B)和对应的羧酸(R
上述化合物(I-B)可以通过使用例如专利文献WO2013/035833中记载的化合物8(实施例4:R
下述化合物(I-A)可以通过例如以下所示的反应式中的对下述化合物(I-C)的一般的烷基化反应而得到。
[式中,R
在上述制造方法中,对上述化合物(I-C),根据需要在乙酸等添加剂的存在下,使对应的醛(R
另外,在DMF或醇等极性溶剂中,对上述化合物(I-C),使对应的烷基化剂(R
此外,对上述化合物(I-C)导入R
上述化合物(I-C)可以通过依照基于例如专利文献WO2013/035833中记载的化合物11(实施例7:R
关于作为本发明提供的化合物的上述通式(I)所示的化合物以外的化合物,也可以通过将上述制造方法、后述实施例中记载的方法、以及上述的专利文献4~6、非专利文献11等组合而制造。
上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在关于对μ、δ和κ阿片受体的功能活性的试验中,对阿片δ受体显示优异的激动活性和选择性(参照实施例40的表6)。
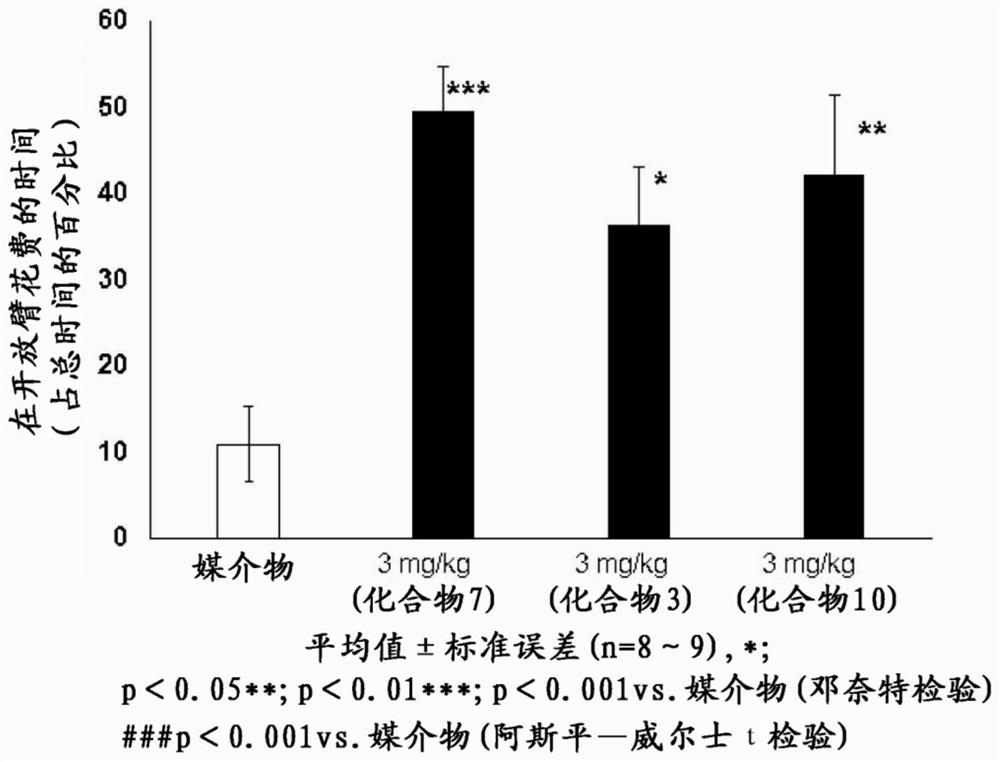
另外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在小鼠、大鼠高架式十字迷宫试验中显著地使开放臂(壁なし走行路)滞留时间百分比增加,发挥抗焦虑样作用(参照实施例41、42的图1~6)。应予说明,高架式十字迷宫试验依据非专利文献6中记载的方法进行。
另外,明确了上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在嗅球摘除模型(OBX)大鼠的情绪过度反应抑制试验中具有优异的抗抑郁作用。(实施例44)
另外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在利血平诱发帕金森氏病模型小鼠中,启示了帕金森氏病的治疗效果。(实施例45)
另外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在使用大鼠脑梗塞诱导的膀胱活动过度模型的试验中,由于排尿间隔和1次排尿量显示用量依赖性地增加的趋势,因此,启示了被验物质的尿频改善作用。(实施例46、表8)
进而,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物如后述实施例43中记载那样,在hERG(人ether-a-go-go相关基因)钾通道抑制试验中仅显示弱的抑制作用。因此,表示上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物的人的心室再极化的延迟和QT间隔的延长的风险低。
此外,明确了上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物显示足以发挥药效的中枢迁移性,在使用人肝微粒体的代谢稳定性试验中,发现化合物显示高稳定性,明确了是能够口服的具有抗焦虑作用、抗抑郁作用、镇痛作用、抗帕金森作用、尿频·尿失禁改善作用的化合物。应予说明,使用人肝微粒体的代谢稳定性可以通过如下操作进行评价:在人肝微粒体中添加已知量的被验化合物,培养一定时间后,使用LC(液相色谱法)等对化合物量进行定量。(实施例47,表9)
因此,若考虑上述的专利文献1~6、非专利文献1~10等,则上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物可以用于抑郁或焦虑的治疗和预防,可以作为DSM-5(美国精神医学会:精神障碍的诊断和统计手册第5版)中所记载的抑郁障碍组、焦虑障碍组、双相障碍组、强迫性障碍和相关障碍组、心理创伤和应激相关障碍组等所含的精神疾病的预防和治疗药(抗抑郁药、抗焦虑药等)以及尿失禁、心肌缺血、脑缺血、慢性咳嗽、高血压、帕金森氏病、癫痫等神经退行性疾病的预防和治疗药使用。
另外,如IOVS,March 2013,Vol.54,No.3;J.Neurochem.(2009)108,741-754等中所记载那样提出了阿片δ激动剂对青光眼的应用。因此,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物可以作为青光眼的预防或治疗药使用。
在本说明书中,抑郁可以为抑郁感、悲哀感、孤独感等情绪障碍、活动意愿降低、思考迟滞、悲观的意念以及兼具睡眠障碍、食欲降低等自主神经障碍的状态。另外,在本说明书中,焦虑可以为在未与可明显确认的刺激相关联的情况下,伴随不安、紧张、心动过速、呼吸困难等,感觉到危险、恐怖的状态。抑郁、焦虑包含上述DSM-5所记载的精神疾病中所见到的抑郁、焦虑的症状(例如,双相障碍组中所见到的抑郁症状、PTSD中所见到的抑郁、焦虑症状)、虽然比DSM-5中所记载的抑郁障碍组症状轻但某种程度上持续的抑郁的状态、以及虽然比DSM-5中所记载的焦虑障碍组症状轻但某种程度上持续的状态。
进而,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物也可以作为辅助上述疾病的治疗的药剂使用。
另外,上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物可以作为伴有急性疼痛和慢性疼痛的疾病的疼痛治疗、风湿性关节炎、变性关节炎、骨瘤等伴有剧烈疼痛的癌性疼痛、糖尿病性神经障碍性疼痛、带状疱疹后神经痛、内脏的疼痛等的预防和治疗药使用。
优选上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物被期待作为抗抑郁药、抗焦虑药。
上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物可以通过口服或非口服这样的适当的给予方法对人进行给予。另外,也可以与其它抗焦虑药、抗抑郁药、镇痛药并用。
为了进行制剂化,可以通过制剂技术领域中的通常的方法制造成片剂、颗粒剂、散剂、胶囊剂、悬浮剂、注射剂、栓剂等剂型。
对于它们的制备,例如在片剂的情况下,可使用通常的赋形剂、崩解剂、粘合剂、润滑剂、色素等。在此,作为赋形剂,可以举出乳糖、D-甘露醇、结晶纤维素、葡萄糖等,作为崩解剂,可以举出淀粉、羧甲基纤维素钙(CMC-Ca)等,作为润滑剂,可以举出硬脂酸镁、滑石等,作为粘合剂,可以举出羟丙基纤维素(HPC)、明胶、聚乙烯基吡咯烷酮(PVP)等。注射剂的制备可使用溶剂、稳定剂、增溶剂、悬浮剂、乳化剂、无痛化剂、缓冲剂、保存剂等。
给予量通常对成人而言,作为有效成分的上述通式(I)所示的化合物、该化合物的互变异构体、立体异构体、或者其药学上允许的盐或它们的溶剂化物在注射剂情况下给予0.1μg~1g/天,优选给予0.001~200mg/天,在口服情况下给予1μg~10g/天,优选给予0.01~2000mg/天,可以根据年龄、症状等而增减。
接着,举出参考例、实施例,对本发明进一步详细地进行说明,但本发明并不限定于这些参考例、实施例。
实施例化合物和参考例化合物的命名是将使用Cambridgesoft公司制ChemDrawver.14描绘的结构式通过该软件搭载的命名算法转换成英语名后进行日语翻译而得到的。
应予说明,实施例1~34的NMR数据、质量分析的实测值(ESI+或ESI-)示于表1~5。
实施例
参考例1-1
(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇的合成
在300mL的圆底烧瓶中加入通过专利文献WO2013/035833实施例67的方法合成的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-甲氧基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚(372mg,1.02mmol),溶解于二氯甲烷(5mL),在0℃剧烈搅拌20分钟后,加入1.0M三溴化硼/二氯甲烷溶液(5mL,5mmol),在室温下搅拌30分钟。在0℃在反应溶液中加入甲醇(10mL),在同温度下搅拌1小时。
将反应溶液在减压下浓缩,将残渣悬浮于氯仿(50mL),用6%氨水溶液(20mL)清洗。将水层用氯仿(30mL)萃取2次,将收集的有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩,以褐色泡沫的形式得到标题化合物(356mg,100%)。
[其它方法]
在500mL的圆底烧瓶中加入通过专利文献WO2013/035833实施例67的方法合成的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-甲氧基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚(3.58g,9.82mmol)和盐酸吡啶(87g,753mmol),在200℃搅拌1小时。反应后返回到室温,向生成的固体加入饱和碳酸钾水溶液使其溶解,用乙酸乙酯和氯仿萃取,使收集的有机层在无水硫酸钠上干燥。过滤出不溶物后,将滤液在减压下浓缩,以褐色泡沫的形式得到标题化合物(3.30g,96%)。
参考例1-2
(1S,3aR,5aS,6R,11bR,11cS)-10-((叔丁基二甲基甲硅烷基)氧基)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚的合成
