通过结构互补激活生物发光
文献发布时间:2024-04-18 19:57:50
本申请是2014年3月13日提交的申请号为201480015213.X,发明名称为“通过结构互补激活生物发光”的发明专利申请的分案申请。
领域
本文提供了由两种或更多种不发光(例如,基本上不发光)的肽和/或多肽单元装配生物发光复合物的组合物和方法。具体而言,经由与另一互补的不发光肽的结构互补赋予不发光多肽以生物发光活性。
背景
生物过程依赖分子、大分子和分子复合物之间的共价和非共价相互作用。为了了解此类过程,并且开发操纵其进行研究应用、临床应用和其它实践应用的技术和化合物,有必要有可用于检测和监测这些相互作用的工具。对这些相互作用的研究,特别是在生理条件下(例如在正常表达水平下以用于监测蛋白质相互作用),需要高灵敏度。
概述
本发明涉及包含互补的不发光氨基酸链(例如,大体上不发光的并非先存蛋白的片段的肽和/或多肽)的组合物、其复合物,及在不发光氨基酸链(例如,肽和/或多肽)缔合后产生任选可检测的生物发光信号的方法。在一些实施方案中,本发明提供了两种或更多种不发光,或大体上不发光的肽和/或多肽,所述肽和/或多肽在集中到一起时,装配成生物发光复合物。在一些实施方案中,一对大体上不发光的肽和/或多肽单元装配成生物发光复合物。在其它实施方案中,三个或更多个大体上不发光的肽和/或多肽单元装配成生物发光复合物(例如,三元复合物、三级复合物等)。本文提供了通过将此类相互作用与其它不发光(例如,大体上不发光)的氨基酸链的生物发光复合物的形成相互关联检测分子实体(例如,蛋白质、核酸、碳水化合物、小分子(例如,小分子文库))之间的相互作用的技术。
在一些实施方案中,装配好的对催化适当底物向高能态的化学反应,并且发光。在一些实施方案中,生物发光复合物在底物(例如腔肠素(coelenterazine)、腔肠素类似物(furimazine)等)的存在下显示发光。
虽然本文所述的实施方案主要描述并是指形成生物发光复合物的互补、不发光氨基酸链,但是应注意到本技术同样可适用于其它可检测的属性(例如,其它酶活性、荧光团的生成、生色团的生成等)。本文所述的涉及发光的实施方案应视为适用于分别缺乏指定可检测活性(例如,酶促活性)的互补、大体上无酶促活性的氨基酸链(例如,并非先存蛋白的片段的肽和/或多肽)或多肽的大体上无酶促活性的亚基、其复合物及在互补、大体上无酶促活性的氨基酸链(例如,肽和/或多肽)缔合后产生可检测活性(例如,酶促活性)的方法。进一步地,本文所述涉及不发光肽和/或多肽的实施方案在一些实施方案中适用于大体上不发光的肽和/或多肽。
本发明还涉及通过将一对不发光肽/多肽的相互作用与目标相互作用分子(例如,瞬时缔合、稳定缔合、复合物形成等)联系起来,检测目标分子之间的分子相互作用的测定。在此类实施方案中,一对不发光元件拴系(例如,融合)到目标分子上并且通过目标分子的分子相互作用操作生物发光复合物的装配。如果目标分子参与足够稳定的相互作用,则形成生物发光复合物,并且产生生物发光信号。如果目标分子不能参与足够稳定的相互作用,则不会形成生物发光复合物或仅较弱地形成,并且生物发光信号不可检测或大幅减少(例如,大体上不可检测、基本上不可检测等)。在一些实施方案中,可检测的生物发光信号的幅度与目标分子之间分子相互作用的量、强度、促进性和/或稳定性成比例(例如,成正比)。
在一些实施方案中,本发明提供了包含与SEQ ID NO:2具有小于100%(例如,20%...30%...40%...50%...60%...70%...80%或更高)序列同一性的氨基酸序列的肽,其中当所述肽接触由SEQ ID NO:440组成的多肽时产生可检测的生物发光信号。在一些实施方案中,本发明提供了包含与SEQ ID NO:2具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的氨基酸序列的肽,其中当所述肽接触由SEQ ID NO:440组成的多肽时产生可检测的生物发光信号。在一些实施方案中,当所述肽接触与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的多肽时产生可检测的生物发光信号。在某些实施方案中,当所述肽与包含SEQ ID NO:440或其一部分或由其组成的多肽缔合时产生可检测的生物发光信号,或生物发光信号大幅增加。在优选实施方案中,所述肽与SEQ ID NO:2的肽相比显示一种或多种特性改变(例如,增强),其中当与由SEQ ID NO:440组成的多肽组合时,所述特性选自:对由SEQ ID NO:440组成的多肽的亲和力、表达、细胞内溶解性、细胞内稳定性和生物发光活性。虽然不限于这些序列,但是所述肽氨基酸序列可选自SEQ ID NO:3-438和2162-2365的氨基酸序列。在一些实施方案中,提供了融合多肽,其包含:(a)上述肽,和(b)第一相互作用多肽,其与第二相互作用多肽在所述第一相互作用多肽和所述第二相互作用多肽接触后形成复合物。在某些实施方案中,提供了生物发光复合物,其包含:(a)上述第一融合多肽和(b)第二融合多肽,其包含(i)第二相互作用多肽和(ii)当与包含与SEQ ID NO:2具有小于100%而大于40%序列同一性的氨基酸序列的所述肽缔合时,发射可检测的生物发光信号的互补多肽;其中所述第一融合多肽和第二融合多肽缔合;并且其中包含与SEQ ID NO:2具有小于100%而大于40%序列同一性的氨基酸序列的所述肽与所述互补多肽缔合。
在一些实施方案中,本发明提供了包含与SEQ ID NO:440具有小于100%序列同一性的氨基酸序列的多肽,其中当所述多肽接触由SEQ ID NO:2组成的肽时产生可检测的生物发光信号。在一些实施方案中,本发明提供了包含与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的氨基酸序列的多肽,其中当所述多肽接触由SEQID NO:2组成的肽时产生可检测的生物发光信号。在一些实施方案中,当所述多肽接触与SEQ ID NO:2具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的肽时产生可检测的生物发光信号。在一些实施方案中,所述多肽与SEQ ID NO:440的肽相比显示一种或多种特性改变(例如,增强),其中当与由SEQ ID NO:2组成的肽组合时,所述特性选自:对由SEQ IDNO:2组成的肽的亲和力、表达、细胞内溶解性、细胞内稳定性和生物发光活性。虽然不限于此类序列,但是所述多肽氨基酸序列可选自SEQ ID NO:441-2156的氨基酸序列之一。在一些实施方案中,当所述多肽与由SE Q ID NO:2组成的肽缔合时产生可检测的生物发光信号。在一些实施方案中,提供了一种融合多肽,其包含:(a)上述多肽,和(b)第一相互作用多肽,其与第二相互作用多肽在所述第一相互作用多肽和所述第二相互作用多肽接触后形成复合物。在某些实施方案中,提供了一种生物发光复合物,其包含:(a)上述第一融合多肽和(b)第二融合多肽,其包含(i)第二相互作用多肽和(ii)使包含与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的氨基酸序列的所述多肽,在二者之间形成缔合时发射可检测的生物发光信号的互补肽;其中所述第一融合多肽和第二融合多肽缔合;并且其中包含与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的氨基酸序列的所述多肽与所述互补肽缔合。
在一些实施方案中,本发明提供了编码本文所述的肽、多肽、融合蛋白等的任何一种的核酸(例如,DNA、RNA等)、寡核苷酸、载体等。在一些实施方案中,提供了包含SEQ IDNO:3-438和2162-2365(编码不发光肽)和/或SEQ ID NO 441-2156(编码不发光多肽)的核酸序列之一或由其组成的核酸。在一些实施方案中,提供了编码SEQ ID NO:3-438和2162-2365和/或SEQ ID NO 441-2156的氨基酸序列的其它核酸序列。
在某些实施方案中,本发明提供了生物发光复合物,其包含:(a)包含与SEQ IDNO:2具有小于100%序列同一性(例如,>99%、<95%、<90%、<80%、<70%、<60%、<50%等)的肽氨基酸序列的肽;和(b)包含与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的多肽氨基酸序列的多肽,其中所述生物发光复合物显示出可检测的发光。在某些实施方案中,本发明提供了生物发光复合物,其包含:(a)包含与SEQ ID NO:2具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的肽氨基酸序列的肽;和(b)包含与SEQ ID NO:440具有小于100%而大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的多肽氨基酸序列的多肽,其中所述生物发光复合物显示出可检测的发光。虽然不限于特定序列,但是在一些实施方案,所述肽氨基酸序列选自SEQ ID NO:3-438和2162-2365中提供的氨基酸序列之一。
在各个实施方案中,提供生物发光复合物,其包含:(a)并非先存蛋白的片段的第一氨基酸序列;和(b)并非先存蛋白的片段的第二氨基酸序列,其中所述生物发光复合物显示出可检测的发光,其中所述第一氨基酸序列和所述第二氨基酸序列缔合。一些此类生物发光复合物还包含:(c)包含相互作用对的第一成员的第三氨基酸序列,其中所述第三氨基酸序列共价连接到所述第一氨基酸序列;和(d)包含相互作用对的第二成员的第四氨基酸序列,其中所述第四氨基酸序列共价连接到所述第二氨基酸序列。在某些实施方案中,在相互作用对的第一成员和第二成员之间没有相互作用时,第一氨基酸序列和第二氨基酸序列之间的相互作用(例如,非共价相互作用(例如,氢键、离子键、范德华力(van der Waalsforce)、疏水相互作用等)、共价相互作用(例如,二硫键)等)未明显地使第一氨基酸序列和第二氨基酸序列缔合。在一些实施方案中,第一多肽链包含第一氨基酸序列和第三氨基酸序列,并且其中第二多肽链包含第二氨基酸序列和第四氨基酸序列。在一些实施方案中,第一多肽链和第二多肽链在细胞内表达。
在一些实施方案中,本发明提供了一种生物发光复合物,其包含:(a)一对不发光元件,其中每个不发光元件均非先存蛋白的片段;(b)相互作用对,其中相互作用对的每个相互作用元件共价连接到不发光元件之一上。
本文所述的各个实施方案提供了检测第一氨基酸序列和第二氨基酸序列之间的相互作用的方法,其例如包括以下步骤:(a)使所述第一氨基酸序列连接到第三氨基酸序列,并且使所述第二氨基酸序列连接到第四氨基酸序列,其中所述第三和第四氨基酸序列并非先存蛋白的片段,其中所述第三和第四氨基酸序列的复合物发射可检测的生物发光信号(例如,分别相对于所述多肽链大幅增加的生物发光),其中所述第三和第四氨基酸序列之间的相互作用(例如,非共价)在没有附加稳定和/或聚集条件时不足以形成或仅较弱地形成所述第三和第四氨基酸序列的复合物,并且其中所述第一氨基酸序列和所述第二氨基酸序列之间的相互作用提供了附加稳定力和/或聚集力以生成所述第三和第四氨基酸序列的复合物;(b)将步骤(a)的所述第一、第二、第三和第四氨基酸序列放置在允许所述第一氨基酸序列和所述第二氨基酸序列之间发生相互作用的条件下;并且(c)检测所述第三和第四氨基酸序列的复合物发射的生物发光信号,其中检测到所述生物发光信号的指示所述第一氨基酸序列和所述第二氨基酸序列之间的相互作用。在一些实施方案中,使所述第一氨基酸序列连接到所述第三氨基酸序列并且使所述第二氨基酸序列连接到所述第四氨基酸序列包括形成包含所述第一氨基酸序列和所述第三氨基酸序列的第一融合蛋白并且形成包含所述第二氨基酸序列和所述第四氨基酸序列的第二融合蛋白。在一些实施方案中,所述第一融合蛋白和所述第二融合蛋白还包含分别介于所述第一和第三氨基酸序列之间及所述第二和第四氨基酸序列之间的接头。在某些实施方案中,由编码所述第一和第三氨基酸序列的第一核酸序列表达所述第一融合蛋白,并且由编码所述第二和第四氨基酸序列的第二核酸序列表达所述第二融合蛋白。在一些实施方案中,单个载体包含所述第一核酸序列和所述第二核酸序列。在其它实施方案中,所述第一核酸序列和所述第二核酸序列在单独的载体上。在某些实施方案中,(a)“连接”和(b)“放置”的步骤包括在细胞内表达第一和第二融合蛋白。
本文提供了建立、生成、产生和/或优化一对不发光元件的方法,包括:(a)比对三种或更多种相关蛋白的序列;(b)确定所述相关蛋白的共有序列;(c)提供与三种或更多种蛋白质相关的蛋白质的第一和第二片段(或提供所述三种或更多种蛋白质之一的第一和第二片段),其中所述片段单独地大体上不发光,但是经所述片段的相互作用后显示发光;(d)使所述第一和第二片段各自在一个或多个位置突变,其中所述突变将所述片段的序列改变成与所述共有序列的相应位置更加相似(例如,其中所述突变产生并非先存蛋白的片段的一对不发光元件),和(e)测试该对不发光元件,当未缔合时没有发光(例如,基本没有、大体上没有等)而在所述不发光对缔合成生物发光复合物后发光。在实施例1-5中描述了此类过程的实例。在一些实施方案中,所述不发光元件与所述第一和第二片段相比显示出一种或多种特性增强,其中所述特性选自:重构亲和力增加、重构亲和力降低、表达增强、细胞内溶解性增加、细胞内稳定性增加和重构发光强度增加。
在一些实施方案中,本发明提供了检测试剂,其包含(a)包含与SEQ ID NO:440具有小于100%而大于40%序列同一性的氨基酸序列的多肽,其中当所述多肽接触由SEQ IDNO:2组成的肽时产生可检测的生物发光信号,和(b)由所述多肽和由SEQ ID NO:2组成的肽生成的生物发光复合物的底物。在一些实施方案中,本发明提供了检测试剂,其包含:(a)包含与SEQ ID NO:2具有小于100%序列同一性的氨基酸序列的肽,其中当所述肽接触由SEQID NO:440组成的多肽时产生可检测的生物发光信号,和(b)由所述肽和由SEQ ID NO:440组成的多肽生成的生物发光复合物的底物。在一些实施方案中,本发明提供了检测试剂,其包含:(a)包含与SEQ ID NO:2具有小于100%而大于40%序列同一性的氨基酸序列的肽,其中当所述肽接触由SEQ ID NO:440组成的多肽时产生可检测的生物发光信号,和(b)由所述肽和由SEQ ID NO:440组成的多肽生成的生物发光复合物的底物。
附图简述
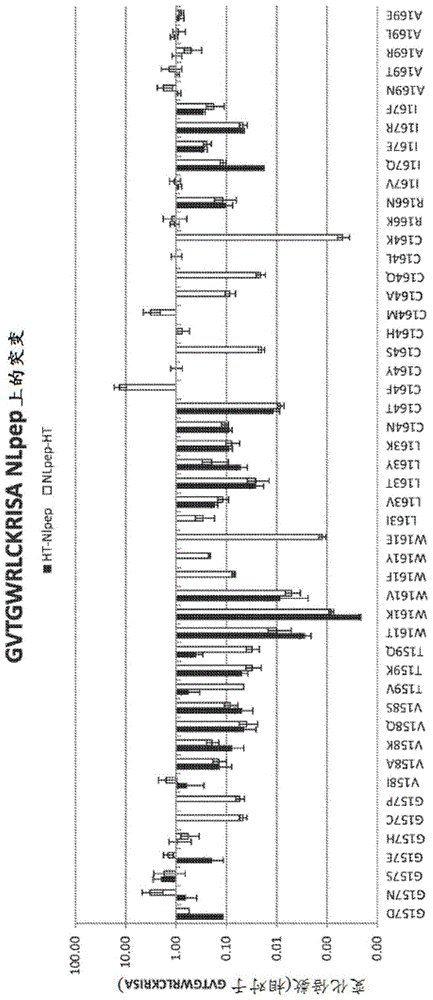
图1显示了描绘GVTGWRLCKRISA(SEQ ID NO:236)肽的各种突变对由与SEQ ID NO:440互补引起的发光的影响的图。
图2显示了描绘SEQ ID NO:440多肽的各种突变对由与GVTGWRLCKRISA(SEQ IDNO:236)或GVTGWRLFKRISA(SEQ ID NO:108)肽互补引起的发光的影响的图。
图3显示了在含有单个甘氨酸向丙氨酸取代的每种不发光的多肽(NLpoly)突变体中检测到的发光(RLU)(上)。还显示了超过野生型的发光的增加倍数(下)。
图4显示了在含有甘氨酸向丙氨酸取代的复合物的每种NLpoly突变体中检测到的发光(RLU)(上)。还显示了超过野生型的发光的增加倍数(下)。
图5显示了描绘在HT-NL肽融合物中检测到的发光(RLU)的图。
图6显示了描绘在HT-NLpep融合物中检测到的发光(RLU)的图。
图7显示了描绘在NL肽-HT融合物中检测到的发光(RLU)的图。
图8显示了在不发光肽(NLpep)的冻融循环之后,由发光复合物产生的发光(RLU)。
图9显示了肽的浓度归一化活性,和用于测定相对浓度的TMR凝胶。
图10显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基R11的各种突变的发光的图。
图11显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基A15的各种突变的发光的图。
图12显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基L18的各种突变的发光的图。
图13显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基F31的各种突变的发光的图。
图14显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基V58的各种突变的发光的图。
图15显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基A67的各种突变的发光的图。
图16显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基M106的各种突变的发光的图。
图17显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基L149的各种突变的发光的图。
图18显示了在NLpep53存在时(上)和没有互补肽时(下)NLpoly-5A2残基V157的各种突变的发光的图。
图19显示了NLpep-HT融合物的发光的图。
图20显示了NLpep-HT融合物的发光的图,和指示其相对表达水平的TMR凝胶。
图21显示了NLpep-HT融合物的发光的图。
图22显示了NLpoly 5A2(上)和NLpoly5A2+R11E在各种NLpep(下)存在下的发光的图。
图23显示了NLpep-HT融合物的发光的图。
图24显示了有NLpep53(上)和无互补肽(下)的NLpoly的发光的图。
图25显示了有NLpep53的各种NLpoly用NANOGLO或DMEM缓冲液和腔肠素类似物或腔肠素底物发光的图。
