防粘连材料
文献发布时间:2023-06-19 10:14:56
技术领域
本发明涉及一种生物组织的防粘连材料(adhesion prevention material),详细而言,涉及一种以具有规定的疏水性基团的明胶衍生物为主剂的生物吸收性的防粘连材料。
背景技术
防粘连材料被定义为生物吸收性的合成材料,其以减轻手术后的粘连为目的,而在手术时直接用于应用部位(“特殊医疗材料及其材料价格(材料价格基准)”日本平成28年医疗卫生和社会保障部公告第402号),通常将这种材料称为“防粘连可吸收隔离物”。防粘连材料具有通过粘着于手术部位而被施用的片材型、和液态或者使用时被制备为液态,通过喷雾而被施用的喷雾型。作为前者的例子,有以透明质酸钠和羧甲基纤维素为主要成分的材料(Seprafilm(商标)、科研制药株式会社)、以明胶为主要成分的材料(Gelfilm(商标)、辉瑞制药株式会社、和专利文献1),作为后者的例子,有由N-羟基琥珀酰亚胺化糊精和碳酸钠/碳酸氢钠的2剂组成的材料(Adspray(商标)、泰尔茂株式会社)。
虽然需要使防粘连材料覆盖组织表面来对其进行保护,但是难以使薄膜状的材料按照手术部位的复杂形状来进行粘着。另外,Seprafilm一旦被水浸湿,其操作性会显著下降,因此,实际上无法修改位置。在这方面,喷雾型的材料能够按照手术部位,而大面积地进行施用。
然而,本发明人等一直在进行一种外科用密封胶的研发,该外科用密封胶通过喷雾将导入了疏水性基团的明胶衍生物(以下,有时称为“疏水化明胶”)和固化剂向组织进行施用(例如专利文献2、专利文献3)。该密封胶由于具有疏水性基团,而与组织的亲和性较高,与不具有该疏水基的明胶、纤维蛋白密封胶相比,具有显著高的密封强度。
现有技术文献
专利文献
专利文献1:日本发明专利公开公报特开2013-226166号
专利文献2:WO2014/112208号
专利文献3:日本发明专利授权特许第5995128号
发明内容
[发明所要解决的技术问题]
由于上述密封胶与组织的高粘接性,因此会担心其与手术部位以外的组织粘接。但是,令人吃惊的是,发现在该密封胶固化而成膜之后,表现出优异的防粘连效果,从而完成了本发明。
[用于解决技术问题的技术方案]
即,本发明为下述的一种防粘连材料:
由(1)含有明胶衍生物的第1剂,和
(2)含有该明胶衍生物的交联剂的第2剂组成,其中,
对于该明胶衍生物而言,
(a)包括由下式表示的结构:
GltnNH-CHR
在上式中,Gltn为明胶残基,R
(b)亚氨基/氨基(摩尔比)为1/99~30/70;
(c)重均分子量为10000~50000。
[发明效果]
由于防粘连材料能够通过喷雾而向组织进行施用,因此,即使复杂形状的手术部位也能够容易地进行施用。该防粘连材料的固化膜表现出优异的防粘连性。由于与手术部位的粘接性也较高,因此也能够将其作为粘接性防粘连材料,或者作为具有防粘连性的外科用密封胶来使用。另外,作为主剂的明胶衍生物能够在水性溶剂中制备,不仅在制造环境和体内中安全,而且能够利用一段工序简单地且以高产率来进行合成。
附图说明
图1是表示细胞粘接数的图表。