在200mL的圆底烧瓶中加入通过参考例1-1的方法合成的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(694mg,1.98mmol),溶解于DMF(20mL),在室温下加入咪唑(241mg,3.54mmol)和叔丁基二甲基氯硅烷(498mg,3.31mmol),在室温下搅拌2小时。由于确认到在反应溶液中残留原料,因此,加入咪唑(529mg,7.77mmol)和叔丁基二甲基氯硅烷(503mg,3.34mmol),在室温下搅拌18小时。在反应溶液中加入水(150m),用乙酸乙酯和己烷的混合溶剂(1:1,100mL)萃取。在水层中加入6%氨水(30mL)形成碱性后,用乙酸乙酯和己烷的混合溶剂(1:1,100mL)萃取2次。将收集的有机层在无水硫酸镁上干燥后,过滤出不溶物,将滤液在减压下浓缩。将残渣利用以甲醇/氯仿(浓度梯度0-50%)、接着包含10%的浓氨水的甲醇/氯仿(浓度梯度20-50%)作为洗脱溶剂的柱色谱(硅胶,25g)进行精制,以黄色浆状物的形式得到标题化合物(456mg,50%),另外,得到原料的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(265mg,38%)。
实施例1
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物的合成
在50mL的圆底烧瓶中加入参考例1中合成的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(31mg,87μmol)、2-羧基吡啶1-氧化物(32mg,0.23mmol)和HATU(125mg,0.33mmol),悬浮于THF(1.5mL)后,加入三乙基胺(70μL,0.50mmol)和DMA(200μL),在室温下搅拌1小时。
在反应液中加入2当量氨/甲醇溶液(2mL),在同温度下搅拌1小时。
将反应溶液在减压下浓缩而得到的残渣悬浮于6%氨水,用乙酸乙酯萃取。将收集的有机层用饱和食盐水清洗后,在无水硫酸镁上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-50%)作为洗脱溶剂的柱色谱(氨基硅胶,16g),以白色固体的形式得到标题化合物(18mg,44%)。
实施例2
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(36mg,0.10mmol)、4-羧基吡啶1-氧化物(42mg,0.30mmol)、三乙基胺(70μL,0.50mmol)和HATU(108mg,0.28mmo)进行反应。将反应溶液直接供于以甲醇和含有5%三乙基胺的乙酸乙酯(浓度梯度:10%-50%)作为洗脱溶剂的柱色谱(硅胶,10g)进行精制。将得到的浆状物溶解于甲醇后,加入氯仿和叔丁基甲基醚进行粉末化后,滤取,以微褐色固体的形式得到标题化合物(30mg,62%)。
实施例3
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(39mg,0.11mmol)、2-氧代-1,2-二氢吡啶-3-羧酸(39mg,0.28mmol)、三乙基胺(70μL,0.50mmol)和HATU(130mg,0.34mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩,将残渣直接供于以甲醇和含有5%三乙基胺的乙酸乙酯(浓度梯度:10%-50%)作为洗脱溶剂的柱色谱(硅胶,10g)进行精制。将得到的残渣由6%氨水进行粉末化,以淡黄色粉末的形式得到标题化合物(13mg,25%)。
实施例4
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(34mg,97μmol)、3-羧基吡啶1-氧化物(40mg,0.29mmol)、三乙基胺(70μL,0.50mmol)和HATU(125mg,0.33mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩,将残渣直接供于以0.1当量氨/甲醇溶液和氯仿(浓度梯度:0%-50%)作为洗脱溶剂的柱色谱(硅胶,25g)进行精制。将得到的浆状物溶解于甲醇后,加入叔丁基甲基醚进行粉末化后,滤取,以微褐色无定形的形式得到标题化合物(14mg,31%)。
实施例5
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(34mg,96μmol)、6-氧代-1,6-二氢吡啶-3-羧酸(40mg,0.29mmol)、三乙基胺(70μL,0.50mmol)和HATU(132mg,0.35mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣直接供于以0.1当量氨/甲醇溶液和氯仿(浓度梯度:1%-50%)作为洗脱溶剂的柱色谱(硅胶,10g)进行精制。为了除去杂质,将得到的化合物悬浮于氯仿后,用6%氨水清洗。将水层用氯仿萃取后,将合并的有机层在无水硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩,以淡黄色粉末的形式得到标题化合物(14mg,30%)。
参考例2
1-甲基-2-氧代-1,2-二氢吡啶-3-羧酸的合成
本化合物通过基于WO2006/107254记载的方法的方法而合成。
在50mL的圆底烧瓶中加入2-氧代-1,2-二氢吡啶-3-羧酸(500mg,3.59mmol),使其悬浮于甲醇(5mL)和水(0.8mL)后,加入氢氧化钾(400mg,7.13mmol),在100℃下搅拌15分钟。将反应溶液返回到室温,加入碘甲烷(2.6mL,41.8mmol),在100℃下搅拌45分钟后,在减压下浓缩至溶剂量成为一半。在反应溶液中加入3当量盐酸(20mL),将产生的固体过滤,用水和乙腈清洗后,使其在减压下干燥,由此以白色粉末的形式得到标题化合物(64.9mg,12%)。
实施例6
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(30mg,86μmol)、参考例2中合成的1-甲基-2-氧代-1,2-二氢吡啶-3-羧酸(29mg,0.19mmol)、二异丙基乙基胺(75μL,0.43mmol)和HATU(72mg,0.19mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以1.4当量氨/甲醇溶液-氯仿(浓度:5%)作为展开溶剂的制备型TLC,以淡黄色无定形的形式得到标题化合物(26.2mg,63%)。
实施例7
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(66mg,0.19mmol)、6-氧代-1,6-二氢吡啶-2-羧酸(83mg,0.59mmol)、三乙基胺(150μL,1.10mmol)和HATU(262mg,0.69mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣直接供于以甲醇和氯仿(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,10g)进行精制。将得到的浆状物溶解于甲醇,加入叔丁基甲基醚进行粉末化,以褐色固体得到标题化合物(83mg,94%)。
实施例8
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-6-甲基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、6-甲基-2-氧代-1,2-二氢吡啶-3-羧酸(19mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(48mg,0.13mmol)进行反应。其中,作为溶剂,使用DMF代替THF和DMA。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩,将残渣供于以1.4当量氨/甲醇溶液-氯仿(浓度:10%)作为展开溶剂的制备型TLC进行精制后,为了进一步除去杂质,将得到的固体悬浮于饱和碳酸钾水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥后,过滤出无机物,将滤液在减压下浓缩,得到标题化合物。得到的化合物为了提供于生物活性试验而依照实施例32形成盐酸盐。
实施例9
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(30mg,86μmol)、1-甲基-6-氧代-1,6-二氢吡啶-3-羧酸(29mg,0.19mmol)、二异丙基乙基胺(75μL,0.43mmol)和HATU(72mg,0.19mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:10%)作为展开溶剂的制备型TLC,以白色无定形的形式得到标题化合物(31.1mg,75%)。
参考例3
1-甲基-6-氧代-1,6-二氢吡啶-2-羧酸的合成
在50mL的圆底烧瓶中加入6-氧代-1,6-二氢吡啶-2-羧酸(500mg,3.59mmol),使其悬浮于甲醇(5mL)和水(0.8mL)后,加入氢氧化钾(400mg,7.13mmol),在100℃搅拌15分钟。将反应溶液返回到室温,加入碘甲烷(2.6mL,41.8mmol),在100℃搅拌1小时后,在减压下浓缩至溶剂量成为一半。在反应溶液加入3当量盐酸,将产生的固体过滤,用水和乙腈清洗后,在减压下干燥,以白色粉末的形式得到标题化合物(339mg,62%)。