图26显示了比较在一定比率的腔肠素类似物与腔肠素的存在下,各种NLpoly和NLpep53的发光的图。
图27显示了比较在一定比率的腔肠素类似物与腔肠素的存在下,各种NLpoly和NLpep53的发光的图。
图28显示了比较在HEK293细胞裂解物中在腔肠素类似物与腔肠素的存在下,各种NLpoly和NLpep53的发光的图。
图29显示了在有腔肠素类似物的DMEM缓冲液中NLpoly和NLpep对的各种组合的发光的图。
图30显示了在有腔肠素类似物的DMEM缓冲液中NLpoly和NLpep对的各种组合的信号/本底发光的图。
图31显示了使用腔肠素类似物或腔肠素作为底物,有NLpep69的各种NLpoly突变体的发光和底物特异性的图。
图32显示了使用腔肠素类似物或腔肠素作为底物,并且在裂解(下图)或活细胞(上图)条件下,有NLpep69的各种NLpoly突变体的发光和底物特异性的比较。
图33显示了使用腔肠素类似物或腔肠素作为底物,并且在裂解(下图)或活细胞(上图)条件下,有NLpep78的NLpoly突变体的发光和底物特异性的比较。
图34显示了使用腔肠素类似物或腔肠素作为底物,并且在裂解(下图)或活细胞(上图)条件下,有NLpep79的各种NLpoly突变体的发光和底物特异性的比较。
图35显示了NLpep78-HT(上)和NLpep79-HT(下)融合物在各种NLpoly存在下的发光的图。
图36显示了各种NLpoly在没有NLpep时的发光的图。
图37显示了在各种NLpoly存在下用腔肠素类似物或腔肠素底物,NLpep78-HT(上)和NLpep79-HT(下)融合物发光的图。
图38显示了有在CHO和HeLa细胞中表达的各种NLpoly的NLpep78-HT的发光的图。
图39显示了来自于与HEK293、Hela和CHO细胞裂解物中表达的萤火虫荧光素酶融合的NLpoly的原始和归一化发光的图。
图40显示了来自于与HEK293、Hela和CHO细胞裂解物中表达的叩头虫红色荧光素酶融合的NLpoly的原始和归一化发光的图。
图41显示了使用NLpoly野生型或5P在活细胞内互补发光的图。
图42显示了有不同NLpoly的NLpep78-HT融合物(上)和NLpep79-HT融合物(下)的无细胞互补发光的图。
图43显示了HeLa、HEK293和CHO细胞裂解物中表达的NLpep和NLpoly的不同组合的结合亲和力的图。
图44显示了在PBS或NANOGLO缓冲液中NLpep和NLpoly的不同组合的结合亲和力的图。
图45显示了NLpoly 5P与HeLa、HEK293和CHO细胞裂解物中表达的NLpep9或NLpep53的结合亲和力的图。
图46显示了在没有NLpep时,不同量的NLpoly的发光的图。
图47显示了各种NLpoly变体的本底发光的图。
图48显示了各种NLpoly变体的本底发光的图。
图49显示了几种NLpoly变体的总裂解物和可溶级分的SDS-PAGE凝胶。
图50显示了(a)NLpoly变体的总裂解物和可溶级分的SDS-PAGE凝胶和(b)NLpoly变体的本底发光。
图51显示了当与10nm(右)或100nM(左)的NLpep78互补时,几种NLpoly变体产生的发光的图。
图52显示了描绘各种NLpoly变体在大肠杆菌(E.coli)裂解物中的本底发光的图。
图53显示了描绘与NLpep78互补的各种NLpoly变体在大肠杆菌裂解物中的发光的图。
图54显示了描绘与NLpep79互补的各种NLpoly变体在大肠杆菌裂解物中的发光的图。
图55显示了与NLpep78或NLpep79互补并且归一化到NLpoly5P的各种NLPoly变体的信背比的图。
图56显示了描绘用NLpep79(右)或NLpep78(左)的本底发光及各种NLPoly变体的信噪比。
图57显示了各种NLpoly 5P变体中总裂解物和可溶级分的SDS-PAGE凝胶。
图58显示了(A)NLpoly 5P和NLpoly I107L的总裂解物和可溶级分的量,(B)无NLpep或有NLpep78或NLpep79的NLpoly 5P和NLpoly I107L产生的发光和(C)NLpoly I107L超出NLpoly 5P的信背比。
图59显示了(A)无互补肽、(B)有NLpep78-HT和(C)有NLpep79-HT的各种NLpoly变体的发光的图。
图60显示了(A)无互补肽、(B)有NLpep78-HT和(C)有NLpep79-HT的各种NLpoly变体的发光的图。
图61显示了(A)无互补肽、(B)有NLpep78-HT和(C)有NLpep79-HT的各种NLpoly变体的发光的图。
图62显示了(A)无互补肽、(B)有NLpep78-HT和(C)有NLpep79-HT的各种NLpoly变体的发光的图。
图63显示了延长型NLpoly变体(C端有附加氨基酸)和缩短型NLpep(N端的氨基酸缺失)之间的结合亲和力。
图64显示了各种NLpoly变体与NLpep78的结合亲和力的图。
图65显示了NLpep80和NLpep87与在哺乳动物细胞(CHO、HEK293T和HeLa)中表达的5P的结合和Vmax。
图66显示了NLpep80和NLpep87与在大肠杆菌中表达的NLpoly5P的结合和Vmax。
图67显示了有延长型NLpep的缩短型NLpoly的发光的图。
图68显示了有各种互补NLpep的HeLa裂解物中NLpoly 5P的Kd和Vmax的图。
图69显示了几种NLpoly变体与NLpep81的结合亲和力的图。
图70显示了几种NLpoly变体与NLpep82的结合亲和力的图。
图71显示了几种NLpoly突变体与NLpep78的结合亲和力的图。
图72显示了几种NLpoly突变体与NLpep78的米氏常数(Michaelis constant)的图。
图73显示了来自于两个NLpep和NLpoly 5P-B9三级互补的发光的图。
图74显示了用NLpep88-HT滴定NLpoly 5P发光的图。
图75显示了有HaloTag(HT)的各种NLpep融合物细胞内定位的图像。
图76显示了NLpoly(wt)和NLpoly(5P)细胞内定位的图像。
图77展示了在通过SDS-PAGE分离并转移到PVDF膜后,经由互补检测目标NLPep缀合蛋白的能力。
图78显示了与NLpoly 5P(没有NLpep时)相比,来自于各种NLpoly变体的相对发光信号的图。
图79显示了与NLpoly 5P(没有NLpep时)相比,来自于各种NLpoly的超过本底的相对发光信号的图。
图80比较了由NLpep78的1或2个重复单元组成的NLpep的解离常数。
图81显示了NLpoly 5A2和NLpep86之间的亲和力。
图82显示了来自于无NLpep、有NLpep78和NLpep79的NLpoly变体的发光的图。
图83-90显示了NLpoly 5A2、5P、8S和11S与96种NLpep变体的解离常数以及Vmax值。
图91显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶图像。
图92显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶图像以及含有相同变体的解离常数的表。
图93显示了有NLpep79的NLpoly 5P和11S的底物特异性并且证明NLpoly 11S对腔肠素类似物的特异性比5P更高。
图94显示了通过结合NLpep78亲和纯化NLpoly 8S后的蛋白质凝胶图像。
图95含有NLpoly WT或11S与NLpepWT、78或79结合的缔合和解离速率常数的表。
图96显示了各对NLpoly/NLpep的Km值。
图97比较了在腔肠素类似物亚饱和及饱和浓度下,NLpoly11S/NLpep79的解离常数。
图98比较了有NLpepWT、78和79的NLpoly 5A2的Km值。
图99显示了在没有NLpep时,来自于演化过程的各个步骤的NLpoly的发光。
图100显示经过演化过程,来自于大肠杆菌来源的NLpoly的发光改善,总体改善约10
图101显示经过演化过程,来自于HeLa表达的NLpoly的发光改善,总体改善约10
图102显示经过演化过程,来自于HEK293细胞表达的NLpoly的发光改善,总体改善约10
图103显示了解离常数并且证明从NLpolyWT:NLpepWT到NLpoly11S:NLpep86结合亲和力提高约10
图104显示了对于来自演化过程各个步骤的NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶图像。
图105显示了在没有NLpep时和在NLpep78和NLpep79存在时,各种NLpoly的发光。
图106显示了在没有NLpep时和在NLpep78和NLpep79存在时,各种NLpoly的发光。
图107显示了在没有NLpep时和在NLpep78和NLpep79存在时,各种NLpoly的发光。
图108显示了在用雷帕霉素(rapamycin)或媒介物处理15分钟后,由表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较。诱导倍数指在雷帕霉素存在下产生的信号与用媒介物产生的信号相比。
图109显示了在用雷帕霉素或媒介物处理60分钟后,由表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较。
图110显示了在用雷帕霉素或媒介物处理120分钟后,由表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较。
图111显示了在用雷帕霉素或媒介物处理120分钟后,由表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较。与NLpoly/NLpep融合的FRB和FKBP的8种可能组合全部经试验并且较少使用总DNA。
图112显示了在没有结合伴侣时表达的FRB或FKBP融合物产生的发光的比较。
图113显示了经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较。
图114显示了在没有结合伴侣时,经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较。
图115显示了经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较。这个实例与图113的不同之处在于使用更低水平的DNA。
图116显示了在没有结合伴侣时,经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较。这个实例与图114的不同之处在于使用更低水平的DNA。
图117显示了在用雷帕霉素处理不同时间长度之后,经不同量的FRB-NLpoly5P和FKBP-NLpep80 DNA转染的细胞产生的发光的比较。
图118显示了在用雷帕霉素处理不同时间长度之后,经不同量的FRB-NLpoly5P和FKBP-NLpep87 DNA转染的细胞产生的发光的比较。
图119显示了表达FRB-NLpoly5P与FKBP-NLpep80/87/95/96/97的不同组合的细胞产生的发光的比较。以两天和三天形式进行测定。
图120显示了表达FRB-NLpoly5A2与FKBP-NLpep80/87/95/96/97的不同组合的细胞产生的发光的比较。以两天和三天形式进行测定。
图121显示了表达FRB-NLpoly5A2或FRB-NLpoly11S与FKBP-NLpep101/104/105/106/107/108/109/110的不同组合的细胞产生的发光的比较。
图122显示了经FRB-NLpoly5A2或FRB-NLpoly11S与FKBP-NLpep87/96/98/99/100/101/102/103的不同组合转染的细胞产生的发光的比较。
图123显示了经不同水平的FRB-NLpoly11S和FKBP-NLpep87/101/102/107DNA转染的细胞产生的发光的比较。
图124显示了经不同水平的FRB-NLpoly5A2和FKBP-NLpep87/101/102/107DNA转染的细胞产生的发光的比较。
图125显示了雷帕霉素剂量响应曲线,其显示了表达FRB-NLpoly5P和FKBP-NLpep80/87DNA的细胞的发光。
图126显示了雷帕霉素剂量响应曲线,其显示了表达FRB-NLpoly5A2或FRB-NLpoly11S和FKBP-NLpep87/101DNA的细胞的发光。
图127显示了表达FRB-11S和FKBP-101并且经底物PBI-4377或腔肠素类似物处理的细胞产生的发光的比较。
图128显示了在存在或没有雷帕霉素时进行的表达FRB-NLpoly11S/5A2和FKBP-NLpep87/101的细胞的雷帕霉素时程,其中手动添加雷帕霉素。
图129显示了在存在或没有雷帕霉素时进行的表达FRB-NLpoly11S/5A2和FKBP-NLpep87/101的细胞的雷帕霉素时程,其中经由仪器注射器添加雷帕霉素。
图130显示了在两台不同的发光读数仪器上测量的FRB-NLpoly11S和FKBP-NLpep101产生的发光。
图131提供了显示表达FRB-NLpoly11S和FKBP-NLpep101的细胞在用雷帕霉素处理后的不同时间发光的图像。
图132提供了显示表达FRB-NLpoly11S和FKBP-NLpep101的单个细胞在用雷帕霉素处理后的不同时间产生的信号的图像J定量。
图133显示了在表达FRB-NLpoly11S和FKBP-NLpep101的不同细胞系中发光的比较。
图134显示了在用雷帕霉素竞争性抑制剂FK506处理后,表达FRB-NLpoly11S和FKBP-NLpep101的细胞产生的发光的比较。
图135显示了(左侧)在用雷帕霉素竞争性抑制剂FK506处理后,表达FRB-NLpoly11S和FKBP-NLpep101的细胞产生的发光,和(右侧)在用FK506处理后剩余发光的百分比。
图136显示了在存在或没有V2R激动剂AVP时,经V2R-NLpoly5A2或V2R-NLpoly11S与NLpep87/101-ARRB2的不同组合转染的细胞产生的发光。
图137显示了AVP处理时程,其显示了在用AVP处理后经V2R-NLpoly11S和NLpep87/101-ARRB2转染的细胞产生的发光,其中手动添加AVP。
图138显示了AVP处理时程,其显示了在用AVP处理后经V2R-NLpoly5A2或V2R-NLpoly11S与NLpep87/101-ARRB2的不同组合转染的细胞产生的发光,其中经由仪器注射器添加AVP。
图139显示了37℃下的AVP处理时程,其显示了在用AVP处理后表达与NLpoly11S和NLpep101融合的V2R和ARRB2的不同构型的细胞产生的发光。
图140显示了在表达V2R-NLpep11S和NLpep101-ARRB2的不同细胞系中发光的比较。
图141显示了60X图像,其显示了在用AVP处理后的不同时间,表达V2R-NLpoly11S和NLpep101-ARRB2的细胞的发光。
图142显示了150X图像,其显示了在用AVP处理后的不同时间,表达V2R-NLpoly11S和NLpep101-ARRB2的细胞的发光。
图143显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶。
图144显示了NLpoly 5P和NLpoly 5P中在位置31、46、75、76和93处的突变组合的解离常数。
图145显示了使用NLpep和氨肽酶进行翻译后修饰酶促活性检测的转移酶实例。
图146显示了使用NLpep和甲基特异性抗体进行翻译后修饰酶促活性检测的水解酶实例。
图147含有与NLpepWT或与缀合至TMR的NLpepWT互补的NLpoly WT的波长扫描。
图148含有与HaloTag融合的NanoLuc(NL-HT)和与带4个附加氨基酸(DEVD)并且与非氯TOM(NCT)缀合的NLPepWT互补的NLpoly 5A2的波长扫描。
图149显示了三级相互作用的示意图,其中与NLpoly和NLpep的能量转移也可用于测量三个分子的相互作用。在示意图中,经NLpoly标记的GPCR和经NLpep标记的GPCR相互作用蛋白在其相互作用时形成生物发光复合物。这样允许测量二元相互作用。如果带有用于能量转移的适当荧光部分的小分子GPCR配体与这种体系相互作用,则会发生能量转移。因此,可在相同实验中测量二元蛋白质-蛋白质相互作用和三元药物-蛋白质-蛋白质相互作用。
图150显示了NLpoly11S与合成NLPep78和NLPep78在融合伴侣(HaloTag)的N或C端的结合亲和力的图和表。
图151显示了NLpoly11S与合成NLPep79和NLPep79在融合伴侣(HaloTag)的N或C端的结合亲和力的图和表。
图152显示了描绘有NLPep86或PBI-4877的NLpoly11S的归一化荧光强度的图。
图153显示了描绘有NLPep86或PBI-5434的NLpoly11S的归一化荧光强度的图。
图154显示了描绘有NLPep86或PBI-5436的NLpoly11S的归一化荧光强度的图。
图155显示了证明NLpoly11S与NLpep86、78、99、101、104、128和114之间的复合物在亲和缓冲液中的腔肠素类似物结合亲和力的图。
图156显示了证明NLpoly11S与NLpep86、78、99、101、104、128和114之间的复合物在NanoGlo测定缓冲液中的腔肠素类似物结合亲和力的图。
图157显示了描绘随着腔肠素类似物底物浓度增加亲和力(NLpoly156/NLPep1和NLpoly11S/NLPep1)的变化的图。
图158显示了描绘随着NLPep1浓度增加亲和力(NLpoly156/NLPep1和NLpoly11S/NLPep1)的变化的图。