图2是细胞渗透性试验所使用的装置的示意图。
图3是表示细胞渗透率的图表。
图4a是手术后1周的手术部位的组织的截面照片。
图4b是手术后2周的手术部位的组织的截面照片。
图5是表示破裂强度的结果的图表。
图6是测定破裂强度后的试验片的截面照片。
图7是蛋白质渗透性试验所使用的装置的示意图。
图8是表示蛋白质渗透比率的图表。
图9是表示溶胀度的经时变化的图表。
图10是表示由酶文献实现的减重的图表。
具体实施方式
本发明的防粘连材料为由第1剂和第2剂组成的2剂型,其中,第1剂作为实质上形成固化膜的主剂,含有明胶衍生物;第2剂含有该明胶衍生物的交联剂(crosslinkingagent)。第1剂和第2剂被分别包装而供给,在使用时将它们混合。以下,对它们的详细情况进行说明。
<第1剂>
在本发明的防粘连材料中,第1剂含有明胶衍生物。该明胶衍生物具有通过亚氨基(imino group),即-NH-而键结的疏水性基团,其包括下式所示的结构。
GltnNH-CHR
在上式中,Gltn为明胶残基,R
在R
对于该明胶衍生物中的衍生化比率而言,以键结有烷基的亚氨基的相对于原料明胶中的氨基量的摩尔%计,为1~20摩尔%,优选为5~10摩尔%。换言之,得到的明胶衍生物中的亚氨基/氨基(摩尔比)为1/99~20/80,优选为5/95~10/90。该衍生化比率能够通过用2,4,6-硝基苯磺酸酸法(TNBS法)来定量原料明胶中的氨基和键结了烷基之后的氨基量,或者通过用NMR等进行烷基的鉴定和定量而求得。
原料明胶可以源自天然、或者通过合成、发酵或基因重组而获得的明胶中的任一种,优选使用源自猪、牛等动物、源自黄线狭鳕(walleye pollack:明太鱼)等鱼的明胶。另外,虽然可以为酸处理明胶、碱处理明胶、基因重组明胶中的任一种,但优选为碱处理明胶,更优选为低内毒素化明胶。另外,对于该明胶的分子量的范围而言,优选使明胶衍生物的重均分子量(Mw)为10000~100000的范围,更优选使其为10000~50000的范围。该分子量能够通过凝胶渗透色谱(GPC)来按照常规方法进行测定。
除了上述明胶衍生物以外,第1剂还可以含有未衍生化的明胶。作为该明胶,可以使用上述的各种明胶。未衍生化的明胶的量为与明胶衍生物的合计重量的0~99wt%,优选为0~50wt%。
第1剂还可以含有用于溶解或分散该明胶衍生物的水性溶剂。从便利性的方面出发,也可以将该明胶衍生物溶解或分散于该水性溶剂而作为水性液(以下,有时简称为“水溶液”)来供给。作为该水性溶剂,可以使用超纯水、生理盐水、含有硼酸、磷酸、碳酸等各种酸及其盐的缓冲液或者它们的混合物。优选使用pH8~11的硼酸缓冲液,更优选使用pH9~10的硼酸缓冲液。以明胶衍生物成为10~80wt/v%,优选成为15~30wt/v%的量来使用该水性溶剂。在含有未衍生化的明胶的情况下,与明胶衍生物的合计重量为成为上述浓度的量。
<第2剂>
在本发明中,第2剂为明胶衍生物的交联剂,通过交联而形成与水、血液等体液具有不溶性的结构体,例如膜。作为该交联剂,使用在分子中至少具有2个以上的、与明胶中的氨基、主要是侧链的伯氨基具有反应性的官能团的交联剂中的至少一种。作为交联剂的例子,可列举出用京尼平、N-羟基琥珀酰亚胺或者N-羟基磺基琥珀酰亚胺活化的多元酸、醛化合物、酸酐和二异硫氰酸酯。