实施例10
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(30mg,86μmol)、通过参考例3的方法合成的1-甲基-6-氧代-1,6-二氢吡啶-2-羧酸(29mg,0.19mmol)、二异丙基乙基胺(75μL,0.43mmol)和HATU(72mg,0.19mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:10%)作为展开溶剂的制备型TLC,以白色无定形的形式得到标题化合物(32.7mg,79%)。
实施例11
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(54mg,0.15mmol)、2-甲氧基异烟酸(54mg,0.35mmol)、三乙基胺(140μL,1.00mmol)和HATU(195mg,0.51mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于氯仿后,用6%氨水清洗。将水层用氯仿萃取,将合并的有机层在无水硫酸镁上干燥后,过滤出不溶物,将滤液在减压下浓缩。将得到的残渣供于以含有10%浓氨水的甲醇和氯仿作为洗脱溶剂的柱色谱(氨基硅胶,16g),以白色固体的形式得到((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-基)(2-甲氧基吡啶-4-基)甲酮(61mg,82%)。
在100mL圆底烧瓶中加入上述得到的((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-基)(2-甲氧基吡啶-4-基)甲酮(48mg,98μmol)、和吡啶盐酸盐(2.88g,25mmol),在200℃加热搅拌10分钟。将反应溶液冷却至室温后,悬浮于6%氨水,用乙酸乙酯萃取。将收集的有机层在无水硫酸镁上干燥后,过滤出不溶物,将滤液在减压下浓缩。将残渣供于以甲醇和氯仿(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,8g),以白色固体的形式得到标题化合物(35mg,75%)。
实施例12
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-2,4(1H,3H)-二酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(32mg,90μmol)、2,4-二氧代-1,2,3,4-四氢嘧啶-5-羧酸·一水合物(35mg,0.20mmol)、三乙基胺(70μL,0.50mmol)和HATU(114mg,0.30mmol)进行反应,在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩,将得到的残渣悬浮于饱和碳酸氢钠水溶液,用氯仿和甲醇的5:1混合溶液萃取3次。将收集的有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以含有10%浓氨水的甲醇和氯仿(浓度:25%)作为展开溶剂的制备型TLC,以白色固体的形式得到标题化合物(16mg,35%)。
实施例13
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-4(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(32mg,90μmol)、4-氧代-1,4-二氢吡啶-3-羧酸(28mg,0.20mmol)、三乙基胺(70μL,0.50mmol)和HATU(114mg,0.30mmol)进行反应,在反应溶液中加入2当量氨/甲醇溶液,停止反应后,悬浮于饱和碳酸氢钠水溶液,用乙酸乙酯萃取3次。将收集的有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以含有10%浓氨水的甲醇和氯仿(浓度:15%)作为展开溶剂的制备型TLC,以白色固体的形式得到标题化合物(19mg,44%)。
实施例14
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-4(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(32mg,90μmol)、4-氧代-1,4-二氢吡啶-2-羧酸(28mg,0.20mmol)、三乙基胺(70μL,0.50mmol)和HATU(114mg,0.30mmol)进行反应,在反应溶液中加入2当量氨/甲醇溶液,停止反应后,悬浮于饱和碳酸氢钠水溶液,用氯仿和甲醇的5:1混合溶液萃取3次。将收集的有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以含有10%浓氨水的甲醇和氯仿(浓度:15%)作为展开溶剂的制备型TLC,以白色固体的形式得到标题化合物(8mg,20%)。
实施例15
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(32mg,90μmol)、1-甲基-2-氧代-1,2-二氢吡啶-4-羧酸(31mg,0.20mmol)、三乙基胺(70μL,0.50mmol)和HATU(114mg,0.30mmol)进行反应,在反应溶液中加入2当量氨/甲醇溶液,停止反应后,悬浮于饱和碳酸氢钠水溶液,用氯仿萃取3次。将收集的有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:5%)作为展开溶剂的制备型TLC,以白色固体的形式得到标题化合物(41mg,94%)。
实施例16
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)哒嗪-3(2H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(30mg,85.9μmol)、6-氧代-1,6-二氢哒嗪-3-羧酸(31mg,0.22mmol)、三乙基胺(70μL,0.50mmol)和HATU(129mg,0.34mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于6%氨水后,用乙酸乙酯萃取,将有机层在无水硫酸镁上干燥。过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,16g),以白色固体的形式得到标题化合物(27mg,66%)。
实施例17
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)喹啉-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(33mg,95μmol)、2-氧代-1,2-二氢喹啉-4-羧酸(50mg,0.26mmol)、三乙基胺(70μL,0.50mmol)和HATU(128mg,0.34mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于6%氨水后,用乙酸乙酯萃取,将有机层在无水硫酸镁上干燥。过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,16g),以白色固体的形式得到标题化合物(28mg,56%)。
实施例18
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-2H-吡喃-2-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、2-氧代-2H-吡喃-5-羧酸(18mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(48mg,0.13mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应开始1小时后,在反应溶液中加入1当量盐酸并进一步搅拌。在反应溶液中加入碳酸钾水溶液,停止反应后,用氯仿萃取,将有机层在硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:5%)作为展开溶剂的制备型TLC,以褐色无定形的形式得到标题化合物(4.0mg,15%)。
实施例19
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-4H-吡喃-4-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、4-氧代-4H-吡喃-2-羧酸(18mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(48mg,0.13mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入2当量甲基胺/甲醇溶液(0.3mL,0.6mmol),停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:10%)作为展开溶剂的制备型TLC,以褐色无定形的形成得到标题化合物(4.4mg,16%)。