图159显示了描绘带的NLPoly156、NLPoly11S和
图160显示了根据NLPep浓度描绘了NLPoly11S和NLPep86、78、79、99、101、104、114、128和wt的RLU的图。
图161显示了描绘与全长
图162显示了描绘β-内酰胺酶SME及其抑制剂BLIPY50A呈未融合蛋白或与NLPoly11S和NLPep114融合时的亲和力比较的图。
图163显示了表达FRB-NLpoly11S与FKBP-NLpep101/111-136的不同组合的细胞产生的发光的比较。
图164显示了表达FRB-NLpoly11S与FKBP-NLpep114和137-143的不同组合的细胞产生的发光的比较。
图165显示了表达FRB-NLpoly11S和FKBP-NLpep78/79/99/101/104/114/128的细胞的雷帕霉素剂量响应曲线。
图166显示了表达FRB-NLpoly11S和FKBP-NLpep78/79/99/101/104/114/128的细胞对雷帕霉素竞争性抑制剂FK506的反应。
图167显示了经不同比率的FRB-NLpoly11S和FKBP-NLpep114转染的细胞产生的发光的比较。
图168显示了在不同方向并以不同接头长度表达FRB/FKBP的NLpoly11S/NLpep114融合物的细胞产生的发光的比较。
图169显示了描绘FRB-NLpoly11S/FKBP-NLpep114或分离萤火虫互补信号的雷帕霉素(A)特定剂量和(B)特定时间诱导的图。
图170显示了描绘FRB-NLpoly11S/FKBP-NLpep114或分离萤火虫互补信号的FK506(A)特定剂量和(B)特定时间诱导的图。
图171显示了描绘在相等水平的转染DNA下,FKBP-NLpep114和FKBP-Fluc(394-544)的相似表达水平的蛋白质印迹。
图172显示了描绘IBET-151对NLpoly11S-BRD4和组蛋白H3.3-NLpep114相互作用的(A)特定剂量和(B)特定时间抑制的图。
图173显示了描绘响应于BRAF抑制剂GDC0879,RAS/CRAF、BRAF/BRAF和CRAF/BRAF二聚作用剂量依赖性增加的图。
图174显示了根据NLPep浓度描绘NLpoly11S和NLpep86、wt和NLpep114的RLU的图。
图175显示了利用发光对的高亲和力肽作为细胞内蛋白质标签和发光对的多肽作为检测试剂的测定的示意图。
图176显示了证明NLpoly11S和MLpep86的亲和力的线性范围的图。
图177显示了证明使用11S检测经高亲和力NLPep标记的蛋白质的灵敏度的图像。该图还比较了使用NLPep/NLPoly的检测与使用荧光标记HaloTag的检测。
图178显示了证明NLpoly11S的稳定性的图。
图179显示了证明NLpoly11S和NLpep78的亲和力的线性范围的图。
图180显示了NLpep序列的汇总。高亲和力(自发)肽是以高亲和力与NLpoly11S结合的那些肽(NLpep)。暗/猝灭肽是可降低由NLpoly11S产生或检测出的光水平的那些肽(NLpep)。
图181显示了将LSP和SSP(即,NLpoly和NLpep)集中到一起产生生物发光信号(图A、B)时,结构互补的概念的示意图。破坏蛋白质相互作用(即,X和Y)后,LSP和SSP分开,导致发光减少(图C)。
图182A显示了将结构互补设计为在X和Y之间的蛋白质相互作用之后信号损失而在破坏X和Y之间的相互作用后获得信号的两个选项(A、B)。选项A表示分子间结构互补,选项B表示分子内结构互补。图182B显示了可适于分子内结构互补的基因构建体列表。
图183显示了(A)各种暗肽对NLpoly11S和NLpep114结合的抑制,和(B)受Lys-162和Gln-162肽的剂量依赖性抑制。
图184A显示受Q-162和A-162的抑制具剂量依赖性。图B显示Q-162以剂量依赖方式独立地产生信号,而A-162的剂量依赖性充其量较微弱。
图185显示了证明带CP Nluc的暗肽的剂量响应的图。
图186显示了描绘带CP Nluc的暗肽的时程的图。
图187显示了在存在和没有雷帕霉素时,对由单独的FRB-NLpoly11S以及在FRB-NLpoly11S和FKBP-NLpep114之间产生的发光的暗肽剂量依赖性抑制。
图188显示了在存在和没有雷帕霉素(RLU)时,对由FRB-NanoLuc(311)或NanoLuc-FRB(307)产生的发光的暗肽剂量依赖性抑制。
图189显示了在存在和没有雷帕霉素时(归一化到无暗肽对照;100%),对由FRB-NanoLuc(311)或NanoLuc-FRB(307)产生的发光的暗肽剂量依赖性抑制。
图190显示暗肽,当与FKBP融合时,可与低(114)和高(80)亲和肽(还有FKBP融合物)竞争并因此减少在活细胞内产生和检测到的总发光。
图191显示了基于Fluc和NLpep86的测定之间对于细胞内Fluc水平的信号比较。
图192显示了证明串联NLpep在竞争Npoly11S中的效用的图。
图193显示了证明NLpoly和NLpep组分不干扰报告蛋白FlucP的细胞内降解的图。
图194显示了证明细胞外蛋白酶促活性测定的示意图。
图195显示了使用ProNLpep测量酶活性的测定的示意图。
图196显示了筛选介导细胞内化的抗体、蛋白质、肽或转运蛋白的测定的示意图。
图197显示了翻译后修饰转移酶测定的示意图。
图198显示了翻译后修饰水解酶测定的示意图。
图199显示了在翻译后修饰测定中将酪氨酸激酶SRC活性与高于本底的发光联系起来的图。
图200显示了描绘3种不同形式的NLpoly11S与12种合成肽的自发互补的图。
图201显示了利用NLpep和NLpoly与单独结合部分A和B的融合物的同种免疫测定形式的示意图。
图202显示了证明:(A)在与GWALFKK和Dabcyl-GWALFKK形成复合物后,来自于NLpoly11S的本底发光减少,和(B)在GWALFKK和Dabcyl-GWALFKK的存在下,NLpep86与NLpoly11S形成复合物的图。
图203显示了证明:(A)VTGWALFEEIL(Trp 11mer)和VTGYALFEEIL(Tyr 11mer)诱导高于本底(单独的NLpoly11S;无肽对照)的发光,并且各自的N端Dabcyl形式提供了对这种信号的明显猝灭,并且(B)在Trp 11mer和Tyr 11mer的Dabcyl形式的存在下,NLpep86与NLpoly11S形成复合物的图。
定义
如本文所使用,术语“大体上”意味着不需要精确地实现所述特征、参数和/或值,但是可以不排除所述特征要提供的影响的量出现偏差或变化,包括例如公差、测量误差、测量精度限制和本领域中技术人员已知的其它因素。大体上不存在的特征或特点(例如,大体上不发光)可为噪声中、低于本底、低于所用测定的检测能力或重要特征(例如,生物发光蛋白或生物发光复合物的发光强度)的一小部分(例如,<1%、<0.1%、<0.01%、<0.001%、<0.00001%、<0.000001%、<0.0000001%)的特征或特点。
如本文所使用,术语“生物发光”指通过由酶、蛋白质、蛋白质复合物或其它生物分子(例如,生物发光复合物)催化或激活的化学反应产生并发射光线。在典型实施方案中,生物发光实体(例如,生物发光蛋白或生物发光复合物)的底物被生物发光实体转化为不稳定形式;所述底物随后发光。
如本文所使用,术语“互补”指两个或更多个结构元件(例如,肽、多肽、核酸、小分子等)能够相互杂交、二聚或以其它方式形成复合物的特征。例如,“互补肽和多肽”能够组合在一起形成复合物。互补元件可能需要帮助以形成复合物(例如,由相互作用元件),例如,使所述元件置于互补的正确构象,使互补元件共同定位,降低互补的相互作用能,等等。
如本文所使用,术语“复合物”指相互直接和/或间接接触的分子(例如,肽、多肽等)的集合物或聚集物。一方面,“接触”或更特别地,“直接接触”意指两个或更多个分子足够靠近,以致吸引性非共价相互作用,例如范德华力、氢键、离子和疏水性相互作用等,在分子相互作用中占优势。在这一方面,在测定条件下形成分子(例如,肽和多肽)的复合物,以致所述复合物在热力学上有利(例如,与其组成分子的非聚集或非复合态相比)。如本文所使用,术语“复合物”,除非另有描述,指两个或更多个分子(例如,肽、多肽或其组合)的集合物。
如本文所使用,术语“不发光”指表现出不发出可检测量的在可见光谱内的光线的特征(例如,在底物的存在下)的实体(例如,肽、多肽、复合物、蛋白质等)。例如,如果在指定测定中未显示出可检测的发光,则可称实体为不发光。如本文所使用,术语“不发光”与术语“大体上不发光”同义。例如,不发光多肽(NLpoly)大体上不发光,例如与NLpoly及其不发光互补肽的复合物相比,显示发光减少10倍或更多(例如,100倍、200倍、500倍、1×10
如本文所使用,术语“不发光肽”(例如,NLpep)和“不发光多肽”(例如,NLpoly)指大体上不显示发光(例如,在底物的存在下),或显示低于噪声的量,或在标准条件下(例如,生理条件、测定条件等)并且用典型仪器(例如,光度计等),与显著信号(例如,发光复合物)相比时,显示10倍或更多(例如,100倍、200倍、500倍、1×10
如本文所使用,术语“相互作用元件”指帮助将一对不发光元件或不发光组集合在一起形成生物发光复合物的部分。在典型实施方案中,一对相互作用元件(亦称“相互作用对”)连接到一对不发光元件(例如,不发光肽/多肽对)上,并且两个相互作用元件之间的吸引性相互作用促进生物发光复合物形成;可是本发明不限于此类机制,并且实践本发明不需要理解所述机制。相互作用元件可通过任何适合机制(例如,使不发光对/组紧密靠近,使不发光对/组置于稳定相互作用的正确构象,减少复合物形成的活化能,其组合等)促进生物发光复合物形成。相互作用元件可为蛋白质、多肽、肽、小分子、辅因子、核酸、脂质、碳水化合物、抗体等。相互作用对可由两个相同的相互作用元件(即,同型对)或两个不同的相互作用元件(即,异型对)构成。在异型对的情况下,相互作用元件可为相同类型的部分(例如,多肽)或可为两个不同类型的部分(例如,多肽和小分子)。在研究由相互作用对形成复合物的一些实施方案中,相互作用对可称为“靶对”或“目标对”,并且单独的相互作用元件称为“靶元件”(例如,“靶肽”、“靶多肽”等)或“目标元件”(例如,“目标肽”、“目标多肽”等)。
如本文所使用,术语“先存蛋白”指在某一事件或日期之前物理存在的氨基酸序列。“并非先存蛋白的片段的肽”是并非在设计和/或合成所述肽之前物理存在的蛋白质(例如,合成或自然发生)的片段或子序列的短氨基酸链。
如本文所使用,术语“片段”指由较大完整实体(例如,蛋白质、多肽、酶等)分解或“片段化”产生的肽或多肽,或制备成具有与此类相同的序列的肽或多肽。因此,片段是由其制成和/或设计片段的完整实体(例如,蛋白质、多肽、酶等)的子序列。并非先存全蛋白的子序列的肽或多肽并非片段(例如,并非先存蛋白的片段)。“并非先存生物发光蛋白的片段”的肽或多肽是并非蛋白质(例如,天然或合成)的子序列的氨基酸链,所述蛋白质:(1)在设计和/或合成所述肽或多肽之前物理存在,并且(2)显示出实质性生物发光活性。
如本文所使用,术语“子序列”指与另一较大肽或多肽具有100%序列同一性的肽或多肽。所述子序列是较大氨基酸链的一部分的完美序列匹配。
如本文所使用,术语“序列同一性”指两个聚合物序列(例如,肽、多肽、核酸等)具有单体亚基相同顺序组成的程度。术语“序列相似性”指两个聚合物序列(例如,肽、多肽、核酸等)具有相似聚合物序列的程度。例如,相似氨基酸是共有相同生物物理特征并且可分为家族的氨基酸,例如酸性(例如,天冬氨酸、谷氨酸)、碱性(例如,赖氨酸、精氨酸、组氨酸)、非极性(例如,丙氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸、色氨酸)和不带电极性(例如,甘氨酸、天冬酰胺、谷氨酰胺、半胱氨酸、丝氨酸、苏氨酸、酪氨酸)。通过以下计算“序列同一性百分比”(或“序列相似性百分比”):(1)在比较窗口(例如,较长序列的长度、较短序列的长度、指定窗口)上比较两个最佳比对序列,(2)测定含相同(或相似)单体(例如,在两个序列中出现的相同氨基酸,在两个序列中出现的相似氨基酸)的位置的数量以得到匹配位置的数量,(3)将匹配位置的数量除以比较窗口(例如,较长序列的长度、较短序列的长度、指定窗口)中位置的总数,并且(4)将结果乘以100得到序列同一性百分比或序列相似性百分比。例如,如果肽A和B长度均为20个氨基酸并且除1个位置外全部具有相同氨基酸,则肽A和B具有95%序列同一性。如果不同位置处的氨基酸共有相同的生物物理特征(例如,均为酸性),则肽A和B将具有100%序列相似性。再如,如果肽C长度为20个氨基酸并且肽D长度为15个氨基酸,并且肽D中15个氨基酸中有14个与肽C的一部分的氨基酸相同,则肽C和D具有70%序列同一性,但是肽D与肽C的最佳比较窗口具有93.3%序列同一性。本文为了计算“序列同一性百分比”(或“序列相似性百分比”),将比对序列中的任何空位处理为该位置的错配。
如本文所使用,术语“生理条件”涵盖与活细胞相适的任何条件,例如主要是温度、pH、盐度、化学组成等与活细胞相适的水性条件。
如本文所使用,术语“样品”在广义上使用。在某种意义上,意在包括由任何来源获得的样本或培养物,以及生物和环境样品。生物样品可由动物(包括人)得到并且涵盖液体、固体、组织和气体。生物样品包括血液制品,例如血浆、血清等。样品也可指本文所述肽和/或多肽的细胞裂解物或纯化形式。细胞裂解物可包括已经用裂解剂裂解的细胞或裂解物,例如兔网织红细胞或麦芽裂解物。样品也可包括无细胞表达系统。环境样品包括环境材料例如表面物质、土壤、水、晶体和工业样品。然而不得将此类实例视为限制适用于本发明的相同类型。
如本文所使用,除非另有说明,术语“肽”和“多肽”指通过主链由肽酰胺键(--C(O)NH--)连接的两个或更多个氨基酸的聚合化合物。术语“肽”通常指短氨基酸聚合物(例如,具有少于25个氨基酸的链),而术语“多肽”通常指较长的氨基酸聚合物(例如,具有多于25个氨基酸的链)。
发明详述
特别是在生理条件下和/或在生理表达水平下,蛋白质相互作用的研究需要高灵敏度。在本文所述的特定实施方案中,基于合在一起形成能够产生可检测信号(例如,发光)的生物发光复合物的两个不发光元件的缔合检测蛋白质与小分子、核酸、其它蛋白质等的相互作用。生物发光复合物的形成取决于监测的蛋白质相互作用。
本文提供了由两种或更多种不发光肽和/或多肽单元(例如,不发光对)装配生物发光复合物的组合物和方法。在一些实施方案中,不发光肽和/或多肽单元并非先存蛋白的片段(例如,并非已知多肽序列的互补子序列)。具体而言,经由与不发光肽的结构互补赋予不发光多肽以生物发光活性。
在一些实施方案中,本文提供了用于检测和监测分子相互作用(例如,蛋白质-蛋白质、蛋白质-DNA、蛋白质-RNA相互作用、RNA-DNA、蛋白质-小分子、RNA-小分子等)的不发光对。本文还提供了在形成各种生物发光复合物后具有可变亲和力和发光(例如,高亲和力/高发光对、中等亲和力/高发光对、低亲和力/中等发光对等)的可交换不发光元件(例如,肽和多肽)的互补群组。利用不发光元件的不同组合提供了包括范围从较低到较高亲和力、发光和其它可变特征的各种配对的可适应系统。这种适应性允许检测/监测微调为特定目标分子的分子相互作用并且使可监测的分子相互作用范围扩大为包括具有极低或极高亲和力的相互作用。本文进一步提供了开发和试验不发光对(或组)及不发光对(或组)的群组的方法。
在一些实施方案中,单独的不发光对的肽/多肽成员之间的相互作用不足以形成生物发光复合物和产生作为结果的生物发光信号。然而,如果相互作用元件连接到不发光对的每个肽/多肽成员上,则相互作用对的相互作用(例如,形成相互作用复合物)促进生物发光复合物形成。在此类实施方案中,来自于生物发光复合物的生物发光信号(或在底物的存在下产生这样一种信号的能力)用作相互作用复合物形成的报道分子。如果形成相互作用复合物,则形成生物发光复合物,并且检测/测量/监测到生物发光信号(例如在底物的存在下)。如果未能形成相互作用复合物(例如,由于条件不利、由于相互作用元件之间的相互作用不稳定、由于相互作用元件不相容),则未形成生物发光复合物,并且未产生生物发光信号。
在某些实施方案中,相互作用对包括两个目标分子(例如,目标蛋白)。例如,可通过将每个分子拴系到不发光对的单独成员上进行测定以检测两个目标分子的相互作用。如果目标分子相互作用(例如,瞬时相互作用、稳定相互作用等),使不发光对呈适合构象紧密靠近并且形成生物发光复合物(并且产生/检测到生物发光信号(在底物的存在下))。在目标分子之间没有相互作用(例如,无复合物形成、甚至无瞬时相互作用等)时,不发光对未以充分方式相互作用,并且未产生或仅较弱地产生生物发光信号。此类实施方案可用于研究抑制剂对复合物形成的影响、突变对复合物形成的影响、条件(例如,温度、pH等)对复合物形成的影响、小分子(例如,潜在治疗剂)与靶分子的相互作用,等等。
不同的不发光对可能需要相互作用复合物的不同强度、持续时间和/或稳定性以引起生物发光复合物形成。在一些实施方案中,产生可检测的生物发光信号需要稳定的相互作用复合物。在其它实施方案中,即使较弱或瞬时的相互作用复合物也引起生物发光复合物形成。在一些实施方案中,相互作用复合物的强度或范围与所产生的生物发光信号的强度成正比。一些不发光对在与具有高毫摩尔解离常数(例如,K
在一些实施方案中,不发光肽/多肽中的一个或多个并非先存蛋白的片段。在一些实施方案中,不发光肽/多肽中的一个或多个并非先存生物发光蛋白的片段。在一些实施方案中,不发光肽/多肽都不/全不是先存蛋白的片段。在一些实施方案中不发光肽/多肽都不/全不是先存生物发光蛋白的片段。在一些实施方案中,装配在一起形成生物发光复合物的不发光肽或不发光多肽都不是先存蛋白的片段。在一些实施方案中,用于本发明实施方案中的不发光元件并非先存蛋白的子序列。在一些实施方案中,用于本文所述实施方案中的不发光对不包含先存蛋白的互补子序列。