作为多元酸,可例示出酒石酸、柠檬酸、苹果酸、戊二酸、谷氨酸、天冬氨酸、草酰乙酸、顺式-乌头酸、2-酮基戊二酸、聚酒石酸、聚柠檬酸、聚苹果酸、聚谷氨酸、聚天冬氨酸、羧甲基化糊精、羧甲基化葡聚糖、羧甲基化淀粉、羧甲基化纤维素、羧甲基化壳聚糖、羧甲基化支链淀粉等,可以使用将它们的羧基活性酯化的化合物,例如二琥珀酰亚胺基戊二酸酯(DSG)、二琥珀酰亚胺基辛二酸酯(DSS)、二琥珀酰亚胺基酒石酸酯(DST)等。
另外,可列举出聚乙二醇(polyethylene glycol)或者聚乙二醇醚的多元酸酯(polyethylene glycol ether polybasic acid ester)、且该多元酸的未与聚乙二醇反应的羧基的至少1个被活性酯化的化合物,例如4,7,10,13,16-五氧杂十九烷二酸双(N-琥珀酰亚胺基)酯和由下式表示的聚乙二醇双(琥珀酰亚胺基琥珀酸酯)(SS-PEG-SS):
[化学式1]
(n为Mw成为约10000~20000的数量);
还可列举出由下式表示的季戊四醇-聚乙二醇醚四琥珀酰亚胺基戊二酸酯(4S-PEG):
(n为Mw成为约3000~30000、优选约5000~27000、更优选约10000~20000的数量)。
作为醛化合物,可例示出在1个分子中导入了2个以上的醛基的醛基导入多糖类,例如醛基导入淀粉、醛基导入葡聚糖、醛基导入糊精和醛基导入透明质酸,作为酸酐,可例示出戊二酸酐、马来酸酐和琥珀酸酐,作为二异硫氰酸酯,可例示出六亚甲基二异硫氰酸酯等。在这些之中,优选使用上述活化聚乙二醇多元酸酯和醛基导入多糖类。
这些交联剂以如下量使用:相对于明胶衍生物的氨基1当量,该交联剂中的官能团例如用N-羟基琥珀酰亚胺活化的酯基成为0.2~3当量,优选成为0.2~2当量,更优选成为0.2~1.0当量,最优选成为0.2~0.6当量。也可以使用2种以上的交联剂的混合物,在该情况下将它们的合计当量设定为成为上述范围的量。
第2剂也可以还含有用于溶解该交联剂的水性溶剂。但是,优选用单独的容器供给该交联剂和该水性溶剂,在进行使用时,大约在使用前的2小时内,将两者适量混合而作为水性溶液(以下,有时简称为“水溶液”)使用。对于该水性溶剂,可以使用针对第1剂所述的水性溶剂。优选使用pH3~8的磷酸缓冲液,更优选使用pH4~6的磷酸缓冲液。最优选的是,以在将第1剂的水溶液与第2剂的水溶液以同体积混合时pH成为8~10的方式,来调整双方的水性溶剂的离子强度。例如,通过使第1剂水溶液为pH9、离子强度0.05~0.1的硼酸缓冲液,使第2剂水溶液为pH4、离子强度0.01~0.03的磷酸缓冲液,在将它们以同体积进行混合时,能够使pH为8~10。或者,也可以使第1剂水溶液为pH10、离子强度0.05~0.1的硼酸缓冲液,使第2剂水溶液为pH4、离子强度0.01~0.07的磷酸缓冲液。