实施例20
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-4(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、4-氧代-4H-吡喃-2-羧酸(18mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(48mg,0.13mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入2当量甲基胺/甲醇溶液(3.0mL,6.0mmol),停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸钾水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-10%)作为洗脱溶剂的柱色谱(氨基硅胶,8g),以微褐色无定形的形式得到标题化合物(19mg,68%)。
实施例21
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡嗪-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、5-氧代-4,5-二氢吡嗪-2-羧酸(18mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(48mg,0.13mmol)进行反应。其中,作为溶剂,使用二氯甲烷代替THF和DMA。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于碳酸钾水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:5%-30%)作为洗脱溶剂的柱色谱(硅胶,10g),以微褐色无定形的形式得到标题化合物(12.2mg,45%)。
实施例22
2-((1S,3aR,5aS,6R,11bR,11cS)-10-乙酰氧基-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物的合成
在10mL的试管中加入实施例1中合成的2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物(52mg,0.11mmol),悬浮于THF(1mL)后,加入三乙基胺(45μL,0.32mmol)和乙酰氯(15μL,0.21mmol),在室温下搅拌1小时。由于确认到在反应液中残留原料,因此,再次加入三乙基胺(45μL,0.32mmol)和乙酰氯(15μL,0.21mmol),在室温下搅拌1小时。在反应溶液中加入饱和碳酸氢钠水溶液和乙酸乙酯,剧烈搅拌20分钟后,将水层分离,用乙酸乙酯萃取。将收集的有机层在无水硫酸镁上干燥后,过滤出不溶物,将滤液在减压下浓缩,以黄色无定形的形式得到标题化合物(51mg,89%)。
实施例23
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用通过专利文献WO2013/035833中记载的化合物297(实施例228)的方法制备的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚(27mg,79μmol)、6-氧代-1,6-二氢吡啶-2-羧酸(18mg,0.16mmol)、三乙基胺(50μL,0.36mmol)和HATU(70mg,0.18mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于6%氨水后,用乙酸乙酯萃取,将有机层在无水硫酸镁上干燥。过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-20%)作为洗脱溶剂的柱色谱(氨基硅胶,8g)。将得到的化合物溶解于甲醇,加入叔丁基甲基醚进行粉末化,以白色固体的形式得到标题化合物(24mg,67%)。
参考例4
3-氧代-3,4-二氢吡嗪-2-羧酸的合成
本化合物通过专利文献WO2009/033084的方法进行合成,1HNMR谱与文献Syn.Commun.2010.40(20).2988-2999记载的数据一致。
在50mL的圆底烧瓶中加入3-氨基吡嗪-2-羧酸(300mg,2.17mmol)和浓硫酸(1.3mL),在冰浴下,滴加溶解于浓硫酸(1.6mL)的亚硝酸钠(149mg,2.16mmol)后,搅拌1小时。将反应溶液加入到冰水中,剧烈搅拌,滤取产生的固体。将得到的固体在减压下在60℃干燥1小时,由此以淡黄色晶体的形式得到标题化合物(166mg,55%)。
实施例24
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡嗪-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、参考例4中合成的3-氧代-3,4-二氢吡嗪-2-羧酸(20mg,0.14mmol)进行反应。其中,使用HOAt(17mg,0.13mmol)代替三乙基胺,使用WSC(24mg,0.13mmol)代替HATU,以及使用DMF代替THF作为溶剂。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,用氯仿萃取,用饱和氯化氨水溶液清洗,接着用饱和碳酸氢钠水溶液清洗。将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:20%)作为展开溶剂的制备型TLC,以淡黄色无定形的形式得到标题化合物(5.9mg,22%)。
实施例25
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-2,4(1H,3H)-二酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、2,6-二氧代-1,2,3,6-四氢嘧啶-4-羧酸(20mg,0.13mmol)进行反应。其中,使用HOAt(17mg,0.13mmol)代替三乙基胺,使用WSC(24mg,0.13mmol)代替HATU,以及使用DMF代替THF作为溶剂。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:5%-30%)作为洗脱溶剂的柱色谱(硅胶,10g)。为了除去杂质,使得到的化合物悬浮于氯仿和氨水后,滤取,以微褐色无定形的形式得到标题化合物(2.5mg,9%)。
参考例5
1-乙基-6-氧代-1,6-二氢吡啶-2-羧酸的合成
在30mL圆底烧瓶中加入6-氧代-1,6-二氢吡啶-2-羧酸(129mg,925μmol)和1,1-二乙氧基-N,N-二甲基甲胺(1.5mL),在100℃搅拌2小时。将反应溶液冷却至室温后,在减压下浓缩。将残渣供于以甲醇和氯仿(浓度梯度0%-20%)作为洗脱溶剂的柱色谱(硅胶,10g),以无色油状物质的形式得到1-乙基-6-氧代-1,6-二氢吡啶-2-羧酸乙酯(104mg,58%)。
将上述得到的1-乙基-6-氧代-1,6-二氢吡啶-2-羧酸乙酯(104mg,533μmol)加入到50mL的圆底烧瓶中,溶解于乙醇(3mL)后,加入5当量氢氧化钠水溶液(200μL,1.0mmol),在55℃搅拌2小时。将反应溶液放冷至室温后,加入5当量盐酸(400μL,2.0mmol)形成酸性后,在减压下浓缩。在残渣中加入乙醇(3mL),在减压下浓缩。将残渣悬浮于乙醇(3mL)后,过滤出不溶物,将滤液在减压下浓缩,以无色结晶性固体的形式得到标题化合物(48mg,54%)。
实施例26
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-乙基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(32mg,92μmol)、参考例5中合成的1-乙基-6-氧代-1,6-二氢吡啶-2-羧酸(33mg,0.19mmol)、三乙基胺(70μL,0.50mmol)和HATU(136mg,0.36mmol)进行反应。在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于6%氨水后,用乙酸乙酯萃取,将有机层在无水硫酸镁上干燥。过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-20%)作为洗脱溶剂的柱色谱(氨基硅胶,8g)。将得到的化合物溶解于甲醇,加入叔丁基甲基醚进行粉末化,以白色固体的形式得到标题化合物(35mg,76%)。
实施例27
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)嘧啶-4(3H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-10-((叔丁基二甲基甲硅烷基)氧基)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚(30mg,65μmol)、6-氧代-1,6-二氢嘧啶-4-羧酸(20mg,0.14mmol)进行反应。其中,使用HOAt(19mg,0.14mmol)代替三乙基胺,使用WSC(27mg,0.14mmol)代替HATU,以及使用DMF代替THF作为溶剂。将残渣悬浮于水后,用乙酸乙酯萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-10%)作为洗脱溶剂的柱色谱(硅胶,10g)。