在一些实施方案中,不发光肽/多肽分离时大体上不发光。在某些实施方案中,当置于适合条件(例如,生理条件)下时,不发光肽/多肽相互作用形成生物发光复合物并且在底物的存在下产生生物发光信号。在其它实施方案中,不添加一个或多个相互作用元件(例如,连接到组分不发光肽和不发光多肽的互补相互作用元件),不发光肽/多肽不能形成生物发光复合物或仅较弱地形成复合物。在此类实施方案中,不发光肽/多肽在彼此单独存在时大体上不发光,但是当被相互作用元件聚集、缔合、定向或以其它方式集合在一起时,产生明显可检测的发光。在一些实施方案中,不添加一个或多个相互作用元件(例如,连接到组分肽和多肽的互补相互作用元件),装配成生物发光复合物的肽和/或多肽在彼此存在时产生低水平的发光,但是当被相互作用元件聚集、缔合、定向或以其它方式集合在一起时,经历可检测发光的明显增加。
在一些实施方案中,本文所述的组合物和方法包括一个或多个相互作用元件。在典型实施方案中,相互作用元件是连接到肽和/或多肽上以装配成生物发光复合物的部分(例如,肽、多肽、蛋白质、小分子、核酸、脂质、碳水化合物等)。相互作用元件通过任何适合机制促进生物发光复合物的形成,包括:与一个或两个不发光元件相互作用,诱导不发光元件内的构象变化,与另一相互作用元件(例如,连接到另一个不发光元件的相互作用元件)相互作用,使不发光元件紧密靠近,为不发光元件定向以进行正确的相互作用,等等。
在一些实施方案中,向含有不发光元件的溶液添加一个或多个相互作用元件,但是未连接到不发光元件上。在此类实施方案中,相互作用元件与不发光元件相互作用以诱导生物发光复合物形成或产生适合生物发光复合物形成的条件。在其它实施方案中,单个相互作用元件连接到不发光对的一个成员上。在此类实施方案中,单独的相互作用元件与一个或两个不发光元件相互作用以产生形成生物发光复合物的有利相互作用。在本发明的典型实施方案中,不发光对的每个成员连接一个相互作用元件。相互作用元件之间的有利相互作用促进不发光元件之间的相互作用。相互作用对可稳定地相互作用,瞬时相互作用,形成复合物等。相互作用对的相互作用通过任何适合机制促进不发光元件的相互作用(和生物发光复合物的形成),包括但不限于:使不发光对成员紧密靠近,由相互作用为不发光对成员正确定向,减小对抗不发光对相互作用的非共价力,等等。
在一些实施方案中,相互作用对包括促进缔合的不发光对的相互作用的任两个化学部分。相互作用对可由例如:两个互补核酸、两个能够二聚的多肽(例如,同型二聚体、杂二聚体等)、蛋白质和配体、蛋白质和小分子、抗体和表位、一对反应性小分子等组成。任一对适合的相互作用分子均可用作相互作用对。
在一些实施方案中,相互作用对包括两个目标分子(例如,目标蛋白)或靶分子。在一些实施方案中,本文的组合物和方法提供了研究一对靶分子之间的相互作用的有用测定(例如,体外、体内、原位、完整动物等)。
在某些实施方案中,一对各自连接到不发光元件之一上的相互作用元件相互作用并由此促进生物发光复合物形成。在一些实施方案中,配体、底物、辅因子或附加相互作用元件(例如,未连接到不发光元件上)的存在对诱导相互作用元件之间的相互作用和促进生物发光复合物形成有必要。在一些实施方案中,检测来自于生物发光复合物的信号指示配体、底物、辅因子或附加相互作用元件或允许与相互作用元件相互作用的条件的存在。
在一些实施方案中,一对相互作用元件和一对不发光元件均存在于单条氨基酸链中(例如,(相互作用元件1)-NLpep-(相互作用元件2)-NLpoly、NLpoly-(相互作用元件1)-NLpep--(相互作用元件2)、NLpoly-(相互作用元件1)-(相互作用元件2)-NLpep等)。在一对相互作用元件和一对不发光元件均存在于单条氨基酸链中的一些实施方案中,对于相互作用对而言形成相互作用复合物和促进生物发光复合物形成需要配体、底物、辅因子或附加相互作用元件。
在某些实施方案中,相互作用元件和不发光元件连接、融合、链接、相连等。在典型实施方案中,第一不发光元件和第一相互作用元件相互连接,并且第二不发光元件和第二相互作用元件相互连接。信号和相互作用元件的连接可通过任何适合机制、化学方法、接头等实现。相互作用和不发光元件通常通过共价相连连接,但是也提供了两种元件的非共价链接。在一些实施方案中,信号和相互作用元件直接相连,而在其它实施方案中,其通过接头相连。
在相互作用元件为肽或多肽的一些实施方案中,信号和相互作用元件含于单条氨基酸链中。在一些实施方案中,单条氨基酸链包含、由或基本上由不发光元件和相互作用元件组成。在一些实施方案中,单条氨基酸链包含、由或基本上由不发光元件、相互作用元件和任选一个或多个N端序列、C端序列、调控元件(例如,启动子、翻译起始位点等)和接头序列组成。在一些实施方案中,信号和相互作用元件含于融合多肽中。信号和相互作用元件(及融合物中要包括的任何其它氨基酸片段)可单独表达,然而,在其它实施方案中,表达包含相互作用和信号序列或由相互作用和信号序列组成的融合蛋白。
在一些实施方案中,包含第一不发光元件和第一相互作用元件的第一融合蛋白以及包含第二不发光元件和第二相互作用元件的第二融合蛋白在相同细胞内表达。在此类实施方案中,从细胞中纯化和/或分离第一和第二融合蛋白,或在细胞内测定融合蛋白的相互作用。在其它实施方案中,第一和第二融合蛋白在单独细胞中表达并且组合(例如,在纯化和/或分离后,或在细胞或部分细胞融合后,或通过从一个细胞到另一个细胞转移融合蛋白,或通过将一种或多种融合蛋白分泌到细胞外基质中)进行信号检测。在一些实施方案中,一种或多种融合蛋白在细胞裂解物(例如,兔网织红细胞裂解物)或无细胞系统中表达。在一些实施方案中,一种或多种融合蛋白由病毒或其它细胞病原体的基因组表达。
在某些实施方案中,提供了编码本发明的肽、多肽、融合多肽、融合蛋白等的核酸、DNA、RNA、载体等。此类核酸和载体可用于表达、转化、转染、注射等。
在一些实施方案中,不发光元件和相互作用元件通过接头相连。在一些实施方案中,接头在元件之间提供所需量的空间/距离的同时,使信号和和相互作用元件相连。在一些实施方案中,接头允许信号和和相互作用元件同时形成其各自的配对(例如,不发光对和相互作用对)。在一些实施方案中,接头帮助相互作用元件促进形成不发光对相互作用。在一些实施方案中,当相互作用对形式时,将每个不发光元件连到其各自的相互作用元件的接头使不发光元件按恰当距离和构象定位以形成生物发光复合物。在一些实施方案中,通过接头保持相互作用元件和不发光元件紧密靠近(例如,<4个单体单元)。在一些实施方案中,接头在信号和相互作用元件之间提供了所需量的距离(例如,1、2、3、4、5、6…10…20个或更多个单体单元)(例如,为防止信号和相互作用元件之间不合需要的相互作用,出于空间考虑,为允许复合物形成由相互作用复合物普及到不发光元件等)。在某些实施方案中,接头在信号和相互作用元件之间提供了适当的连接化学性质。接头也可改进制备信号和相互作用元件的合成过程(例如,允许其作为单一单元合成,允许两个元件合成后相连等)。
在一些实施方案中,接头是能够将不发光元件链接、连接或拴系到相互作用元件上的任何适合化学部分。在一些实施方案中,接头是一个或多个重复或非重复单体单元(例如,核酸、氨基酸、含碳聚合物、碳链等)的聚合物。当不发光元件和相互作用元件是融合蛋白的一部分时,接头(当存在时)通常是氨基酸链。当不发光元件和相互作用元件在单独元件表达之后拴系在一起时,接头可包含在任一端具有与信号和相互作用元件上的官能团分别有反应性的官能(或反应性)基团的任何化学部分。能够拴系信号和相互作用元件的任何适合部分均可用作接头。
可使用各种各样的接头。在一些实施方案中,所述接头为单个共价键。在一些实施方案中,所述接头包含具有1-20个非氢原子(例如,C、N、P、O和S)的直链或支链、环状或杂环、饱和或不饱和结构并且由烷基、醚、硫醚、亚胺、羧基、胺、酯、甲酰胺、磺胺、酰肼键和芳香族键或芳香杂环键组成。在一些实施方案中,接头长于20个非氢原子(例如,21个非氢原子、25个非氢原子、30个非氢原子、40个非氢原子、50个非氢原子、100个非氢原子等)。在一些实施方案中,接头包含选自1-50个选自C、N、P、O和S的非氢原子(除氢原子外)(例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49或50个非氢原子)。
本发明不受可用接头的类型限制。信号和相互作用元件直接地(例如接头由单个共价键组成)链接或经由适合的接头链接。本发明不限于任何特定接头类别。考虑到了多种接头类别,并且适合的接头可包括但不限于烷基、亚甲基碳链、醚、聚醚、烷基酰胺接头、肽接头、经修饰的肽接头、聚(乙二醇)(PEG)接头、链霉亲和素-生物素或抗生物素蛋白-生物素接头、聚氨基酸(例如聚赖氨酸)、官能化PEG、多糖、糖胺聚糖、树枝状聚合物(WO93/06868和Tomalia等在Angew.Chem.Int.Ed.Engl.29:138-175(1990)中,以引用的方式整体并入本文)、PEG-螯合剂聚合物(W94/08629、WO94/09056和WO96/26754,以引用的方式整体并入本文)、寡核苷酸接头、磷脂衍生物、烯基链、炔基链、二硫化物或其组合。在一些实施方案中,接头可裂解(例如,酶促(例如,TEV蛋白酶位点)、经化学、光诱导等)。
在一些实施方案中,大体上不发光的肽和多肽与现有荧光素酶(例如,以引用的方式整体并入本文的美国专利申请2010/0281552和美国专利申请2012/0174242中所述的萤火虫荧光素酶、海肾荧光素酶、刺虾荧光素酶、增强型刺虾荧光素酶)的任何部分具有小于100%序列同一性和/或相似性。本发明的某些实施方案涉及与整个SEQ ID NO:2157(例如,完整的NANOLUC序列)或其一部分(例如,对于肽而言为8个或更多个氨基酸,小于约25个氨基酸)具有小于100%序列同一性的不发光肽和多肽的生物发光复合物形成。本发明的某些实施方案涉及与整个SEQ ID NO:2157(例如,完整的NANOLUC序列)或其一部分(例如,对于肽而言为8个或更多个氨基酸,小于约25个氨基酸)具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性的不发光肽和多肽的生物发光复合物形成。在一些实施方案中,不发光肽和多肽与SEQ ID NO:2157(例如,相互作用以形成生物发光复合物的肽和多肽)的一部分(例如,对于肽而言为8个或更多个氨基酸,小于约25个氨基酸)具有小于100%序列相似性。在一些实施方案中,不发光肽和多肽与SEQ ID NO:2157(例如,相互作用以形成生物发光复合物的肽和多肽)的一部分(例如,对于肽而言为8个或更多个氨基酸,小于约25个氨基酸)具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列相似性。提供了与SEQ ID NO:2157的约25个氨基酸或更小的部分具有小于100%序列同一性和/或相似性的不发光肽,其中此类肽在适当条件下(例如,通过相互作用对稳定)和与另一部分SEQ ID NO:2157具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性/或相似性的多肽组合时形成生物发光复合物。提供了与SEQ ID NO:2157的约25个氨基酸或更小的部分具有小于100%序列同一性和/或相似性的不发光肽,其中此类肽在适当条件下(例如,通过相互作用对稳定)和与另一部分SEQID NO:2157具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性和/或相似性的多肽组合时形成生物发光复合物。提供了与SEQ ID NO:2157的约25个氨基酸或更小的部分具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性和/或相似性的不发光肽,其中此类肽在适当条件下(例如,通过相互作用对稳定)和与另一部分SEQ ID NO:2157具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性和/或相似性的多肽组合时形成生物发光复合物。类似地,提供了与SEQ ID NO:2157的一部分具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性或相似性的不发光多肽,其中此类多肽在适当条件下(例如,通过相互作用对稳定)和与另一部分SEQ ID NO:2157具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性和/或相似性的肽组合时形成生物发光复合物。在一些实施方案中,提供了与SEQ ID NO:2具有小于100序列同一性或相似性的不发光肽。在一些实施方案中,提供了与SEQ ID NO:2具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性或相似性的不发光肽。在一些实施方案中。提供了与SEQ ID NO:440具有小于100序列同一性或相似性的不发光多肽。在一些实施方案中,提供了与SEQ ID NO:440具有小于100%但大于40%(例如,>40%、>45%、>50%、>55%、>60%、>65%、>70%、>75%、>80%、>85%、>90%、>95%、>98%、>99%)序列同一性或相似性的不发光多肽。
在一些实施方案中,用于本发明实施方案中的不发光肽包括来自GVTGWRLCKRISA(SEQ ID NO:236)的具有一个或多个氨基酸取代、缺失或添加的肽。在一些实施方案中,本发明提供了包含表1的一个或多个氨基酸序列的肽,和/或包含表1的核酸序列的核酸(其编码表1的肽序列)。
表1.肽序列
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
在某些实施方案中,提供了来自表1的肽。在某些实施方案中,肽包含与GVTGWRLCKRISA(SEQ ID NO:236)和/或表1中所列任何肽的单个氨基酸差异。在一些实施方案中,肽包含与GVTGWRLCKRISA(SEQ ID NO:236)和/或表1中所列任何肽的两个或更多个(例如,2、3、4、5、6、7、8、9、10个等)氨基酸差异。在一些实施方案中,提供了包含SEQ ID NO:3-438和2162-2365的氨基酸序列之一的肽。在一些实施方案中,提供了包含SEQ ID NO:3-438和2162-2365的氨基酸序列之一,带一个或多个添加、取代和/或缺失的肽。在一些实施方案中,肽或其一部分与SEQ ID NO:3-438和2162-2365的一个或多个氨基酸序列包含大于70%序列同一性(例如,71%、75%、80%、85%、90%、95%、99%)。在一些实施方案中,提供了包含SEQ ID NO:3-438和2162-2365的核酸编码序列之一的核酸。在一些实施方案中,提供了包含SEQ ID NO:3-438和2162-2365的核酸序列之一,带一个或多个添加、取代和/或缺失的核酸。在一些实施方案中,核酸或其一部分与SEQ ID NO:3-438和2162-2365的一个或多个核酸序列包含大于70%序列同一性(例如,71%、75%、80%、85%、90%、95%、99%)。在一些实施方案中,提供了编码SEQ ID NO:3-438和2162-2365的氨基酸序列之一的核酸。在一些实施方案中,提供了编码SEQ ID NO:3-438和2162-2365的氨基酸序列之一,带一个或多个添加、取代和/或缺失的核酸。在一些实施方案中,提供了编码与SEQ ID NO:3-438和2162-2365的一个或多个氨基酸序列具有大于70%序列同一性(例如,71%、75%、80%、85%、90%、95%、99%)的氨基酸的核酸。
在某些实施方案中,提供了来自表1的核酸。在一些实施方案中,提供了编码表1的肽的核酸。在一些实施方案中,本发明的核酸编码包含与MGVTGWRLCERILA(SEQ ID NO:2)和/或表1中所列任何肽的单个氨基酸差异的肽。在一些实施方案中,核酸编码包含与MGVTGWRLCERILA(SEQ ID NO:2)和/或表1中所列任何肽的两个或更多个(例如,2、3、4、5、6、7、8、9、10个等)氨基酸差异的肽。在一些实施方案中,提供了包含表1的核酸之一的序列的核酸。在一些实施方案中,提供了包含表1的核酸之一,带一个或多个添加、取代和/或缺失的核酸。在一些实施方案中,核酸或其一部分与表1的一个或多个核酸包含大于70%序列同一性(例如,71%、75%、80%、85%、90%、95%、99%)。
在一些实施方案中,用于本发明实施方案中的不发光多肽包括与SEQ ID NO:440相比的具有一个或多个氨基酸取代、缺失或添加的多肽。在一些实施方案中,本发明提供了包含表2的一个或多个氨基酸序列的多肽,和/或包含表2的核酸序列的核酸(其编码表2的多肽序列)。
表2.多肽序列
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
表2的多肽和核酸编码序列(SEQ ID NO:441-1298)全部含有N端Met残基(氨基酸)或ATG起始密码子(核酸)。在一些实施方案中,表2的多肽和核酸编码序列没有N端Met残基或ATG起始密码子(SEQ ID NO:1299-2156)。