对第2剂中的交联剂浓度进行调整,从而使第2剂中的官能团的当量相对于第1剂中的氨基的当量、即(第2剂中的官能团当量/第1剂中的氨基当量)成为上述范围。也可以使用2种以上的交联剂的混合物,在该情况下使它们的合计的量成为上述范围。
<添加剂>
上述第1剂和/或第2剂还可以以不阻碍本发明的目的的量含有各种添加剂。作为该添加剂,可列举出着色料、pH调节剂、粘度调节剂、保存剂等。优选在第1剂或者第2剂水溶液中添加着色料,例如亮蓝,从而易于识别防粘连材料的应用部位。添加量例如可以为10~100μg/mL。
<制造方法>
本发明的防粘连材料可以通过将第1剂和第2剂单独地制备并包装,根据需要进行灭菌而获得。
[第1剂的制备法]
(1)原料明胶水性溶液的制备
将原始材料的明胶以成为5~50wt/v%的量,在40~90℃下进行加热而将其溶解于水性溶剂。作为该水性溶剂,使用水与水溶性有机溶剂的混合物。作为该水溶性有机溶剂,可以使用碳原子数1~3的醇、酯等,优选使用乙醇。
(2)衍生化
在由工序(1)所获得的明胶水溶液中添加具有导入的烷基的衍生化药剂,搅拌规定时间而使其反应。作为该衍生化药剂,可以使用具有上述烷基的醛或酮,例如十二醛、十四醛、癸基乙基酮。反应温度为30~80℃,反应时间为0.5~12小时,通常仅仅通过进行搅拌就能够获得在明胶的氨基上通过席夫碱(GltnN=CR
接下来,对该席夫碱进行还原。作为还原剂,可以使用氰基氢化硼钠(NaBH
(3)纯化
在由工序(2)所获得的反应溶液中加入大幅过量的不良溶剂,例如冷乙醇,使明胶衍生物沉淀。在对该沉淀进行过滤分离后,用乙醇等进行清洗,从而获得最终产物。
(4)第1剂的制备
由工序(3)所获得的明胶衍生物以粉末状的形态,或者以硼酸缓冲液等水性溶剂中的水溶液的形态,填充于容器中进行供给。作为容器,可以使用玻璃制或塑料制的、安瓿瓶、瓶、分配器、注射器等。也可以根据需要,而添加未衍生化的明胶、其他的添加剂。在以粉末状供给明胶衍生物的情况下,单独将硼酸缓冲液等水性溶剂填充于容器中进行供给。在以水溶液供给明胶衍生物的情况下,可以填充于在将防粘连材料应用于患部时使用的、能够在顶端部将两剂混合的双注射器型分配器等的一方。此外,本发明的防粘连材料当然可以含有作为附属品的双注射器型分配器、混合用的安瓿瓶、备用的水性溶剂等。
[第2剂的制备法]
作为第2剂例示出的上述各交联剂既可以用公知的方法进行合成,又可以使用市售品。将该交联剂和用于将其溶解的、例如磷酸缓冲液等水性溶剂装入各自的容器进行供给,例如将交联剂装入玻璃制的安瓿瓶,将水性溶剂装入塑料瓶。根据需要,也可以添加添加剂。第1剂的量和该第2剂的量以(该交联剂的官能团的当量/该明胶衍生物的氨基的当量)成为0.2~2的量进行供给,但也可以将第1剂和第2剂以可以分别独立地补充的形态进行供给。
<灭菌>