在100mL圆底烧瓶中加入上述得到的固体、甲醇(2mL)和氨水溶液,在室温下搅拌3天。将反应溶液浓缩后,将残渣悬浮于氯仿后,过滤出不溶物,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度:20%)作为展开溶剂的制备型TLC,以白色无定形的形式得到标题化合物(1.7mg,6%)。
参考例6
1-乙基-6-氧代-1,6-二氢吡啶-3-羧酸的合成
对溶解于二氯甲烷(3.3mL)和THF(3.3mL)的2-氧代-2H-吡喃-5-羧酸(200mg,1.43mmol)和DMAP(17.5mg,143μmol)加入WSC(274mg,1.43mmol)和苄醇(148μL,1.43mmol),在室温下搅拌2小时。在反应溶液中加入水,过滤出不溶物后,用己烷萃取,用饱和碳酸氢钠水溶液清洗。将收集的有机层在硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩。
使得到的残渣与乙基胺盐酸盐(112mg,1.37mmol)一起溶解于甲醇(10mL),加入三乙基胺(520μL,3.73mmol),在室温下搅拌16小时。反应后在减压下浓缩,在得到的残渣中加入饱和碳酸氢钠水溶液,用氯仿萃取,用饱和食盐水清洗。将收集的有机层在无水硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩。将残渣供于以乙酸乙酯和己烷(浓度梯度10%-60%)作为洗脱溶剂的硅胶柱色谱(10g),以淡黄色无定形的形式得到1-乙基-6-氧代-1,6-二氢吡啶-3-羧酸苄酯(126mg,2步骤34%)。
使上述得到的1-乙基-6-氧代-1,6-二氢吡啶-3-羧酸苄酯溶解于甲醇(2mL)和乙酸乙酯(2mL),加入10%钯碳后,在氢环境下在室温下搅拌2小时。反应后将不溶物进行硅藻土过滤,将得到的溶液浓缩,以淡黄色无定形的形式得到标题化合物(73mg,89%)。
实施例28
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-乙基吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(15mg,43μmol)、参考例6中合成的1-乙基-6-氧代-1,6-二氢吡啶-3-羧酸(16mg,94μmol)、二异丙基乙基胺(37μL,0.21mmol)和HATU(36mg,94μmol)进行反应。其中,作为溶剂,仅使用THF。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在无水硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(硅胶,10g),以白色无定形的形式得到标题化合物(13.3mg,62%)。
实施例29
2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物盐酸盐的合成
在50mL的圆底烧瓶中加入实施例1中合成的2-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶1-氧化物(79mg,0.17mmol),溶解于乙醇(2mL)后,加入2当量盐酸(1mL),将得到的溶液在减压下浓缩。将得到的残渣在减压下在80℃下干燥18小时,以白色无定形的形式得到标题化合物(85mg,99%)。
实施例30
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮盐酸盐的合成
在50mL的圆底烧瓶中加入实施例3中合成的3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮(44mg,93μmol),溶解于2当量盐酸(2mL),将得到的溶液在减压下浓缩。将得到的残渣在减压下在100℃下干燥18小时,以黄色固体的形式得到标题化合物(40mg,84%)。
实施例31
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮盐酸盐的合成
在10mL的试管中加入实施例6中合成的3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮(26mg,54μmol)和乙酸乙酯,使用1当量盐酸进行萃取,将水层在减压下浓缩。将得到的残渣在减压下在60℃下干燥1小时,以淡黄色无定形的形式得到标题化合物(23mg,83%)。
实施例32
3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-6-甲基吡啶-2(1H)-酮盐酸盐的合成
在10mL的试管中加入实施例8中合成的3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-6-甲基吡啶-2(1H)-酮和乙酸乙酯,使用1当量盐酸进行萃取,将水层在减压下浓缩。将得到的残渣在减压下干燥,以淡黄色无定形的形式得到标题化合物(11mg,从实施例8起2工序39%)。
实施例33
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮盐酸盐的合成
在10mL的试管中加入实施例9中合成的5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮(31mg,64μmol)和乙酸乙酯,使用1当量盐酸进行萃取,将水层在减压下浓缩。将得到的残渣在减压下在60℃下干燥2小时,以淡黄色无定形的形式得到标题化合物(22mg,67%)。
实施例34
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮盐酸盐的合成
在10mL的试管中加入实施例10中合成的6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基吡啶-2(1H)-酮(33mg,67μmol)和乙酸乙酯,使用1当量盐酸进行萃取,将水层在减压下浓缩。将得到的残渣在减压下在60℃下干燥2小时,以微褐色无定形的形式得到标题化合物(33mg,94%)。
[表1]
[表2]
[表3]
[表4]
[表5]
参考例7-1
(1S,3aR,5aS,6R,11bR,11cS)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羧酸2,2,2-三氯乙酯的合成
在100mL的茄形烧瓶中加入通过WO2014136305、实施例34(1)记载的方法合成的(1S,3aR,5aS,6R,11bR,11cS)-10-甲氧基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羧酸2,2,2-三氯乙酯(972.7mg,2.00mmol),溶解于二氯甲烷(20mL)。将反应溶液冷却至0℃后,一边剧烈搅拌一边加入1M三溴化硼/二氯甲烷溶液(6mL)后,一边升温至室温一边搅拌1小时。
在反应溶液中加入饱和碳酸氢钠水溶液(30mL)后,用氯仿(20mLx3)进行萃取。将收集的有机层在无水硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩,以白色泡沫状物质的形式得到标题化合物(1.04g,>100%)。粗产物在不进行以上的精制的情况下直接用于以下的反应。
参考例7-2
(1S,3aR,5aS,6R,11bR,11cS)-10-羟基-14-(2,2,2-三氟乙酰基)-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羧酸2,2,2-三氯乙酯的合成
在100mL的茄形烧瓶中加入参考例7-1中合成的(1S,3aR,5aS,6R,11bR,11cS)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羧酸2,2,2-三氯乙酯(1.04g),溶解于THF(20mL)。在得到的溶液中加入三乙基胺(2.79mL,20mmol)和三氟乙酸酐(1.41mL,10mmol),在室温下搅拌1小时。将反应溶液在减压下浓缩。将残渣用饱和碳酸氢钠水溶液(50mL)稀释后,用乙酸乙酯(30mLx2)进行萃取。将收集的有机层在无水硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩,以白色泡沫状物质的形式得到标题化合物(1.46g,>100%)。粗产物不在进行以上的精制的情况下直接用于以下的反应。
参考例7-3
2,2,2-三氟-1-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-14-基)乙烷-1-酮的合成