在某些实施方案中,提供了SEQ ID NO:441-2156的氨基酸聚合物之一的多肽。在一些实施方案中,多肽包含与SEQ ID NO:440的单个氨基酸差异。在一些实施方案中,多肽包含与SEQ ID NO:440和/或SEQ ID NO:441-2156的任何氨基酸聚合物的两个或更多个(例如,2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30…35…40…45…50个或更多个)氨基酸差异。在一些实施方案中,提供了包含SEQ IDNO:441-2156的氨基酸聚合物之一的序列,带一个或多个添加、取代和/或缺失的多肽。在一些实施方案中,多肽或其一部分与SEQ ID NO:441-2156的一个或多个氨基酸聚合物包含大于70%序列同一性(例如,>71%、>75%、>80%、>85%、>90%、>91%、>92%、>93%、>94%、>95%、>96%、>97%、>98%或>99%)。
在某些实施方案中,提供了来自表2的核酸。在一些实施方案中,提供了编码来自表2的多肽的核酸。在一些实施方案中,本发明的核酸编码包含与SEQ ID NO:440和/或SEQID NO:441-2156的任何氨基酸聚合物的单个氨基酸差异的多肽。在一些实施方案中,核酸编码包含与SEQ ID NO:440和/或表2中所列任何多肽的两个或更多个(例如,2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30…35…40…45…50个或更多个)氨基酸差异的多肽。在一些实施方案中,提供了包含SEQ ID NO:441-2156的核酸聚合物之一的序列的核酸。在一些实施方案中,提供了包含SEQ ID NO:441-2156的核酸聚合物之一的序列,带一个或多个添加、取代和/或缺失的核酸。在一些实施方案中,核酸或其一部分与SEQ ID NO:441-2156的一个或多个核酸聚合物包含大于70%序列同一性(例如,>71%、>75%、>80%、>85%、>90%、>91%、>92%、>93%、>94%、>95%、>96%、>97%、>98%或>99%)。在一些实施方案中,核酸或其一部分编码与SEQ ID NO:441-2156的一个或多个核酸聚合物包含大于70%序列同一性(例如,>71%、>75%、>80%、>85%、>90%、>91%、>92%、>93%、>94%、>95%、>96%、>97%、>98%或>99%)的多肽。在一些实施方案中,提供了编码SEQ ID NO:441-2156的多肽之一的核酸。在一些实施方案中,提供了编码SEQ ID NO:441-2156的多肽之一,带一个或多个添加、取代和/或缺失的核酸。
在一些实施方案中,不发光肽或多肽和/或相互作用元件包括合成肽、含一个或多个非天然氨基酸的肽、肽模拟物、缀合合成肽(例如,与官能团(例如,荧光团、发光底物等)缀合)。
本发明提供了用于各种领域,包括基础研究、医学研究、分子诊断等的组合物和方法。虽然本文所述的试剂和测定不限于任何特定应用,并且任何有用的应用均应视为在本发明的范围之内,但是下面是利用当前要求保护的发明的示例性测定、试剂盒、领域、试验装置等。
利用本发明实施方案的典型应用涉及监测/检测蛋白质二聚化(例如,杂二聚体、同型二聚体等)、蛋白质-蛋白质相互作用、蛋白质-RNA相互作用、蛋白质-DNA相互作用、核酸杂交、蛋白质-小分子相互作用或分子实体的任何其它组合。第一目标实体连接到不发光对的第一成员上并且第二目标实体连接到不发光对的第二成员上。如果在特定测定条件下产生可检测信号,则推断第一和第二实体的相互作用。此类测定用于监测在任何适合条件(例如,体外、体内、原位、完整动物等)的分子相互作用,并且用于,例如药物发现、阐明分子途径、研究复合物装配的平衡或动力学方面、高通量筛选、近程式传感器等。
在一些实施方案中,已知特征(例如,光谱特征、配对的相互亲和力)的不发光对用于阐明目标相互作用对的亲和力,或了解其相互作用。在其它实施方案中,表征清楚的相互作用对用于确定不发光对的特征(例如,光谱特征、配对的相互亲和力)。
本文所述的实施方案可用于药物筛选和/或药物开发。例如,在一种或多种相关条件(例如,生理条件、疾病条件)下监测小分子药物或整个小分子文库与目标靶蛋白(例如,治疗靶标)的相互作用。在其它实施方案中,测定了小分子药物或整个小分子文库增强或抑制两个实体(例如,受体和配体、蛋白质-蛋白质等)之间的相互作用的能力。在一些实施方案中,药物筛选应用以高通量形式进行以允许检测成千上万个不同分子与靶标的结合,或测试那些分子对其它实体结合的影响。
在一些实施方案中,本发明提供了对活体(例如,细菌、酵母、真核生物、哺乳动物、灵长类动物、人等)和/或细胞内分子相互作用的检测。在一些实施方案中,包含信号和相互作用(靶标)多肽的融合蛋白在细胞或完整生物体内共表达,并且检测信号并与相互作用复合物的形成联系起来。在一些实施方案中,细胞经编码不发光元件、相互作用元件、融合蛋白(例如,包含信号和相互作用元件)等的载体瞬时和/或稳定地转化或转染。在一些实施方案中,产生编码进行本文所述测定的必需融合蛋白的转基因生物。在其它实施方案中,将载体注入完整生物体内。在一些实施方案中,转基因动物或细胞(例如,表达融合蛋白)用于监测拴系(例如,缀合或基因融合)到NL肽序列上将形成复合物的小分子或生物制剂在其集中的亚细胞隔室和/或组织内的生物分布。
在一些实施方案中,采用发光复合物的肽(例如,不发光肽)部分作为蛋白质标签(例如,在细胞内)。在此类实施方案中,将发光复合物的多肽(例如,不发光多肽)部分(例如,能够与不发光肽形成发光复合物)涂到细胞上(例如,作为试剂的一部分)以检测/定量经不发光肽标记的蛋白质的存在。例如,目标蛋白与高亲和力NLpep(例如,NLpep86)融合。然后将NLpep转染至目标细胞内,再向细胞+培养基内添加含NanoGlo+NLpoly11S的试剂,并检测发光。图175中展示了这种测定方案。在一些实施方案中,小尺寸的肽用于蛋白质标记。在一些实施方案中,用于此类系统中的不发光多肽足够稳定而存在于适合缓冲液中更长时间(例如,在腔肠素类似物底物的存在下)。在某些实施方案中,不发光多肽在没有竞争肽时(例如,甚至在腔肠素类似物底物的存在下)具有最低限度的可检测发光。在一些实施方案中,利用优化缓冲条件以满足蛋白质标记所必需的标准。自发性高亲和力多肽和肽用于此类系统中,并且在例如免疫测定、病毒粒子检测、活细胞内蛋白质动力学的研究等中具有实用性。在一些实施方案中,此类系统提供了极小的蛋白质标签(例如,11个氨基酸),其提供高灵敏度检测、稳定性(例如,特别是在变性条件下)和/或宽广的动态范围。
本文提供的组合物和方法,以及以其为基础的任何技术或工艺用于各种应用和领域中,实例应用的非限制性列表如下:
●无抗体蛋白质印迹:例如,使目标蛋白与不发光肽融合(例如,通过基因工程)并通过任何适合方式表达。分离蛋白质(例如,通过PAGE)并转移到膜上。然后用互补的不发光多肽洗涤所述膜(例如,使发光复合物形成),并置于成像仪(例如,利用CCD相机)上,在膜上面有腔肠素类似物(PBI-3939),并且检测目标蛋白(例如,经由发光复合物的发光)。
●“LucCytochemistry”:例如,使目标蛋白与不发光肽或多肽表达融合,然后以与免疫细胞化学类似的方式用互补的不发光多肽或肽检测。
●蛋白质定位测定:例如,向不发光多肽或多肽添加定位信号(例如,经由基因工程)并且在细胞内表达(例如,添加的核定位信号将导致不发光多肽在细胞核内表达)。互补的不发光肽或多肽与目标蛋白融合(例如,经由基因工程)并且在细胞内与不发光多肽或肽一起表达。如果目标蛋白定位在与信号定位的不发光多肽相同的亚细胞隔室(例如,细胞核)内,则产生发光。
●蛋白质稳定性测定:例如,使目标蛋白与不发光肽或多肽融合(例如,经由基因工程)并且在一种或多种目标条件下孵育。添加互补的不发光多肽或肽(例如,在不同时间点),并将发光用于定量目标蛋白的量(例如,稳定性的代表)。
●蛋白质检测/定量:例如,使目标蛋白与不发光肽或多肽融合(例如,经由基因工程)并且通过任何方法表达和/或操纵。然后添加互补的不发光多肽或肽以检测和/或定量目标蛋白。
●蛋白质纯化:例如,使目标蛋白与不发光肽或多肽融合(例如,经由基因工程)并通过任何方法表达。使蛋白质的混合物通过固定的互补不发光多肽或肽(例如,在珠粒、柱、芯片上等),用适合缓冲液洗涤并洗脱(例如,用高离子强度或低pH的缓冲液)。不激活互补的不发光肽或多肽发光的突变形式的不发光肽或多肽可用于洗脱目标蛋白。
●下拉(pull-down):例如,使用固定、互补的不发光多肽分离与不发光肽融合(例如,经由基因工程)的目标蛋白(和相互作用蛋白)。
●G蛋白偶联受体(GPCR)内化测定:例如,使不发光肽或多肽与目标GPCR融合(例如,经由基因工程)并且在细胞表面表达。向细胞培养基中添加互补的不发光多肽或肽并用于检测细胞表面的GPCR。添加配体以刺激GPCR的内化,并且观察到发光减少。
●针对细胞活力的膜完整性测定:例如,当表达不发光多肽的细胞的细胞膜受损时,不发光肽进入细胞(例如,以其它方式不能穿过细胞膜的肽),从而形成发光复合物,并产生发光。
●5-羟甲基胞嘧啶检测:例如,向不发光肽添加胞嘧啶并与DNA和甲基转移酶孵育。甲基转移酶催化仅向经5-羟甲基化的胞嘧啶残基添加硫醇(半胱氨酸)。然后从DNA分离未并入的肽(使用任何可能方法),并且添加不发光多肽以检测与DNA缀合的肽。
●甲酰基胞嘧啶检测:例如,与以上5-羟甲基胞嘧啶检测类似,这种检测方法使用对甲酰基胞嘧啶具有特定反应性的化学性质。
●病毒并入:编码不发光肽或多肽的核酸并入病毒基因组中,并且互补的不发光多肽或肽在靶细胞内组成性表达。在靶细胞转染和不发光肽表达之后,形成生物发光复合物并且检测到信号(例如,在底物的存在下)。
●蛋白质的化学标记:使不发光肽融合或拴系到反应性基团(例如,生物素、琥珀酰亚胺酯、马来酰亚胺等)上。通过反应性基团与目标蛋白的结合,目标蛋白(例如,抗体)经不发光肽标记。因为所述肽较小,所以不影响目标蛋白的功能性。向系统添加互补的不发光多肽,并且在多肽与肽结合后生成发光复合物。
●蛋白酶测定:例如,为目标蛋白酶所识别的肽序列可按防止在暴露于NLPoly后的生物发光的这种方式与NLPep接合。实现这一目的的方式包括将发光猝灭剂连接到蛋白酶识别序列上或使蛋白酶识别序列以阻碍互补的这种方式与NLPep结合。受蛋白酶裂解识别序列的活性后,记录NLPoly与NLPep互补并发光的能力,并且因此所述系统为灵敏性蛋白酶测定。
●RNA检测。
●生物分子接头表征:例如,可在一组条件下通过经由目标接头将NLPep连接到分子上,评价与生物分子例如抗体连接的接头的稳定性。随时间推移,可通过添加NLPoly和腔肠素类似物并且定量产生的生物发光监测通过接头降解生成游离NLPep。
●突变测定:例如,在体外或体内引入的点突变、移码突变等导致来自于互补对的信号增加或信号丢失。此类测定可用于,例如,试验化合物的诱变性。
●肽抑制剂的靶标结合:使用低亲和力NLpep缀合肽(在细胞内表达)监测肽基抑制剂的靶标结合。NLpoly拴系到目标靶标上。结合导致来自于发光复合物的信号丢失。
●信号蛋白酶增益生物传感器:蛋白酶裂解位点在NLpoly和暗肽NLpep(低亲和力)之间表达。裂解释放允许高亲和力NLpep互补NLpoly的暗肽。
●功能蛋白酶增益测定:在蛋白酶(例如,半胱天冬酶、ADAM等)全长底物的裂解位点的近侧工程化NLpep的序列。只要底物保持完整并且肽“隐藏”,所述肽就保持在空间上难以接近。基因工程化蛋白酶底物和NLpoly(例如,NLpoly11S)共转染至靶细胞系内。在诱导导致底物裂解并且其中一个片段N或C端的激活肽暴露的蛋白酶活性后,诱导出荧光素酶活性。这种原理也可扩展以检测构象变化和/或蛋白质修饰。
●使用重组细胞内抗体的细胞内分析物定量:抗体片段在细胞内作为NLpoly或NLpep融合物表达。互补亚基与目标分析物基因融合。当分析物存在时,抗体结合并形成发光复合物。所述应用可扩展到细胞内PTM(例如,磷酸化)生物传感器,其中细胞内抗体仅在其已经磷酸化(或另外被修饰特异性Ab结合)时与分析物结合。
本发明的组合物和方法的以上应用并非限制性并且可在仍属于本发明范围的同时以任何适合方式修改。
本发明还提供了设计和/或优化不发光对/组及由其形成的生物发光复合物的方法。设计与本文所述实施方案一致的不发光对/组,或其群组的任何适合方法均在本发明的范围之内。
在某些实施方案中,不发光对/组重新设计为单独地没有发光并且在缔合后显示发光。在此类实施方案中,不发光元件之间相互作用的强度不足以在没有促进生物发光复合物形成的相互作用元件时产生生物发光信号。
在其它实施方案中,使用生物发光蛋白(例如,SEQ ID NO:2157)作为起始点合理地设计不发光元件和/或不发光对。例如,此类方法可包括:(a)比对三种或更多种相关蛋白的序列;(b)确定相关蛋白的共有序列;(c)提供与测定共有序列的蛋白质相关的生物发光蛋白的第一和第二片段,其中所述片段单独地大体上不发光,但是经所述片段的相互作用后显示发光;(d)使第一和第二片段各自在一个或多个位置突变(例如,在体外、经电脑模拟等),其中所述突变将所述片段的序列改变成与共有序列的相应位置更加相似,其中所述突变产生并非先存蛋白的片段的不发光对,及(e)测试所述不发光对,当未缔合时没有发光而在不发光对缔合后发光。在其它实施方案中,提供、突变并试验了用于测定共有序列的其中一种蛋白质的第一和第二片段。
在一些实施方案中,发光对的肽为“暗肽”,或与其补体(例如,NLpoly)(例如,以低或高亲和力)结合,但是产生最低限度的发光或不产生发光的肽(见图180-182)。在一些实施方案中,高亲和力暗肽用于反向互补,或信号增益测定以测量抑制剂。在一些实施方案中,低亲和力暗肽用于降低NLpoly11S在用于检测高亲和力肽标签(例如,NLpep86)的试剂中的本底。图180中提供了示例性暗肽。
在一些实施方案中,发光对的肽为“猝灭肽,”或含有猝灭部分(例如,DAB)的肽,并且猝灭剂吸收分离的NLpoly(例如,独立于互补NLpep产生的信号)和NLpoly-NLpep复合物(例如,由于复合物形成而产生的信号)产生的光/能。示例性暗猝灭肽将具有适合的吸收光谱并且包括DAB-161(DAB-GWRLFKK)、DAB-162(DAB-GWALFKK)、DAB-163(DAB-VTGWALFEEIL)、DAB-164(DAB-VTGYALFQEIL)、DAB-165(DAB-VTGYALFEQIL)和DAB-166(DAB-VTGYALFEEIL);其中DAB=Dabcyl(475nm猝灭剂)+dPEG4间隔区。
在一些实施方案中,以上方法不限于设计和/或优化不发光对。进行相同步骤以生成成对的单独缺乏指定功能性(例如,酶促活性),但是在缔合时展示出此类功能性的元件。在这些情况的任一种下,不发光对元件之间相互作用的强度可经由突变而改变以确保其在没有促进生物发光复合物形成的相互作用元件时,不足以产生功能性。
实验
实施例1
肽的生成
通过三种方法之一生成肽构建体:使5’-磷酸化寡核苷酸退火,接着连接到pF4Ag-Barnase-HALOTAG载体(Promega Corporation;用SgfI和XhoI切割)或pFN18A(PromegaCorporation;用SgfI和XbaI切割),使用来自于Agilent的Quik Change Lightning Multi试剂盒定点诱变或将克隆外包给Gene Dynamics。
实施例2
肽制备
通过将用编码肽的质粒转化的KRX大肠杆菌细胞的单个菌落(PromegaCorporation)接种到2-5ml的LB培养基中并且在37℃下使其生长过夜,制备实施例1中生成的肽。然后将过夜培养物(10ml)稀释到1L的LB中并且在37℃下使其生长3小时。然后通过向1L培养物中添加10ml 20%鼠李糖诱导培养物并且在25℃下诱导18小时。
诱导后,在4℃下以5000×g离心各800ml的培养物30分钟。然后使产生的沉淀重新悬浮在80ml肽裂解缓冲液(25mM HEPES pH 7.4)、0.1×被动裂解缓冲液(PromegaCorporation)、1ml/ml溶菌酶和0.03U/μl RQ1 DNA酶(Promega Corporation))中并且在室温下孵育15分钟。然后在干冰上冷冻裂解的细胞15分钟,然后在室温浴中解冻15分钟。然后在4℃下以3500×g离心细胞30分钟。将上清液等分成10ml样品,将50μl的一个等分试样置于1.5ml管中。
向50μl样品中,添加450μl H
实施例3
肽分析
实施例1-2中生成的所有肽均含对肽序列:GVTGWRLCKRISA(SEQ ID NO:236)的单个突变。所有肽均与HALOTAG蛋白(Promega Corporation)融合。鉴定为“HT-NLpep”的肽指示所述肽位于HALOTAG蛋白的C端处。在这种情况下,编码所述肽的基因包括终止密码子,但是不包括起始翻译的甲硫氨酸。鉴定为“NLpep-HT”的肽指示所述肽位于HALOTAG蛋白的N端处。在这种情况下,所述肽的确包括起始翻译的甲硫氨酸,但是不包括终止密码子。
为测定所述肽激活发光的能力,用编码来自于实施例1的肽的质粒转化KRX大肠杆菌细胞的单独菌落(Promega Corporation),接种于200μl基本培养基(1×M9盐、0.1mMCaCl
在第一次过夜生长之后,将10μl培养物稀释至190μl新鲜基本培养基中并使其再在37℃下生长过夜。
在第二次过夜生长之后,将10μl培养物稀释至190μl自动诱导培养基(基本培养基+5%葡萄糖和2%鼠李糖)中。然后在25℃下诱导培养物约18小时。
诱导之后,测定小肽突变体培养物的活性。将含WT 1-156片段的培养物汇合,与10ml的2×裂解缓冲液(50mM HEPES pH 7.4、0.3×被动裂解缓冲液和1mg/ml溶菌酶)混合并在室温下孵育10分钟。然后将30μl裂解的WT 1-156培养物等分至白色圆底96孔测定板(Costar 3355)的各孔内。向测定板的各孔中,添加20μl肽培养物,并且在室温下孵育所述板10分钟。孵育之后,添加50μl NANOGLO荧光素酶测定试剂(Promega Corporation),并且在室温下孵育样品10分钟。在GLOMAX光度计上以0.5秒积分测量发光。