接下来,对填充于分配器等的水溶液的形态的第1剂或填充于安瓿瓶等的明胶衍生物粉末与填充于瓶等的水性溶剂组合的形态的第1剂、以及填充于安瓿瓶等的粉末形态的交联剂与填充于瓶等的用于溶解该交联剂的水性溶剂组合的形态的第2剂分别进行灭菌。明胶衍生物粉末和粉末形态的交联剂的灭菌法优选放射线灭菌。作为该放射线,可列举出电子束、γ射线、连续X射线,优选电子束灭菌。作为总吸收辐射剂量,只要为现有技术中广泛使用的(第十四次修订的日本药典、第二部、参考信息、第1235页、右栏、2.2放射线法)20kGy以上即可,优选为25kGy~45kGy。只要使总吸收辐射剂量为20kGy以上即可,即使为了防止对明胶衍生物、交联剂造成损伤,而分多次来照射电子束,也不会改变灭菌效果。例如,在以总吸收辐射剂量30kGy进行灭菌的情况下,也可以进行照射三次10kGy的照射。明胶衍生物水溶液的灭菌法优选高压釜或者过滤器灭菌。
<对组织的应用方法等>
本发明还提供包括对病患对象的患部施用第1剂和第2剂的工序的防粘连材料膜的制造方法、以及包含该防粘连材料膜的制造方法的、病患对象的防粘连方法其中病患对象为人或人以外的动物,。本发明的防粘连材料可以应用于皮肤、血管、腱、神经、肠和淋巴管等管状组织、肝脏、胰脏和心脏等脏器的断裂部分。其中,优选被应用于湿润组织,例如血管、肺等。作为防粘连材料的应用方法,如上所述优选在即将使用前将第2剂制成水溶液。此时的交联剂的浓度如已述那样。将得到的第2剂水溶液填充于已填充有第1剂水溶液的双注射器型分配器的空置的一方的注射器而应用,或者用具有双注射器的空气辅助式喷雾器,进行喷雾而施用于患部。施用后,用几分钟~10分钟左右形成膜,从而可以将患部密封。
实施例
以下,通过实施例来说明本发明,但本发明并不限定于此。
<明胶衍生物的制备>
用专利文献3(日本发明专利授权特许第5995128号)中所记载的方法制备了明胶衍生物。作为例子,示出使用辛醛制备了明胶衍生物1的方法。
在将源自黄线狭鳕的明胶(Mw33000、新田明胶(株)制造)30g在50℃下溶解于105mL水中,并向其中加入将相当于该明胶的氨基的10摩尔%的化学计量量的正辛醛139μL溶解于40mL的乙醇而得到的溶液,在50℃下搅拌1小时。向其中加入将2-甲基吡啶硼烷143μL溶解于5mL的乙醇而得到的溶液。将所得到的反应混合物在50℃下搅拌17小时,之后,将该反应混合物滴加到反应混合物的10倍体积量的冷乙醇中,从而使明胶衍生物沉淀,并进行了吸滤。将所得到的沉淀物用冷乙醇清洗3次后,进行了过滤分离。将所得到的过滤物进行减压干燥,从而获得明胶衍生物1。
对明胶衍生物1的红外光谱(FTIR-8400、岛津制作所株式会社)和
用以下的方法测定了衍生化比率。将明胶衍生物1溶解于体积比1:1的二甲基甲酰胺/水混合溶剂中,得到了0.1w/v%溶液。在该溶液中加入0.1v/v%三乙基胺水溶液和0.1w/v%硝基苯磺酸水溶液,搅拌约1分钟而使硝基苯磺酸与该明胶衍生物中的氨基反应,之后,用酶标仪(SPARK 10M、帝肯日本株式会社)测定了340nm的吸光度。明胶衍生物1的衍生化比率为7.2摩尔%。
除了将正辛醛替换为正癸醛或正十二醛、以及替换为Mw38000的明胶以外,用与上述明胶衍生物的制备方法同样的方法,制备了表1所示的明胶衍生物。此外,在表1等中,例如“C8”表示由C8醛衍生化的明胶(在式(1)中,R
[表1]
将上述各明胶衍生物以15w/v%的浓度溶解于pH9.5的0.1M硼酸缓冲液,制成了第1剂。
<第2剂的制备>