在100mL的茄形烧瓶中加入参考例7-2中合成的(1S,3aR,5aS,6R,11bR,11cS)-10-羟基-14-(2,2,2-三氟乙酰基)-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羧酸2,2,2-三氯乙酯(1.46g),溶解于乙酸(25mL)。在得到的溶液中加入锌粉末(1.31g,20mmol),在室温下搅拌2小时。将反应溶液进行硅藻土过滤,馏去过量的锌粉末。将滤液在减压下浓缩后,与甲苯共沸。将残渣用饱和碳酸氢钠水溶液(30mL)稀释后,用氯仿(30mLx3)进行萃取。将收集的有机层在无水硫酸钠上干燥后、过滤出不溶物,将滤液在减压下浓缩。将得到的残渣供于以乙酸乙酯和甲醇(浓度梯度0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶:16g),以淡黄色泡沫状物质的形式得到标题化合物(215mg,3工序总收率27%)。
1H NMR CDCl
参考例8-1
3-氧代-2,3-二氢-1H-吡唑-4-羧酸乙酯的合成
本化合物的合成依照WO2011/090935记载的方法来合成。
在500mL的茄形烧瓶中加入20%乙醇钠/乙醇溶液(60mL)和2-(乙氧基亚甲基)丙二酸乙酯(10.5mL,524mmol),在室温下搅拌10分钟。在得到的混合物中加入肼一水合物(5.1mL,104mmol),在80℃加热搅拌18小时后,将得到的黄色的悬浮液冷却到0℃。在剧烈搅拌的反应液中在同温度下缓慢地加入1N盐酸(180mL),得到黄色溶液。在得到的溶液中加入乙酸乙酯(150mL),在室温下搅拌1小时。将有机层分离后,将水层用乙酸乙酯(100mLx2)萃取。将收集的有机层在无水硫酸钠上干燥,过滤出不溶物。将滤液在减压下浓缩,将得到的残渣使用乙酸乙酯和己烷进行结晶化,以黄色晶体(互变异构体的混合物)的形式得到标题化合物(2.82g,35%)。质量分析ES M-H=155
参考例8-2
3-甲氧基-1-甲基-1H-吡唑-4-羧酸的合成
在50mL的圆底烧瓶中加入3-氧代-2,3-二氢-1H-吡唑-4-羧酸乙酯(200mg,1.28mmol)、碘甲烷(397μL,6.40mmol)和DMF(5mL),加入氢化钠(60%,分散于液体石蜡)(256mg,6.40mmol),在室温下搅拌22小时。在冰冷下,在反应溶液中加入水,用乙酸乙酯萃取3次,将收集的有机层在硫酸钠上干燥后,过滤出不溶物,将滤液在减压下浓缩。将残渣供于以乙酸乙酯和己烷(浓度梯度5%-60%)作为洗脱溶剂的硅胶柱色谱(25g),以白色固体的形式得到3-甲氧基-1-甲基-1H-吡唑-4-羧酸乙酯(51mg,22%)。
在50mL的圆底烧瓶中加入上述得到的3-甲氧基-1-甲基-1H-吡唑-4-羧酸乙酯(51mg,0.279mmol),溶解于乙醇(1mL)后,加入5当量氢氧化钠水溶液(0.5mL,2.50mmol),在室温下搅拌3天。在反应溶液中加入1当量盐酸(2.7mL),将溶液在减压下浓缩。将得到的残渣溶解于THF,使用硅藻土过滤出不溶物,将滤液在减压下浓缩,由此以白色粉末的形式得到标题化合物(43mg,100%)。
1H NMR DMSO-d6,11.91(br s,1H),7.99(s,1H),3.80(s,3H),3.69(s,3H).
实施例35
6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
在10mL的试管中加入参考例7-3中合成的2,2,2-三氟-1-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-14-基)乙烷-1-酮(54mg,136μmol)、6-氧代-1,6-二氢吡啶-2-羧酸(67mg,0.48mmol)和HATU(197mg,0.52mmol),悬浮于THF(2mL)后,加入三乙基胺(100μL,0.72mmol)和DMA(100μL),在室温下搅拌1.5小时。
在反应液中加入乙醇胺(100μL)和甲醇(2mL),在同温度下搅拌1小时。
将反应溶液在减压下浓缩,将得到的残渣溶解于氯仿(30mL),用6%氨水(10mLx3)清洗。将收集的水层用氯仿(20mL)萃取。将收集的有机层在无水硫酸镁上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和氯仿(浓度梯度:10%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,16g),以白色泡沫状物质的形式得到6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-14-(2,2,2-三氟乙酰基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮(M+H=514.26)。
将上述得到的6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-14-(2,2,2-三氟乙酰基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮在100mL的茄形烧瓶中溶解于甲醇(5mL),加入硼氢化钠(124mg,3.26mmol),在室温下搅拌2小时。将反应溶液在减压下浓缩,将残渣悬浮于6%氨水(20mL),用氯仿(20mLx2)清洗。将水层在减压下浓缩,将残渣供于以甲醇和氯仿(浓度梯度:10%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,12g)进行精制,得到6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-14-(2,2,2-三氟乙酰基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮和标题化合物6-((1S,3aR,5aS,6R,11bR,11cS)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的混合物。
将得到的上述混合物在50mL的茄形烧瓶中溶解于浓氨水(3mL),使用橡胶栓以封管状态在80℃加热搅拌18小时。
将反应混合物在减压下浓缩,将残渣供于以甲醇和氯仿(浓度梯度:10%-50%)作为洗脱溶剂的柱色谱(氨基硅胶,7g)。将得到的粗产物使用甲醇(0.2mL)和叔丁基甲基醚(3mL)进行粉末化,得到标题化合物(23mg,41%)。
1H NMR DMSO-d
实施例36
4-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1-甲基-1,2-二氢-3H-吡唑-3-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(30mg,86μmol)、3-甲氧基-1-甲基-1H-吡唑-4-羧酸(29mg,0.19mmol)、二异丙基乙基胺(75μL,0.43mmol)和HATU(72mg,0.19mmol)进行反应。其中,仅使用THF(2mL)作为溶剂。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和乙酸乙酯(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(硅胶,10g)进行精制,以淡黄色无定形的形式得到((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-基)(3-甲氧基-1-甲基-1H-吡唑-4-基)甲酮(33.3mg,80%)。
1H NMR CD
7.69(s,0.7H),7.55(s,0.3H),6.90-6.96(m,1H),6.63(d,0.7H,J=2.8Hz),6.53-6.58(m,1.3H),2.78-5.02(m,8H),3.90(s,3H),3.73(s,2.1H),3.68(s,0.9H),2.53-2.57(m,1H),2.31-2.33(m,2H),1.90-2.09(m,2H),1.66-1.76(m,1H),1.51-0.78(m,7H),0.45-0.48(m,2H),0.09-0.12(m,2H).
在30mL圆底烧瓶中加入上述得到的((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-1,2,3a,4,5,6,7,11c-八氢-3H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-基)(3-甲氧基-1-甲基-1H-吡唑-4-基)甲酮(15mg,31μmol),溶解于二氯甲烷(1mL),在冰冷下加入1.0M三溴化硼/二氯甲烷溶液(153μL,0.15mmol),在室温下搅拌1小时。加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以含有氨水的甲醇和氯仿(浓度:10%)作为展开溶剂的制备型TLC,以淡黄色无定形的形式得到标题化合物(10.6mg,73%)。
1H NMR DMSO-d
11.47(s,0.1H),11.37(s,0.9H),9.11(s,1H),8.09(s,0.9H),7.48(s,0.1H),6.94(d,1H,J=8.2Hz),6.60(d,1H,J=2.3Hz),6.54(dd,1H,J=8.2,2.3Hz),4.33-4.50(m,1H),2.50-4.07(m,12H),2.19-2.34(m,2H),1.80-2.00(m,2H),1.58-1.65(m,1H),0.70-1.43(m,6H),0.38-0.53(m,2H),0.02-0.16(m,2H).