结果(见表3和图1)证明了在与野生型不发光多肽互补后,肽中(相对于SEQ IDNO:1)改变(例如,增加、减少)发光的各种突变。认为发光增加源于5个主要因素之一(或其组合),其中任何因素均有益:不发光肽和不发光多肽之间的亲和力、所述肽的表达、细胞内溶解性、细胞内稳定性和生物发光活性。可是本发明不限于任何特定作用机制并且实践本发明不需要理解作用机制。
表3
/>
/>
实施例4
不发光多肽的生成
使用pF4Ag-NanoLuc1-156(WT 1-156)作为模板,使用来自于Clontech的多样化PCR随机诱变试剂盒进行易错PCR(epPCR)。用SgfI和XbaI消化所得PCR产物并连接到pF4Ag-Barnase(Promega Corporation)上,可商购获得的一种含T7和CMV启动子的pF4A载体形式(Promega),并且经修饰为含有大肠杆菌核糖体结合位点。在42℃下通过热休克转化至KRX大肠杆菌细胞(Promega Corporation)内后,使用单独菌落接种透明、平底96孔板(Costar3370)内的200μl培养物。
实施例5
不发光多肽分析
为测定实施例4中生成的不发光多肽突变体的发光,根据实施例3中使用的程序使经含有来自于实施例4的不发光多肽突变体之一的质粒转化的KRX大肠杆菌细胞(PromegaCorporation)的单独菌落生长。还根据实施例3中使用的程序诱导细菌培养物。
为测定每种不发光多肽突变体诱导的培养物,将30μl测定裂解缓冲液(25mMHEPES pH 7.4、0.3×被动裂解缓冲液(Promega Corpo ration))、0.006U/μl RQ1 DNA酶(Promega Corporation)和含肽片段GVTGWRLCKRISA(SEQ ID NO:18)或GVTGWRLFKRISA(SEQID NO:106)的1×肽溶液(如实施例2中解释那样测定所述肽的相对浓度;从测定的相对浓度,于裂解缓冲液中将所述肽稀释至1×)等分到96孔测定板(Costar 3355)的孔内。向测定板的孔内,添加20μl经诱导的不发光多肽突变体培养物,并且在室温下孵育所述板10分钟。孵育之后,添加50μl的NANOGLO荧光素酶测定试剂(Promega Cor poration),并且在室温下孵育样品10分钟。在GLOMAX光度计上以0.5秒积分测量发光。
结果(表4和图2)证明了提高不发光多肽在与两种不同肽互补之后的发光的许多点突变。与所述肽中的突变类似,不发光多肽中的这些突变可源于各种因素,其全部总体上对所述系统有益。
表4
*表4中的单位为RLU(突变体)/RLU(WT)
实施例6
不发光多肽中的甘氨酸至丙氨酸取代
以下实施例鉴定了不发光多肽内可取代为丙氨酸以提供改良(例如,发光信号更强)的不发光多肽的甘氨酸残基。单独地(见图3),或在复合物(图2)中产生取代。如是实施例1所述生成含甘氨酸至丙氨酸取代的不发光多肽。
将每个单一突变菌落接种于200μl基本培养基(1×M9盐、0.1mM CaCl
为测定每种突变体培养物,添加30μl测定裂解缓冲液(50mM HEPES pH 7.5、0.3×被动裂解缓冲液(Promega Corporation))和含有不发光肽(NLpep9-HT的1:10稀释液(NLpep9为SEQ ID NO:17和18;HT为HaloTag大肠杆菌澄清裂解物))的0.006U/μl RQ1 DNA酶(Promega Corporation))。在室温下振荡样品10分钟,然后添加50μlNANOGLO荧光素酶测定试剂(Promega Corporation)。在室温下孵育样品10分钟,并且在GLOMAX光度计上以0.5秒积分测量发光。
为生成NLpep9-HT大肠杆菌澄清裂解物,用NLpep9-HT的单个大肠杆菌菌落接种5ml LB并且在37℃下孵育过夜。然后将500μl过夜培养物稀释于50ml LB中并且在37℃下孵育3小时。添加500μl的20%鼠李糖并且在25℃下孵育18小时。在3000×g下离心表达培养物30分钟,并且使细胞沉淀重新悬浮于5ml肽裂解缓冲液(25mM HEPES(pH 7.5)、0.1×被动裂解缓冲液、1mg/ml溶菌酶和0.3U/μl RQ1DNA酶)中并在室温下孵育10分钟。将裂解样品置于干冰上15分钟,在室温水浴中解冻并且在3500×g下离心30分钟。上清液为澄清裂解物。
图3和4证明了突变对发光的影响。
实施例7
不发光肽内的突变
在以下实施例中,根据与其它脂肪酸结合蛋白(FABP)的比对在不发光肽内产生突变并根据高概率(FABP中的频率)选择突变以鉴定保持/提高活性的突变(例如,NLpep2、4和5)或确定在该位置不大可能耐受突变(例如NLpep3)。NLpep1-5含单个突变(见表1),并且NLpep6-9是NLpep2、4和5中突变的复合集(见表1)。如实施例1所述生成突变体。
将每个突变体菌落接种于200μl基本培养基中并且在37℃下振荡孵育20小时。然后将10μl培养物添加到190μl新鲜基本培养基中并且再在37℃下振荡孵育20小时。然后将第二个10μl培养物添加到190μl自动诱导培养基中并且在25℃下振荡孵育18小时以允许不发光肽突变体表达。
为测定每种突变体培养物,添加30μl测定裂解缓冲液(50mM HEPES pH 7.5、0.3×被动裂解缓冲液(Promega Corporation))和含有不发光多肽(野生型不发光多肽大肠杆菌澄清裂解物的1:10稀释液)的0.006U/μl RQ1 DNA酶(Promega Corporation))。在室温下振荡样品10分钟,然后添加50μl NANOGLO荧光素酶测定试剂(Promega Corporation)。在室温下孵育样品10分钟,并且在GLOMAX光度计上以0.5秒积分测量发光。图1显示了在每种不发光肽突变体中检测到的发光(RLU)。结果证明了能够耐受突变且发光无大幅损失的各个位置,以及改善发光的一些特定突变。
实施例8
融合标签定向对发光的影响
在以下实施例中,比较了带N或C端HaloTag蛋白的不发光肽产生的发光。
根据实施例7中使用的程序使每种肽-HT融合物的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。根据实施例7中使用的程序测定和检测发光。图6和7展示了在每种肽-HT融合物中检测到的发光(RLU)。结果证明了产生与NLpep1相似的发光的突变的组合。
实施例9
多个冻融循环对不发光肽的影响
1ml的NLpep9-HT于干冰上冷冻5分钟,然后于室温水浴中解冻5分钟。然后取出60μl用于测定。然后再重复冻融程序10次。每个冻融循环之后,取出60μl样品用于测定。
为了测定,将各20μl的冻融样品与30μl的SEQ ID NO:2混合并且在室温下孵育10分钟。添加50μl的NANOGLO荧光素酶测定试剂,并且在室温下孵育样品10分钟。在GLOMAX光度计上以0.5秒积分测量发光。结果描述于图8并且证明NLpep可经受多个冻融循环且活性(发光)无损失。
实施例10
不发光肽中突变的区别
在以下实施例中,使用TMR凝胶分析归一化不发光肽突变体的浓度以区别改变表达的突变与改变发光的突变(例如,改变的发光可源于改变的结合亲和力)。
用单个突变体肽菌落接种5ml的LB并且在37℃下振荡孵育20小时。将50μl过夜培养物稀释至5ml新鲜LB中并且在37℃下振荡孵育3小时。然后添加50μl的20%鼠李糖并且在25℃下振荡孵育18小时。
对于TMR凝胶分析而言,将各79μl的诱导培养物与10μl 10×Fast Break裂解缓冲液(Promega Corporation)、10μl的HALOTAG TMR配体(Promega Corporation)不发光多肽的1:100稀释液和10μl的RQ1DNA酶混合并且在室温下孵育10分钟。添加33.3μl的4×SDS上样缓冲液,并且在95℃下孵育样品5分钟。根据生产商的指示将各15μl的样品上样到SDS凝胶上并进行电泳。然后在Typhoon上扫描凝胶。根据TMR-凝胶强度稀释每种培养物以归一化浓度。然后将各20μl的稀释培养物与30μl含不发光多肽(SEQ ID NO:2大肠杆菌澄清裂解物的1:10稀释液)的测定裂解缓冲液混合并且在室温下振荡孵育10分钟。添加50μl的NANOGLO荧光素酶测定试剂,并且在室温下孵育样品10分钟。在GLOMAX光度计上以0.5秒积分测量发光(见图9)。
实施例11
不发光多肽中的定点饱和
在以下实施例中,由筛选野生型不发光多肽中的随机突变文库将位置11、15、18、31、58、67、106、149和157鉴定为目标位点。比较了这些位置处的全部20种氨基酸(基于实施例6中生成的5A2不发光突变体(SEQ ID NO:539和540)建立以用5A2突变体中的其它突变验证)以确定该位置处的最佳氨基酸。如先前在实施例1中所述生成突变体不发光多肽。根据实施例6中使用的程序使每种不发光多肽突变体的单菌落生长。还根据实施例6中使用的程序诱导细菌培养物。除使用1:11.85稀释的NLpep53大肠杆菌澄清裂解物外,根据实施例6中使用的程序测定和检测发光。
图10-18展示了突变对有和无NLpep的情况下产生发光的能力的影响。
实施例12
不发光肽中半胱氨酸对比脯氨酸作为第一氨基酸的比较
在以下实施例中,进行了不发光肽中使用半胱氨酸或脯氨酸作为第一氨基酸(在必要甲硫氨酸之后)的比较。如先前在实施例1中所述生成突变体不发光多肽。根据实施例7中使用的程序使每种不发光多肽突变体的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。根据实施例7中使用的程序测定和检测发光。图19证明半胱氨酸和脯氨酸均可用作NLpep的第一氨基酸并产生发光。
实施例13
不发光肽突变最佳复合集的鉴定
在以下实施例中,鉴定了不发光肽突变的最佳复合集。如先前在实施例1中所述生成突变体不发光多肽。
1)对于不发光肽复合突变体NLpep53、NLpep66、NLpep67和NLpep68而言,根据实施例10中使用的程序使各自的单菌落生长。还根据实施例10中使用的程序诱导细菌培养物。根据实施例10中使用的程序测定和检测TMR凝胶分析和发光。图20中的结果展示了含多个突变的NLpep的发光以及大肠杆菌表达。
2)对于不发光肽复合突变体NLpep53和NLpeps 66-74而言,根据实施例7中使用的程序使各自的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。根据实施例7中使用的程序测定和检测发光。图21中的结果展示了含多个突变的NLpep的发光。
3)对于不发光肽复合突变体NLpep53和NLpeps 66-76而言,根据实施例7中使用的程序使各自的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。除不发光多肽为5A2或5A2+R11E(大肠杆菌澄清裂解物的1:10稀释液)外,根据实施例7中使用的程序测定和检测发光。图22中的结果展示了含多个突变与5A2或5A2+R11E的NLpep的发光。这些结果还展示了当NLpoly突变R11E与含E作为第9个残基的NLpep(NLpep72、75和76)互补时的较低发光。
4)对于不发光肽复合突变体NLpep1、NLpep69、NLpep78和NLpep79而言,根据实施例7中使用的程序使各自的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。除不发光多肽为WT(大肠杆菌澄清裂解物的1:10稀释液)外,根据实施例7中使用的程序测定和检测发光。图23中的结果展示了含多个突变的NLpep的发光。
实施例14
复合不发光多肽突变体
在以下实施例中,将来自于文库筛选的9种突变组合成复合克隆(NLpoly1,SEQ IDNO:941、942),然后突变之一恢复为原氨基酸(NLpoly2-10,SEQ ID NO:943-960)以便鉴定最佳复合集。基于NLpoly1-10的先前结果,出于相同目的设计并测试NLpoly11-13(SEQ IDNO:961-966)。如先前在实施例1中所述生成突变体NLpoly。根据实施例6中使用的程序使每种不发光多肽突变体的单菌落生长。还根据实施例6中使用的程序诱导细菌培养物。除使用以1:11.85稀释的NLpep53大肠杆菌澄清裂解物外,根据实施例6中使用的程序测定和检测发光。
图24展示了含多个突变的NLpoly的发光。
实施例15
不发光多肽突变体的底物特异性
以下实施例研究了不发光多肽突变体的底物特异性。由各种不发光多肽突变体(作为底物的腔肠素类似物或腔肠素)和各种不发光肽形成的发光复合物产生发光。
按100,000个细胞/ml将HEK 293细胞涂铺到24孔板的含有0.5ml DMEM+10% FBS的孔内(50,000个/孔)。在37℃、5% CO
在图25-26中,除使用以1:10稀释的NLpep53大肠杆菌澄清裂解物并且使用了于NanoGlo荧光素酶测定缓冲液或DMEM中的腔肠素类似物或腔肠素外,根据实施例6中使用的程序测定和检测发光。该数据展示了在有腔肠素类似物或腔肠素作为底物的NANOGLO和DMEM中NLpoly的发光。这指示了在NANOGLO和DMEM中NLpoly的底物特异性(腔肠素类似物对比腔肠素)。
在图27中,除使用以1:10稀释的来自于各种不发光肽(NLpep1、NLpep9、NLpep48、NLpep53、NLpep69或NLpep76)的大肠杆菌澄清裂解物外,根据实施例6中使用的程序测定和检测发光。另外,使用了于NanoGlo荧光素酶测定缓冲液中的腔肠素类似物或腔肠素。该数据展示了NLpoly/NLpep对的底物特异性。
在图28中,通过单独地将NLpep53-HT融合物1:10和不发光多肽裂解物1:10稀释于DMEM+0.1% Prionex中测定和检测发光。然后合并20μl不发光肽和20μl不发光多肽并且在室温下孵育10分钟。然后向样品中添加40μl含100μM腔肠素类似物的NanoGlo缓冲液或含0.1% Prionex和20μM腔肠素类似物的DMEM,并且在GloMax Multi上检测发光。该数据展示了在HEK293细胞内表达的NLpoly的底物特异性。
在图29中,通过单独地将NLpep1-HT、NLpep53-HT、NLpep69-HT或NLpep76-HT融合物1:10和不发光多肽裂解物1:10稀释于DMEM+0.1% Prionex中测定和检测发光。然后合并20μl不发光肽和20μl不发光多肽并且在室温下孵育10分钟。然后向样品中添加40μl含100uM腔肠素类似物的NanoGlo缓冲液或含0.1% Prionex和20uM腔肠素类似物的DMEM,并且在GloMax Multi上检测发光。该数据展示了在哺乳动物细胞内表达并且用各种NLpep测定的NLpoly的发光。
实施例16
用腔肠素类似物或腔肠素的不发光多肽突变体的信背比
以下实施例研究了不发光多肽突变体的信背比。使用腔肠素类似物或腔肠素作为底物以及用各种不发光肽测量由各种不发光多肽突变体产生的发光。
按15,000个细胞/孔将HEK 293细胞涂铺到96孔板孔内的100μlDMEM+10% FBS中。在37℃、5% CO
实施例17
发光和底物特异性
以下实施例用NLpep69并且使用腔肠素类似物或腔肠素作为底物研究了各种不发光多肽突变体的发光和底物特异性。
按20,000个细胞/孔将CHO细胞涂铺到96孔板孔内100μl的DMEM+10% FBS中。在37℃、5% CO2孵育箱内孵育细胞过夜。通过在OptiMem中添加各0.66ug的用于表达不发光多肽突变体的质粒DNA和不发光肽突变体质粒至31μl的最终体积制备转染复合物。向每种转染复合物中添加2μl Fugene HD并且在室温下孵育15分钟。对于每种肽/多肽组合而言,向96孔板的6个孔中添加5μl转染复合物并使其在37℃下于CO
实施例18
活细胞和裂解条件之间的发光和底物特异性
以下实施例用NLpep69、NLpep78或NLpep79,使用腔肠素类似物或腔肠素作为底物并且在裂解或活细胞条件下研究了各种不发光多肽突变体的发光和底物特异性。
按15,000个细胞/孔将HEK 293细胞涂铺到96孔板孔内的100μlDMEM+10% FBS中。在37℃、5% CO2孵育箱内孵育细胞过夜。通过在OptiMem中添加各0.66ug的用于表达不发光多肽突变体的质粒DNA和不发光肽突变体质粒至31μl的最终体积制备转染复合物。向每种转染复合物中添加2μl Fugene HD并且在室温下孵育15分钟。对于每种NLpoly-NLpep组合而言,向96孔板的6个孔中添加5μl转染复合物并使其在37℃下于CO2孵育箱内生长过夜。孵育过夜之后,去除生长培养基并更换含20uM腔肠素或20uM腔肠素类似物的非CO2非依赖型培养基。在37℃下孵育样品10分钟,并且在1小时时程中在37℃下于GloMax Multi+上进行动力学测量。图32-34展示了以活细胞和裂解形式在哺乳动物细胞内与NLpep69、78或79共表达的NLpoly的底物特异性。
实施例19
大肠杆菌内表达的不发光多肽突变体的比较
根据实施例7中使用的程序使每种不发光多肽的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。除使用以1:1,000稀释的NLpep78-HT或NLpep79-HT外,根据实施例7中使用的程序测定和检测发光。图35展示了在大肠杆菌中表达并且用NLpep78或79测定的NLpoly的发光。
实施例20
在无互补不发光肽的情况下不发光多肽克隆产生发光的能力
根据实施例7中使用的程序使每种不发光多肽的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。除不向测定缓冲液中添加不发光肽外,根据实施例7中使用的程序测定和检测发光。图36展示了在大肠杆菌中表达并且在没有NLpep时测定的NLpoly的发光。
实施例21