作为第2剂,使用了季戊四醇-聚乙二醇醚四琥珀酰亚胺基戊二酸酯(4S-PEG、日油(株)社制造)。在即将进行下述各评价之前,将4S-PEG溶解于pH4.0的0.01M磷酸缓冲液中,使以同体积与第1剂进行混合时相对于第1剂的残存氨基1当量为0.4当量(实施例4中为0.5当量)。
<膜的制备法>
用双注射器型分配器将同体积的第1剂和第2剂挤到各试验中的基材上,而将实施例的膜涂布于组织上。另外,如表2所示,作为比较例,分别按照附件文件使用了衍生化前的明胶(比较例1:Org)、纤维蛋白(比较例2:fibrin;商品名BOLHEAL、化学和血清疗法研究所制造)、Seprafilm(比较例3:HA/CMC Film;商标、科研制药株式会社)。比较例2和3是以成为与实施例同面积的量使用的。
[表2]
此外,在图3等中,比较例2标记为“Fibrin”,比较例3标记为“HA/CMC”。
为了评价防粘连性,在体外进行了细胞粘接试验、细胞渗透试验和体内防粘连试验。
<细胞粘接性>
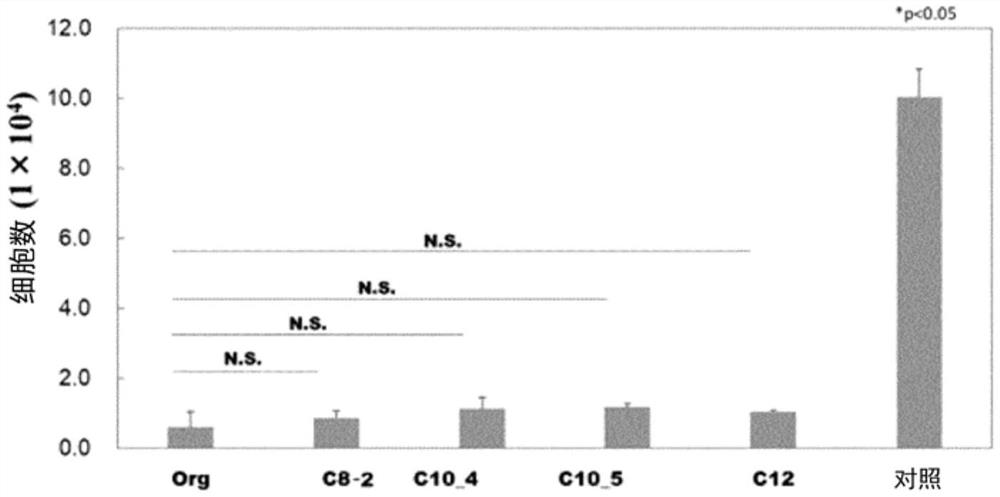
在48孔板的底面上,分别施用150μL实施例2~5和比较例1的第1剂和第2剂而形成膜,照射紫外线1小时进行了灭菌。在膜上接种L929成纤维细胞(1.0×105细胞/孔),在二氧化碳下,在含有10%胎牛血清和1%青霉素/链霉素溶液的RPMI-1640培养基中,在37℃下进行了培养。24小时后,将细胞用D-PBS进行清洗,使用WST-8分析试剂盒来测算附着于膜上的细胞数。作为对照,使用了没有膜的材料。
结果在图1中示出。根据该图可知,与对照相比,所有的实施例中细胞的粘接均较少,表现出体外的优异的防粘连性。
<细胞渗透性>
如图2中示意地所示,使用细胞培养小室(Transwell insert、PTFE膜片口径8.0μm,康宁国际株式会社),评价了细胞渗透性。在小室的底面上,将实施例1和比较例1的第1剂和第2剂分别施用100μL,并且施用成为同面积的量的比较例2、3而形成膜,照射紫外线1小时进行了灭菌。在膜上接种L929成纤维细胞(1.0×106细胞/孔),在二氧化碳下,在含有10%胎牛血清和1%青霉素/链霉素溶液的RPMI-1640培养基中,在37℃下进行了培养。24小时后,测算渗透至孔下部的细胞数,求得相对于未形成膜的未处理的情况的比率(%)。结果在图3中示出。
根据图3可知,实施例与Seprafilm相比,细胞的渗透性显著较低,认为这有助于防粘连性。
<体内防粘连性评价>
开腹大鼠(Wistar、7周雌性、日本维通利华株式会社)而准备了盲肠腹壁膜缺损模型。用医用纱布擦拭盲肠壁(Cecum)而使其出血,从而使盲肠壁损伤。用医用刀具将与该损伤部位相向的腹膜壁(Abdominal wall)切除规定的尺寸(1.0×2.0cm),据此,准备了腹膜壁缺损。对腹膜壁缺损和盲肠损伤双方以覆盖创伤的整个范围的方式涂布实施例1和比较例1~3的防粘连材料,之后,缝合了开腹部。