实施例37
5-氯-3-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(20mg,57μmol)、5-氯-2-氧代-1,2-二氢吡啶-3-羧酸(22mg,0.13mmol)、二异丙基乙基胺(50μL,0.29mmol)和HATU(72mg,0.13mmol)进行反应。其中,作为溶剂,仅使用THF(1mL)。在反应溶液中加入1.4当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于饱和碳酸氢钠水溶液后,用氯仿萃取,将有机层在硫酸钠上干燥,过滤出不溶物后,将滤液在减压下浓缩。将得到的残渣供于以甲醇和乙酸乙酯(浓度梯度:0%-80%)作为洗脱溶剂的柱色谱(氨基硅胶,8g)进行精制,以褐色无定形的形式得到标题化合物(11.6mg,40%)。
1H NMR DMSO-d
11.99(br s,1H),9.06(br s,1H),7.68(s,0.7H),7.59(s,0.3H),7.48(d,1H,J=2.3Hz),6.89(d,0.7H,J=8.2Hz),6.85(d,0.3H,J=8.2Hz),6.40-6.56(m,2H),4.25-4.32(m,0.7H),3.93-3.98(m,0.3H),3.78-3.84(m,0.3H),2.11-3.62(m,10.7H),1.68-1.91(m,2H),1.48-1.63(m,1H),0.87-1.46(m,4H),0.50-0.79(m,2H),0.29-0.47(m,2H),0.06-0.12(m,2H).
实施例38
5-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-羟基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)-1,3-二甲基嘧啶-2,4(1H,3H)-二酮的合成
依照与实施例1同样的方法,使用(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-10-醇(35mg,98μmol)、1,3-二甲基-2,4-二氧代-1,2,3,4-四氢嘧啶-5-羧酸(35mg,0.19mmol)、三乙基胺(70μL,0.50mmol)和HATU(145mg,0.38mmol)进行反应后,在反应溶液中加入2当量氨/甲醇溶液,停止反应后,在减压下浓缩。将残渣悬浮于6%氨水(20mL),用乙酸乙酯(15mLx2)进行萃取。将收集的有机层用饱和食盐水(10mL)清洗后,在无水硫酸镁上干燥。过滤出不溶物后,将滤液在减压下浓缩。将残渣供于以甲醇和乙酸乙酯(浓度梯度:0%-30%)作为洗脱溶剂的柱色谱(氨基硅胶,10g)进行精制。将得到的浆状物状物质溶解于甲醇(0.2mL)后,加入叔丁基甲基醚(3mL)进行粉末化后,滤取,以白色粉末的形式得到标题化合物(39mg,76%)。
1H NMR CD
7.82(s,1H),6.92-6.98(m,1H),6.52-6.65(m,2H),4.53-4.62(m,1H),4.02-4.18(m,1H),3.50-3.80(m,2H),3.42(s,2H),3.37(s,1H),3.33(s,2H),3.31(s,1H),2.81-3.18(m,5H),2.57-2.59(m,1H),2.30-2.38(m,2H),1.93-2.09(m,2H),1.67-1.78(m,1H),1.43-1.59(m,2H),1.10-1.29(m,2H),0.81-0.95(m,2H),0.44-0.53(m,2H),0.08-0.17(m,2H).
实施例39
6-((1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-甲氧基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚-3-羰基)吡啶-2(1H)-酮的合成
实验依照与实施例1同样的方法进行。
使用依照WO2013035833记载的实施例67的方法制备的(1S,3aR,5aS,6R,11bR,11cS)-14-(环丙基甲基)-10-甲氧基-2,3,3a,4,5,6,7,11c-八氢-1H-6,11b-(桥亚胺基桥亚乙基)-1,5a-亚甲基萘并[1,2-e]吲哚(82mg,0.23mmol)、三乙基胺(200μL,1.43mmol)和HATU(167mg,0.44mmol)进行反应后,在反应溶液中加入乙醇胺(200μL)和甲醇(1mL),停止反应后,用乙酸乙酯(50mL)稀释后,用6%氨水(50mL)清洗。将水层用氯仿(30mLx2)萃取,将收集的有机层在无水硫酸钠上干燥。过滤出不溶物后,将滤液在减压下浓缩。将残渣供于以甲醇和乙酸乙酯(浓度梯度:10%-50%)作为洗脱溶剂的柱色谱(氨基硅胶,7g)进行精制。将得到的浆状物质溶解于甲醇(0.2mL)后,加入叔丁基甲基醚(3mL)进行粉末化。将得到的粉末在100℃在减压下干燥16小时,以白色无定形状物质的形式得到标题化合物(87mg,100%)。
1H NMR DMSO-d
7.5(br s,1H),6.97-7.03(m,1H),6.45-6.73(m,4H),4.40-4.45(m,0.7H),3.84-3.89(m,0.3H),3.69(s,3H),3.55-3.62(m,1H),2.95-3.22(m,4H),2.79-2.84(m,2H),2.13-2.62(m,4H),1.79-1.87(m,2H),1.26-1.60(m,3H),0.99-1.14(m,3H),0.70-0.74(m,1H),0.54-0.61(m,1H),0.39-0.40(m,2H),0.00-0.07(m,2H).
实施例40
调查本发明提供的化合物对μ、δ和κ阿片受体的功能活性。
方法:使用Lance Ultra cAMP试剂盒(Perkinelmer公司),依照规定的方法实施。在激动剂活性的评价中,将各人阿片受体(δ、μ和κ:登录号和目录号如下)表达CHO细胞和被验化合物分别在10μM毛喉素存在下在分析缓冲液(1×HBSS,1M HEPES,pH7.4,250mM IBMX(异丁基甲基黄嘌呤),7.5%BSA)中反应30分钟。接下来,添加试剂盒中的cAMP检测试剂,在1小时后使用EnVision平板读数器(Perkinelmer公司)进行时间分解荧光测定。被验化合物和各对照物(δ:SNC80、μ:DAMGO、κ:U-69593)在10
SNC80:
(+)-4-[(αR)-α-((2S,5R)-4-烯丙基-2,5-二甲基-1-哌嗪基)-3-甲氧基苄基]-N,N-二乙基苯甲酰胺
DAMGO:
[D-Ala
U-69593:
(+)-(5α,7α,8β)-N-甲基-N-[7-(1-吡咯烷基)-1-氧杂螺[4.5]癸-8-基]苯乙酰胺
δ:目录号CT4607,登录号NM_000911.2
μ:目录号CT4605,登录号NM_000914
κ:目录号CT4606,登录号NM_000912
(ChanTest公司)
[表6]
N.C.:由于在最高浓度(10μM)未达到最大反应,因此,未算出ED
*:由于在最高浓度未达到最大反应,因此,示出作为参考值的在最高浓度的反应率。
如表6所示,确认了本发明的化合物对阿片δ受体具有强效的激动剂活性且对μ和κ受体不具有激动剂活性或仅显示极弱的激动剂活性。
实施例41
(试验方法)