在大肠杆菌中表达的不发光多肽突变体的底物特异性
根据实施例7中使用的程序使每种不发光多肽的单菌落生长。还根据实施例7中使用的程序诱导细菌培养物。除将腔肠素类似物或腔肠素与NANOGLO测定缓冲液混合外,根据实施例中使用的程序测定和检测发光。图37展示了在大肠杆菌中表达并且用NLpep78或79测定的NLpoly的底物特异性。
实施例22
带NLpep78的不发光多肽突变体的改善的发光
在CHO和Hela细胞中证明了不发光多肽突变体与NLpep78-HT的互补。
向24孔板的孔内使用Fugene HD,用5ng不发光多肽突变体5A2或5P或用野生型不发光多肽转染CHO和Hela细胞(CHO:在转染前一天接种100,000个;Hela:在转染前一天接种50,000个)并且在37℃下孵育过夜。过夜孵育之后,将培养基更换为无酚红的DMEM,并且在-80℃下冷冻细胞30分钟。然后解冻细胞并转移到1.5ml管内。然后用无酚红的DMEM 1:10稀释细胞裂解物,20μl与NLpep78混合(以1:1,000稀释于无酚红的DMEM中的NLpep78-HT7大肠杆菌裂解物)并在室温下振荡10分钟。添加40μl无酚红的DMEM和20uM腔肠素类似物并且在GloMax上以0.5秒积分测量发光。图38展示了在哺乳动物细胞中表达并且用NLpep78测定的NLpoly的发光。
实施例23
不发光多肽融合和归一化不发光多肽浓度
进行来自于与萤火虫荧光素酶(图39)或叩头虫红色荧光素酶(图40)融合的不发光多肽的原始和归一化发光的比较以提供对例如在表达、溶解性和/或稳定性上,源于不发光多肽的浓度以及作为融合不发光多肽互补的益处程度的了解。
根据实施例22中的程序,用5ng 5P NLpoly-萤火虫荧光素酶融合物、5P NLpoly-叩头虫荧光素酶融合物、野生型5P-萤火虫荧光素酶融合物或野生型5P-叩头虫荧光素酶融合物转染HEK293、Hela或CHO细胞。还根据实施例22制备裂解物。然后用无酚红的DMEM 1:10稀释细胞裂解物,20μl与NLpep78(以1:1,00稀释于无酚红的DMEM中;大肠杆菌裂解物)混合并在室温下振荡10分钟。添加40μl含20uM腔肠素类似物或Bright-Glo(PromegaCorporation)的NanoGlo并且在GloMax上以0.5秒积分测量发光。图39和40展示了在哺乳动物细胞内表达并且用NLpep78测定的5P对比野生型NLpoly的比活性。
实施例24
在活细胞内的互补
该实施例使用野生型或5P NLpoly证明了在活细胞内的互补。将Hela细胞涂铺到涂铺的96孔板的孔内,用0.5ng野生型或5P不发光多肽质粒DNA使用Fugene HD转染并且在37℃下孵育过夜。过夜孵育之后,然后用0.5ng NLpep78-HT质粒DNA使用Fugene HD转染细胞并且在37℃下孵育3小时。然后将培养基更换为非CO
实施例25
在无细胞提取物内的互补
为证明在无细胞提取物内的互补,将0.5ug NLpep78-HT和0.5ug不发光多肽突变体质粒DNA与TNT兔网织红细胞裂解物主混合物(Promega Corporation)混合并且在30℃下孵育1小时。将25μl的无细胞表达提取物与25μl NanoGlo荧光素酶测定试剂混合并且在室温下孵育10分钟。在GloMax上以0.5秒积分测量发光。图42展示了来自于以无细胞形式表达的互补NLpoly/NLpep对的发光。
实施例26
在哺乳动物细胞内表达的不发光多肽与合成不发光肽的结合亲和力
为证明不发光多肽和不发光肽对之间的结合亲和力,如先前所述由Hela、HEK293和CHO细胞制备不发光多肽裂解物并1:10稀释于PBS+0.1% Prionex中。在PBS+0.1%Prionex中制备4×浓度的不发光肽(合成)。将20μl不发光多肽裂解物与20μl不发光肽混合并且在室温下振荡10分钟。添加40μl含腔肠素类似物的NanoGlo荧光素酶测定试剂或PBS+0.1% Prionex并且在室温下振荡10分钟。在GloMax上以0.5秒积分检测发光。使用Graphpad Prism、单点特异性结合(One Site-Specific Binding)测定Kd值。图43和44展示了在不同缓冲条件下(PBS用于互补,然后NanoGlo用于检测;PBS用于互补和检测;NanoGlo用于互补和检测)测量的解离常数。
实施例27
当不发光肽突变体中半胱氨酸突变为苯丙氨酸时提高的结合亲和力
为证明在肽的第8个残基处具有突变的半胱氨酸的不发光肽突变体中的结合亲和力提高,如先前所述由Hela、HEK293和CHO细胞制备不发光多肽突变体裂解物并以1:10稀释于PBS+0.1% Prionex中。在PBS+0.1% Prionex+10mM DTT中制备4×浓度的不发光肽(NLpep)。将20μl不发光多肽裂解物与20μl不发光肽混合并且在室温下振荡10分钟。添加40μl NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上以0.5秒积分检测发光。图45证明NLpep C8F突变显著提高了5P的结合亲和力。
实施例28
Hela细胞中无不发光肽的多肽变体的可检测发光
为证明在无不发光肽的不发光多肽中的发光,使用Fugene HD,用不同量的不发光多肽+pGEM-3zf载体DNA转染96孔板孔内的Hela细胞(在转染前一天接种10,000个)至总计50ng并且在37℃下孵育过夜。孵育之后,将培养基更换为非CO
实施例29
另外的不发光多肽变体的生成
如下所述生成另外的不发光多肽变体:Ile-11(Ile在残基11处)、Val-11、Tyr-11、Glu-11、Glu-157、Pro-157、Asp-157、Ser-157、Met-149、Leu-106、NLpoly11和NLpoly12,并分析其表达。在5A2不发光多肽背景下制备另外的不发光多肽变体。
挑取另外每种不发光多肽变体的单独新鲜菌落(KRX)并使其在30℃下于LB+氨苄青霉素(100μg/ml)中生长过夜,然后1:100稀释于LB+氨苄青霉素中并且使其在37℃下生长2.5小时(OD600约0.5)。添加鼠李糖至0.2%的最终浓度,并且将细胞分成三份并使其在25℃下生长过夜约18小时。在环境温度下使用0.5×Fast Break裂解细胞30分钟,于干冰上速冻并储存在-20℃下。快速解冻后,通过在4℃下10K离心15分钟制备可溶级分。在TecanInfinite F-500光度计上测定样品的发光。
图49展示了如通过SDS-PAGE分析的每种不发光多肽变体的总裂解物和可溶级分。数据提供了关于另外的不发光多肽变体的表达、溶解性和稳定性的信息。大多数另外的不发光多肽变体生成比野生型更多的蛋白质(总蛋白质和可溶蛋白质),但是在许多情况下,差异细微。NLpoly11和NLpoly12的表达提高更明显。
实施例30
另外的不发光多肽变体的本底发光
通过在室温下将25μl不发光多肽变体裂解物与25μl DMEM孵育10分钟测量实施例29中生成的另外的不发光多肽变体的本底发光。然后添加50μl NanoGlo荧光素酶测定试剂,并且在5和30分钟时在Tecan Infinite F500上测量发光。使用单独的NLpep53(Pep 53)和单独的DMEM(DMEM)作为对照。图47证明大多数另外的不发光多肽变体显示出提高的本底发光。
实施例31
另外的不发光多肽变体互补后的发光
通过在室温下将25μl不发光多肽变体裂解物与25μl NLpep-53孵育10分钟测量实施例28中生成的另外的不发光多肽变体的发光,然后添加50μl NanoGlo荧光素酶测定试剂,并且在5和30分钟时在Tecan Infinite F500上测量发光。使用单独的NLpep53(Pep 53)和单独的DMEM(DMEM)作物对照。图48证明不发光多肽变体Val-11、Glu-11、Glu-157、Pro-157、Asp-157、Ser-157和Met-149产生比亲本5A2明显更多的发光。
实施例32
在缺乏不发光肽时不发光多肽的增加的本底发光与可溶性部分中蛋白质的量之间的相关性
挑取不发光多肽变体3P、3E、5P、5E、6P和6E的单独菌落并使其在30℃下于LB+氨苄青霉素中生长过夜,然后1:100稀释于LB+氨苄青霉素中并使其在37℃下生长2.5小时(OD600约0.5)。添加鼠李糖至0.2%的最终浓度,并且将细胞分成三份并使其在25℃下生长约18小时。在环境温度下使用0.5×Fast Break裂解细胞30分钟,于干冰上速冻并储存在-20℃下。快速解冻后,通过在4℃下10K离心15分钟制备可溶级分。在Tecan Infinite F-500上测定样品的发光。图50A显示了每种不发光多肽变体的总裂解物和可溶级分。图50B显示了每种不发光多肽变体的本底发光。图51显示了当与LB培养基中的10或100nM NLpep78(NVSGWRLFKKISN)互补时,每种不发光多肽变体产生的发光。
实施例33
不发光多肽的延长和缺失
在C端通过添加残基VAT、AA、VTG、VT、VTGWR、VTGW、V、A、VA、GG、AT、GTA、ATG或GT或者在5P的C端缺失1-7个残基(例如D1=1个残基缺失,D2=2个残基缺失等)延长不发光多肽变体5P,测量在大肠杆菌裂解物(图52)中的本底发光和在与NLpep78(图53;NVSGWRLFKKISN)或NLpep79(图54;NVTGYRLFKKISN)互补后产生的发光。图55显示了不发光多肽5P变体的信背比。图56提供了发光结果的总结。图57显示了每种不发光多肽5P变体中总裂解物和可溶级分的量。
实施例34
5P和I107L不发光多肽变体的比较
图58显示了5P和I107L的总裂解物和可溶级分的量(A),无不发光肽或有NLpep78或NLpep79的5P或I107L产生的发光(B)及I107L比5P提高的信背比(C)。
实施例35
5P不发光多肽突变体的生成
如先前所述产生在筛选5P不发光多肽变体中的随机突变中鉴定的突变。将每个单一5P不发光多肽突变体菌落接种于200μl基本培养基中并且在37℃下振荡孵育20小时。然后向190μl的新鲜基本培养基中添加10μl培养物并且再在37℃下振荡孵育20小时。然后向190μl自动诱导培养基(基本培养基+5%葡萄糖+2%鼠李糖)中添加第二个10μl的培养物并且在25℃下振荡孵育18小时以允许不发光多肽突变体表达。将10μl的5P不发光多肽突变体表达培养物添加到40μl含NLpep78-HT(1:386稀释)或NLpep79-HT(1:1,000稀释)的测定裂解缓冲液中并且在室温下振荡10分钟。添加50μl含100μM腔肠素的NanoGlo测定缓冲液并且在室温下振荡10分钟。在GloMax上以0.5秒积分测量发光。图59-62A显示了本底发光,而图59-62B和C显示了在与NLpep78或NLpep79互补之后产生的发光。
实施例36
延长型不发光多肽变体和缺失型不发光肽之间的结合亲和力
延长型不发光多肽变体(即,在C端含附加氨基酸)和缺失型不发光肽(即,在N端缺失的氨基酸)之间的结合亲和力。
如先前所述制备的表达不发光多肽5P/+V/+VT/+VTG的大肠杆菌的裂解物以1:2000稀释于PBS+0.1% Prionex中。在室温下用25μl的NLpep78、NLpep80、NLpep81或NLpep82(于稀释缓冲液中稀释的0-500nM)孵育25μl的稀释裂解物5分钟。向每份样品添加50μl用NanoGlo测定缓冲液稀释至1×的腔肠素类似物并且在室温下孵育10分钟。在GloMaxMulti上以0.5秒积分时间测量发光。图63展示了在C端带附加氨基酸的NLpoly与氨基酸从N端缺失的NLpep之间的结合亲和力。
实施例37
大肠杆菌中表达的不发光多肽与合成不发光肽之间的结合亲和力
制备不发光多肽LB裂解物并且1:100稀释于PBS+0.1% Prionex中。于PBS+0.1%Prionex中制备合成NLpep78的2×稀释液。将25μl的稀释不发光多肽裂解物与各25μl的不发光肽稀释液混合并且在环境温度下孵育3分钟。添加50μl的NanoGlo荧光素酶测定试剂,在室温下孵育5分钟,并且在GloMax Multi+上测量发光。图64显示了使用单点特异性结合计算的Kd值。
实施例38
哺乳动物细胞中表达的5P不发光多肽与NLpep80或NLpep87之间的结合亲和力
将表达NLpoly 5P的CHO、HEK293T或HeLa细胞的裂解物1:1000稀释于稀释缓冲液(PBS+0.1% Prionex)中。在室温下用25μl的NLpep80/87(于稀释缓冲液中稀释成0-5μM)孵育25μl的稀释裂解物5分钟。向每孔添加50μl的腔肠素类似物(用NanoGlo缓冲液稀释至1×),并且在室温下孵育所述板10分钟。然后在GloMax Multi上以0.5秒积分时间读取发光(图65)。
实施例39
大肠杆菌中表达的5P不发光多肽与NLpep80或NLpep87之间的结合亲和力
将表达NLpoly 5P的大肠杆菌的裂解物1:2000稀释于稀释缓冲液(PBS+0.1%Prionex)中。在室温下用25μl的NLpep80/87(于稀释缓冲液中稀释成0-5μM)孵育25μl的稀释裂解物5分钟。向每孔添加50μl的腔肠素类似物(用NanoGlo缓冲液稀释至1×),并且在室温下孵育所述板10分钟。然后在GloMax Multi上以0.5秒积分时间读取发光(图66)。
实施例40
缺失型不发光多肽与延长型不发光肽之间的互补
进行缺失型不发光多肽(即,从C端缺失的氨基酸)和延长型不发光肽(即,向N端添加的氨基酸)之间的互补。如先前在实施例6中所述制备NLpep-HT大肠杆菌澄清裂解物。经由HaloTag融合定量NLpep-HT的量。简言之,将10μl的澄清裂解物与10μl HaloTag-TMR配体(1:100稀释)和80μl水混合并且在室温下孵育10分钟。添加33.3μl4×SDS上样缓冲液并且在95℃下孵育5分钟。将15μl上样到SDS-PAGE凝胶上并且在Typhoon上成像。基于来自于SDS-PAGE凝胶的强度,将不发光肽稀释于PBS+0.1% Prionex不发光肽中以产生相等浓度。然后将不发光多肽裂解物1:100稀释于PBS+0.1%Prionex中。混合20μl稀释不发光多肽和20μl稀释不发光肽并且在室温下振荡10分钟。添加40μl NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上使用0.5秒积分测量发光。图67展示了带有在N端有附加氨基酸的NLpep的从C端去除了氨基酸的NLpoly的发光。
实施例41
Hela细胞中表达的5P不发光多肽与NLpep78或截短
NLpep78(NLpep80-87)之间的结合亲和力
如先前所述由Hela细胞制备5P不发光多肽裂解物并且于PBS+0.1% Prionex中1:10稀释制备。于PBS+0.1% Prionex中制备4×浓度(在初步滴定实验中确定的范围)的不发光肽(合成肽;通过Peptide2.0(Virginia);以5、10或20mg级制成;最后通过乙酰化和酰胺化封端,并通过净肽含量分析验证)。混合20μl 5P不发光多肽和20μl不发光肽并且在室温下振荡10分钟。添加40μl的NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上以0.5秒积分测量发光。图68展示了5P和截短形式的NLpep78之间的结合亲和力和相应发光。当从N端、C端去除1个氨基酸,或从每端去除1个氨基酸时,结合亲和力增加。从任一端去除1个以上氨基酸使亲和力降低,但是并非总是使Vmax降低到相同程度。
实施例42
延长型不发光多肽和截短不发光肽之间的结合亲和力
测定延长型不发光多肽(即,在C端具有2个额外氨基酸的不发光多肽)与截短不发光肽(即,从N端去除了2个氨基酸的不发光肽)(NLpep81)之间的结合亲和力。
如先前所述制备不发光多肽裂解物并且于PBS+0.1% Prionex中1:100稀释制备。于PBS+0.1% Prionex中制备NLpep81的2×稀释液(合成肽;通过肽2.0(Virginia);以5、10或20mg级制成;最后通过乙酰化和酰胺化封端,并通过净肽含量分析验证)。混合25μl不发光多肽和各25μl的不发光肽稀释液并且在室温下振荡3分钟。添加50μl的NanoGlo荧光素酶测定试剂并且在室温下振荡5分钟。在GloMax上以0.5秒积分测量发光。图69显示了使用单点特异性结合计算的Kd值。
实施例43
延长型不发光多肽和截短不发光肽之间的结合亲和力
测定延长型不发光多肽(即,在C端具有3个额外氨基酸的不发光多肽)与截短不发光肽(即,从N端去除了3个氨基酸的不发光肽)(NLpep82)之间的结合亲和力。
制备不发光多肽裂解物并且于PBS+0.1% Prionex中1:100稀释制备。于PBS+0.1% Prionex中制备NLpep82的2×稀释液(合成肽;通过Peptide 2.0(Virginia);以5、10或20mg级制成;最后通过乙酰化和酰胺化封端,并通过净肽含量分析验证)。混合25μl不发光多肽和各25μl的不发光肽稀释液并且在室温下振荡3分钟。添加50μl的NanoGlo荧光素酶测定试剂并且在室温下振荡5分钟。在GloMax上以0.5秒积分测量发光。图70显示了使用单点特异性结合得到的计算的Kd值。
实施例44
大肠杆菌中表达的不发光多肽克隆与合成NLpep78之间的结合亲和力
使不发光多肽变体在M9基本培养基上生长。接种单独菌落并使其在37℃下生长过夜。将样品1:20稀释于M9基本培养基中并使其在37℃下生长过夜。将样品再次1:20稀释于M9诱导培养基中并使其在25℃下生长过夜。汇合样品,并用400μl的PLB裂解缓冲液裂解100μl的汇合细胞并且在室温下孵育10分钟。将裂解物1:100稀释于PBS+0.1% Prionex中。于PBS+0.1% Prionex中制备合成NLpep78的2×稀释液。将25μl的不发光多肽稀释液与各25μl的不发光肽稀释液混合并且在室温下孵育3分钟。添加50μl的NanoGlo荧光素酶测定试剂,在室温下孵育5分钟,并且在GloMax Multi+上读取发光。图71显示了使用单点特异性结合得到的计算Kd值。