在手术1周后和2周后,使大鼠死亡,并采集手术部位的组织3处的截面的试验片,在10%福尔马林中性缓冲液中进行固定,进行苏木精-伊红染色,通过光学显微镜进行了观察。1周后的照片在图4a中示出,2周后的照片在图4b中示出。图中,“Untreated”表示对缺损部未应用任何防粘连材料。按表3的基准评价了各试验片的粘连(Adhesion)的结果在表4、5中示出。分数低的试验片数量越多,则表示粘连越少。
[表3]
[表4]
[表5]
如表4、5所示,在未处理试样和应用了纤维蛋白的组织中发现显著地粘连,但实施例1即使在2周后也完全不粘连,确认到优异的防粘连性。
<密封强度>
按照ASTM(F2392-04),使用猪大肠
如图5所示,本发明的防粘连材料与其他的试样相比,表现出显著高的密封强度,再次确认了专利文献3所示的结果。另外,如图6所示,纤维蛋白膜和Seprafilm在膜与猪大肠的界面处发生了剥离,但本发明的防粘连材料是防粘连材料膜本身内聚破坏。据此,可知本发明的防粘连材料的粘接强度也优异。
<蛋白质渗透性>
如图7所示,使用细胞培养小室(Transwell insert、PTFE膜片口径8.0μm、康宁国际株式会社),按照D.Nwa et al.,J.Biomed.Mater.Res.B,2013,101B,1251-1258所记载的方法,考察了蛋白质渗透性。在小室的底面上,将第1剂和第2剂分别施用100μL而形成膜,照射紫外线1小时进行了灭菌。将小室浸渍于0.9mL的D-PBS中。将荧光黄异氰酸酯标记白蛋白的0.1w/v%水溶液的0.1mL滴加于该膜上。使用该小室的背面的荧光测定和图像解析系统(IVIS Lumina II、珀金埃尔默公司)来求得24小时后的渗透率。结果在图8中示出。
如图8所示,实施例1示出与纤维蛋白膜和Seprafilm相比显著低的蛋白质渗透率(约20%),大部分的蛋白质残留于表面上。认为该蛋白质渗透抑制作用有助于防粘连性。未衍生化的明胶膜的渗透率也低,但蛋白质遍及整个厚度。
<溶胀度>
分别使实施例1、比较例1的第1剂和第2剂各1000μL流入到以厚度0.5mm的有机硅橡胶为间隔物的2片玻璃板之间,制成了板状的防粘连材料固化物。通过将所得到的防粘连材料固化物用直径4mm的冲头打穿,而制备了厚度0.5mm、直径4mm的圆形膜。对冷冻干燥后的重量(W
溶胀度(%)={(W
在图9中示出溶胀度随时间的变化。在进行试验的时间内,没有发现两者的试样存在显著性差异。
<酶分解性>
针对实施例1和比较例1,分别使第1剂和第2剂各1000μL流入到以厚度1.0mm的有机硅橡胶为间隔物的2片玻璃板之间,制成了板状的防粘连材料固化物。通过将所得到的防粘连材料固化物用直径10mm的冲头打穿,而制备厚度1.0mm、直径10mm的圆形膜,对初始重量(W
重量(%)=(W
在上式中,W
如图10所示,确认到实施例的膜与比较例相比分解时间较长,从而能够更长时间地保护手术部位。
产业上的可利用性
本发明的防粘连材料能够应用于复杂形状的手术部位,非常适合作为具有粘接性的防粘连材料,或者作为具有防粘连性的外科用密封胶而在临床中使用。另外,本发明的防粘连法能够简单地施用于现有的方法中难以应用的部位。
- 一种可喷涂使用的防粘连材料和防粘连组织密封胶及其制备方法
- 防粘连材料和防粘连方法