试验使用5-6周龄的C57BL/6N系雄性小鼠。在由开放臂(宽度6cm、长度30cm)和闭合臂(壁あり走行路)(宽度6cm、长度30cm、壁的高度15cm)构成的高度40cm的十字迷宫装置中,将小鼠朝向闭合臂侧放置,使其自发地侵入十字迷宫。被验物质溶解于生理盐水或0.005NHCl-生理盐水,在试验开始30分钟前给予到背部皮下。在试验开始时开始录像机的录像,将小鼠侵入十字迷宫的时刻作为试验开始,拍摄记录5分钟的探索行为。以影像为基础,求出在各臂的滞留时间,算出开放臂滞留时间百分比(%)。
(试验结果)
如图1和2所示,在本实验中,将化合物1(实施例1记载的化合物)和7(实施例7记载的化合物)分别以3mg/kg和10mg/kg皮下给予时显著地使开放臂滞留时间百分比增加,确认到显示抗焦虑样作用。另外,关于化合物3(实施例3记载的化合物)、9(实施例9记载的化合物)和10(实施例10记载的化合物),确认到开放臂滞留时间百分比的延长趋势(图3~5)
实施例42
使用大鼠高架式十字迷宫试验对本发明提供的化合物的抗焦虑作用进行调查。
(试验方法)
试验使用7-9周龄的Wistar系雄性大鼠。在由开放臂(宽度10cm、长度50cm)和闭合臂(宽度10cm、长度50cm、壁的高度30cm)构成的高50cm的十字迷宫装置中,将大鼠朝向闭合臂侧放置,使其自发地侵入十字迷宫,观察5分钟的探索行为。被验物质溶解于4.5%环糊精水溶液,在试验开始2小时前经口给予。试验数据使用视频图像行为解析软件(PanLab公司制的Smart3.0、PanLab S.L.)进行自动解析,算出开放臂滞留时间百分比(%)。
(试验结果)
如图6所示,在本实验中,化合物7(实施例7记载的化合物)、3(实施例3记载的化合物)和10(实施例10记载的化合物)以3mg/kg经口给予时,显著地使开放臂滞留时间百分比增加,确认到显示抗焦虑样作用。
实施例43
(试验方法)
试验使用hERG通道稳定表达CHO细胞(从Channelopathy Foundation公司购入),通过Port-a-Patch自动膜片钳装置(Nanion TechnoloGies)进行。hERG电流如下确认,即,将细胞的膜电位保持在-80mV后,以10秒1次的频率赋予+20mV 1.5秒的去极化脉冲、接着赋予-50mV 1.5秒的测试脉冲,通过由测试脉冲诱导的尾电流来确认。被验化合物溶解于细胞外液(137mM NaCl、4mM KCl、1.8mM CaCl
(试验结果)
将试验结果示于表7。
表中,化合物1、3、7、9、10分别为实施例1、3、7、9、10中记载的化合物。
由表7可知,试验化合物均仅显示弱的抑制作用。
另一方面,发现在WO2013/35833(专利文献4)记载的化合物中存在hERG抑制作用强的化合物。
[表7]
比较化合物1:WO2013/35833的实施例93(化合物104)
比较化合物2:WO2013/35833的实施例205(化合物267)
实施例44
(试验方法)
依照斋藤等的方法(Saitoh A,Yamada M,Yamada M,Takahashi K,Yamaguchi K,Murasawa H,Nakatani A,Tatsumi Y,Hirose N,Kamei J:Antidepressant-like effectsof the delta-opioid receptor agonist SNC80([(+)-4-[(alphaR)-alpha-[(2S,5R)-2,5-dimethyl-4-(2-propenyl)-1-piperazinyl]-(3-methoxyphenyl)methyl]-N,N-diethylbenzamide)in an olfactory bulbectomized rat model.BrainRes.20081208:160-169.],OBX大鼠通过在摘除大鼠嗅球部位的手术之后进行隔离饲养而制作。情绪过度反应性的评价在术后第14天的分组前和给予第1、4、7、10、14天的给予2小时后,依照五味田等制作的情绪过度反应性评价基准(五味田等:7-Chloro-1-methyl-5-phenyl-1H-1,5-benzodiazepine-2,4-(3H,5H:)-dione(Clobazam)的行为药理学以及脑波学性研究日药理志82,267(1983))进行评价。药物1天1次连续14天给予到皮下。应予说明,作为阳性对照物,使用作为选择性血清素再摄取抑制剂(SSRI)的氟西汀。另外,溶剂使用1%环糊精(CD)。
(试验结果)
以0.1mg/kg给予被验物质(上述实施例7记载的化合物)时,从给予第4天开始,与溶剂给予组相比,显著地使OBX大鼠的情绪过度反应性降低,在给予第7天,恢复至与假手术组大鼠相同程度的水平。以1mg/kg给予被验物质时,从给予第1天开始,与溶剂给予组相比,显著地使OBX大鼠的情绪过度反应性降低,在给予第4天,恢复至与假手术组大鼠相同程度的水平。另外,这些效果持续至第14天。另一方面,以10mg/kg给予氟西汀时,在给予第14天,与溶剂给予组相比,显著地使OBX大鼠的情绪过度反应性降低。
根据以上的研究,启示了被验物质与SSRI不同,有可能由单次给予显示抗抑郁样作用。进而,启示了被验物质的抗抑郁样作用没有产生耐性的可能性。
实施例45
(试验方法)
获得ICR系雄性小鼠(5周龄:日本SLC),在设定驯化期(5-12天)后使用。
PD模型的制作参考Hille等的报告(Exp Neurol.2001,172:189)来制作。通过在试验开始18-24小时前在腹腔内处置利血平(5mg/kg)而制作。试验通过如下操作实施:在当天皮下给予被验化合物,立即放入自发运动活动笼中,测定60分钟的移动距离。
(试验结果)
以10mg/kg给予被验物质(上述实施例7记载的化合物)时,显示探索行为的显著增加作用,另外,虽然不显著(P=0.16)但显示站立行为的增加趋势,因此,启示了被验物质有帕金森氏病的治疗效果。
实施例46
(试验方法)
使用8周龄的SD系雄性大鼠,在异氟醚吸入麻醉下制作暂时性大脑中动脉闭塞模型。第二天,再次在异氟醚吸入麻醉下将颈部切开小切口,在颈静脉留置给药用的导管,导向背部。另外,进行膀胱测压手术,将插入到膀胱内的插管的另一端导向背部,与swivel(シ一ベル)连接。
从脑缺血手术起4天后,实施在无麻醉无约束下的膀胱测压测定。膀胱内压在稳定期间测定后,静脉内给予媒介物,对被验物质给予前值测定约30分钟。然后,以约30分钟间隔从低用量依次累积静脉内给予被验物质,对各给予后值测定约30分钟。在给予前测定中,采用被判断为尿频(排尿间隔为10分钟以下)的动物,算出各时刻的静止时压力、排尿时压力、排尿间隔和1次排尿量。
(试验结果)
将测定结果示于表8。
由表8可知,被验物质(上述实施例7记载的化合物)在任一用量下均未对静止时压力和排尿时压力造成影响。另一方面,排尿间隔和1次排尿量显示用量依赖性增加的趋势,因此,启示了被验物质的尿频改善作用。
[表8]
平均值±标准误差(n=5)
实施例47
(试验方法)
使人肝微粒体和被验物质反应一定时间(0~60分钟),测定反应试样中的被验物质的未变化体残留量,求出残留率。将反应时间0小时的未变化体残留率设为100%,将培养后的残留率相对于时间作对数一直线图,求出回归直线(y=100e
CL
*:Davies,B.and Morris,T.:Physiological parameters in laboratoryanimals and humans.Pharm.Res.,10(7):1093-1095,1993.
(试验结果)
将试验结果示于表9。
[表9]
比较化合物1:WO2013/35833的实施例93(化合物104)
由表9可知本发明化合物具有优异的代谢稳定性。另一方面,发现在WO2013/35833(专利文献4)记载的化合物中存在代谢稳定性差的化合物。
符号说明
在图1~6中,纵轴表示在开放臂的滞留时间的比例,横轴表示被验药物和其给予量。