实施例45
突变对Km影响的测定
使用来自于实施例11的稀释汇合裂解物,对于每份样品而言将25μl不发光多肽稀释裂解物(1:100于PBS+0.1% Prionex中)与25μl的500nM NLpep78混合并且在室温下孵育5分钟。制备腔肠素类似物于NanoGlo荧光素酶测定缓冲液中的2×稀释液,并且将50μl的不发光肽和不发光多肽样品与50μl的NanoGlo/腔肠素类似物稀释液混合。在室温下孵育5分钟后测量发光。图72显示了使用Michaelis-Menten得到的计算Kd。
实施例46
证明三组分互补
证明使用2个NLpep和NLpoly 5P不发光多肽的三级互补。制备NLpoly 5P-B9(残基147-157缺失的5P)和NLpep B9-HT(Met+与HT7的N端融合的残基147-157)裂解物。
A)用NLpep78滴定NLpoly 5P-B9+NLpoly B9
用NLpep78滴定NLpoly 5P-B9+NLpoly B9。将20μl 5P-B9(未稀释)与20μl肽B9-HT(未稀释)混合。于PBS+0.1% Prionex中制备NLpep78的稀释液(合成肽,最高浓度=100uM)。将20μl NLpep78添加到40μl的5P-B9+肽B9-HT混合物中并且在室温下振荡10分钟。添加60μl NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上以0.5秒积分测量发光。
B)用NLpepB9-HT滴定NLpoly 5P-B9+NLpep78
将20μl NLpoly 5P-B9(未稀释)与20μl NLpep78(100μM)混合。于PBS+0.1%Prionex中制备肽B9-HT的稀释液(最高浓度=未稀释)。将20μl肽B9-HT添加到40μl的5P-B9+NLpep78混合物中并且在室温下振荡10分钟。添加60μl NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上以0.5秒积分测量发光。
图73展示了由2个不同的NLpep和截短NLpoly组成三元系统的可行性。因为所有3个组分在无另外2个组分时不发光,所以可构造该系统,使得每个NLpep与在高浓度下使用的结合部分和截短NLpoly融合(经合成或基因工程)以仅在结合部分之间的相互作用的存在下发光,或使得3个组分中的每一个均与结合部分融合以仅在三元复合物形成的情况下发光。
实施例47
与NLpep88(Gly代替Arg作为第6个残基的NLpep78)的互补
如先前所述制备NLpep88-HT和5P大肠杆菌澄清裂解物。于PBS+0.1% Prionex中制备NLpep88-HT裂解物的连续稀释液。混合20μl的5P裂解物和20μl NLpep88-HT裂解物并且在室温下振荡10分钟。添加40μl的NanoGlo荧光素酶测定试剂并且在室温下振荡10分钟。在GloMax上以0.5秒积分测量发光。图74展示了在NLpep第6位的精氨酸残基的重要性。虽然在较低浓度的NLpep88下发光没有高于单独的5P的增加,但是高浓度的NLpep使发光增加,表明催化受损的复合物并非缺乏在5P和NLpep88之间的相互作用。
实施例48
NLpep78和79作为与HaloTag的N端融合物的亚细胞定位
涂铺U2OS细胞并任其在37℃下恢复过夜。然后用HaloTag单独的DNA构建体或HaloTag-NanoLuc肽DNA构建体(全部受CMV启动子控制):使用FuGENE HD用载体DNA(pSI)1:10稀释的P1-HT、P78-HT或P79-HT转染细胞并且在37℃下孵育24小时。然后用HaloTag-TMR配体通过生产商的标准快速标记方案标记细胞并成像。图75证明NLpep78和79并未改变HaloTag蛋白的细胞内定位。
实施例49
不发光多肽(WT和5P)的亚细胞定位
涂铺U2OS细胞并任其在37℃下恢复过夜。保持细胞为非转染对照或用NanoLucDNA构建体:使用FuGENE HD用载体DNA(pSI)1:10稀释的FL、NLpoly(野生型)或NLpoly(5P)转染并且在室温下孵育24小时。固定细胞并且随后加工用于ICC。在4℃下使用1:5000GS(PRO)一抗过夜,接着用Alexa488山羊抗兔二抗进行ICC。图76证明NLpolyWT和NLpoly 5P均匀地定位于细胞内。
实施例50
证明不发光多肽可容易且快速地检测出与目标蛋白缀合的不发光肽
将99μl的NLpep53-HT大肠杆菌澄清裂解物与24.75μl 4×SDS上样缓冲液混合。制备裂解物-上样缓冲液混合物的1:10连续稀释液并且在95℃下孵育5分钟。将15μl上样到SDS-PAGE凝胶上。凝胶完成后,使用iBlot将其转移到PVDF上并在室温下用10mL NLpolyL149M大肠杆菌澄清裂解物洗涤30分钟。然后将膜置于LAS4000成像仪上并添加2mL
实施例51
在5P的情况下不发光多肽位置31、46、108、144和157的定点饱和
在根据以下表5的位点向NLpoly 5P(pF4Ag载体背景)上构建单氨基酸变化变体。实际上,对于总计95种变体,将天然残基改变为19种替代氨基酸的每一种。
表5
使单独菌落在LB+amp中生长并且在30℃下孵育过夜。还包括5P对照。使用过夜培养物接种新鲜的LB+amp(1:100),并且这些培养物在37℃下生长2小时45分钟。添加鼠李糖至0.2%,并任培养物在25℃下生长/诱导过夜。诱导18小时后,用0.5×FastBreak裂解细胞(环境温度30分钟),于干冰上速冻,并储存在-20℃下。快速解冻之后,在没有和存在Pep87(亦称NLpep 87)时测定样品。
对于(-)肽反应而言,将30uL裂解物与30uL PBS pH 7.5孵育10分钟,然后添加60uL
将(-)肽样品的发光(RLU)数据归一化到5P对照的读数,并且在图78中呈现了这些结果。也将(+)肽样品的发光(RLU)数据归一化到5P,但是然后还归一化到图76中的值以便表示信背比(S/B;图79)。
实施例52
NLpoly和NLpep之间的高亲和力用于蛋白质纯化/下拉的用途
如下,平衡MAGNEHALOTAG珠粒(Promega Corporation;G728A):a)将1mL珠粒置于磁铁上约30秒,并去除缓冲液;b)将珠粒从磁铁上移除,重新悬浮于1mL PBS+0.1%Prionex中,并且在室温下振荡5分钟;及c)再重复步骤a)和b)两次,通过使珠粒重新悬浮在1mL NLpep78-HT澄清裂解物中,在室温下振荡1小时并置于磁铁上约30秒,使NLpep78-HaloTag(大肠杆菌澄清裂解物)与MAGNEHALOTAG珠粒结合。去除裂解物(流过)并储存用于分析。通过使珠粒重新悬浮在1.5mL 8S裂解物中,在室温下振荡1小时并置于磁铁上约30秒,使NLpoly 8S(大肠杆菌澄清裂解物)与来自以上步骤的NLpep78结合的MagneHaloTag珠粒结合。去除裂解物(流过)并储存用于分析。使珠粒重新悬浮在1mL PBS+0.1% Prionex中,在室温下振荡5分钟,置于磁铁上约30秒,并去除PBS(洗涤)。再洗涤珠粒三次。
为洗脱结合的肽/多肽,使珠粒重新悬浮在500uL 1×SDS缓冲液中并且在室温下振荡5分钟。然后将珠粒置于磁铁上约30秒;去除SDS缓冲液(洗脱)并储存用于分析。再重复洗脱一次。
然后用凝胶分析样品。将37.5uL的样品(除洗脱物外)与12.5uL 4×SDS缓冲液混合并且在95℃下孵育5分钟。将5uL上样到Novex 4-20% Tris-甘氨酸凝胶上并且在约180V下电泳约50分钟。用SimplyBlue Safe Stain为凝胶染色并且在LAS4000成像仪上成像。
图94说明NLpoly和NLpep的亲和力足以允许从大肠杆菌裂解物中纯化。当从大肠杆菌裂解物纯化NLpoly 8S时,期望可按类似方式纯化与NLpoly 8S(或本文所述其它变体)融合的蛋白是合理的。虽然在该实施例中固定NLpep并且用于纯化NLpoly,但是如果固定NLpoly,则期望类似结果是合理的。
实施例53
NLpoly/NLpep结合的动力学
制备2×浓度的合成NLpep并且在PBS+0.1% Prionex中稀释2.7倍9次(10个浓度)。用于测定的最终浓度为30uM-3.9nM。将野生型NLpoly(大肠杆菌澄清裂解物;1:10,000)或11S(1:10,000,000)稀释于NanoGlo+100uM腔肠素类似物(Fz)中。将50uL的NLpep置于白色96孔测定板的孔内。在
实施例54
NLpoly/NLpep底物亲和力
如下,将NLpoly稀释于PBS+0.1% Prionex中:WT按1:10
实施例55
底物对NLpoly/NLpep亲和力的影响
按1:10
实施例56
NLpoly 5A2:NLpep的Km
按1:10
实施例57
无NLpep的NLpoly的发光
如先前对于NLpoly野生型、5A2、5P、8S和11S所述,制备大肠杆菌澄清裂解物。混合各50uL的裂解物和50uL NanoGlo+Fz并且在室温下孵育5分钟。在
实施例58
整个演化过程中在大肠杆菌内的发光改善
在200uL基本培养基中接种WT、5A2、5P、8S或11S的单个NLpoly菌落并使其在37℃下在振荡器上生长20小时。将10uL过夜培养物稀释至190uL的新鲜基本培养基中并使其在37℃下在振荡器上生长20小时。将10uL这种过夜培养物稀释至190uL自动诱导培养基(先前所述)中并使其在25℃下在振荡器上生长18小时。稀释经自动诱导的培养物50倍(4uL稀释至196uL测定裂解缓冲液中),向40uL含NLpep(合成;1nM;野生型、NLpep78、NL79或NLpep80)的测定裂解缓冲液中添加10uL表达培养物并且在室温下振荡10分钟。添加50uL NanoGlo+Fz,并且在室温下振荡样品5分钟。在GloMax光度计上以0.5秒积分测量发光。图100说明了在演化过程中,来自于大肠杆菌源NLpoly的发光的改善,总体改善约10
实施例59
整个演化过程中在HeLa细胞内的改善的发光
使用FugeneHD将50ng表达NLpolyWT、5A2、5P、8S或11S的质粒DNA转染至进入12孔板孔内的HeLa细胞中。然后在37℃/5%CO
实施例60
整个演化过程中在HEK293细胞内的改善的发光
使用FugeneHD将50ng表达NLpolyWT、5A2、5P、8S或11S的质粒DNA转染至进入12孔板孔内的HEK293细胞中。然后在37℃/5%CO
实施例61
整个演化中提高的结合亲和力
如下,将NLpolyWT、5A2、5P、8S或11S(大肠杆菌澄清裂解物)稀释于PBS+0.1%Prionex中:野生型1:10
实施例62
NLpoly发光
用200uL基本培养基接种单个NLpoly变体菌落并使其在37℃下在振荡器上生长20小时。将10uL过夜培养物稀释至190uL新鲜基本培养基中并使其在37℃下在振荡器上生长20小时。然后将10uL这种过夜培养物稀释至190uL自动诱导培养基(先前所述)中并使其在25℃下在振荡器上生长18小时。将10uL这种表达培养物与40uL无NLpep或NLpep78-HT(1:3,860稀释)或NLpep79-HT(1:10,000稀释)的测定裂解缓冲液(先前所述)混合并且在室温下振荡10分钟。添加50uL的NanoGlo+Fz并且在室温下再振荡10分钟。在
实施例63
NLpoly变体的溶解性
向5mL LB培养物中接种单个NLpoly变体菌落(见图143)并且在37℃下振荡孵育过夜。将过夜培养物1:100稀释至新鲜LB中并且在37℃下振荡孵育3小时。向培养物中添加鼠李糖至0.2%并且在25℃下振荡孵育过夜。将900ul这些过夜培养物与100uL 10XFastBreak裂解缓冲液(Promega Corporation)混合并且在室温下孵育15分钟。从每种培养物中取出75ul等分试样(总)并储存用于分析。在4℃下于台式微量离心机中以14,000×rpm离心来自于每份样品的剩余培养物15分钟。从每份样品取出75ul等分试样的上清液(可溶)并储存用于分析。向储存的等分试样中添加25ul的4×SDS缓冲液并且在95℃下孵育5分钟。将各5ul样品上样到4-20% Tris-甘氨酸SDS凝胶上并且在约190V下电泳约50分钟。用SimplyBlueSafe Stain为凝胶染色并且在LAS4000上成像。图143显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶。
实施例64
解离常数
将NLpoly变体裂解物(见图144;如先前所述制备)1:10稀释至PBS+0.1% Prionex中。于PBS+0.1% Prionex中制备4×浓度的NLpep78(合成NLpep78)。混合20uL NLpoly变体裂解物和20uL NLpep并且在室温下振荡10分钟。添加40uL NanoGlo/Fz并且在室温下振荡10分钟。在
实施例65
表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较
用表达NLpoly5P和/或NLpep80/87的N或C端融合物的1μgpF4A Ag FKBP或1μgpF4A Ag FRB载体,使用FuGENE HD按1:4的DNA:FuGENE HD比率,反向转染HEK293T细胞(400,000)。转染后24小时,使细胞胰蛋白酶化并按10,000个细胞/孔的密度重新涂铺于不透明的96孔测定板中。涂铺后24小时,用PBS洗涤细胞,然后在无酚红的OptiMEMI中用或不用20nM雷帕霉素孵育15、60或120分钟。向每个孔直接添加于OptiMEM中有或无20nM雷帕霉素的10μM腔肠素类似物底物并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分测量发光。图108(15分钟诱导)、109(60分钟诱导)和110(120分钟诱导)说明随时间推移诱导普通增强,NLpoly5P和NLpep80组合产生最多的发光。单独组分最低限度地有助于信号。
实施例66
表达与NLpoly5P和NLpep80/87融合的FRB和FKBP的不同组合的细胞产生的发光的比较
虽然与实施例65相似,但是该实施例试验了与NLpoly/NLpep融合的FRB和FKBP的全部8种可能组合以及使用更少的总DNA。用总计0.001μg pF4A Ag FRB-NLpoly5P和0.001μg pF4A Ag FKBP-NLpep80/NLpep87,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板上并且再孵育24小时。用PBS洗涤细胞,然后在有0或50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有0或50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图111说明NLpep80组合产生最高的发光并且所有构型均对雷帕霉素处理响应。
实施例67
在没有结合伴侣时表达的FRB或FKBP融合物产生的发光的比较
用总计0.001μg pF4A Ag FRB-NLpoly5P或pF4A Ag FKBP-NLpep80/NLpep87,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有0或50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有0或50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图112说明单独组分产生低基础水平的对雷帕霉素处理无响应的发光。
实施例68
经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较
用总计2、0.2、0.02或0.002μg pF4A Ag FRB-NLpoly5P和pF4AAg FKBP-NLpep80,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到2μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有或无20nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有或无20nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图113说明用更少的DNA转染减少了总体发光,但是增加了诱导倍数。
实施例69
在没有结合伴侣时经不同量的FRB-NLpoly5P或FKBP-NLpep80/87
DNA转染的细胞产生的发光的比较
用总计2、0.2、0.02或0.002μg pF4A Ag FRB-NLpoly5P或pF4AAg FKBP-NLpep80,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到2μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有或无20nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有或无20nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图114说明较低的DNA水平不会改变经单独组分转染的细胞的总体发光。
实施例70
经不同量的FRB-NLpoly5P和FKBP-NLpep80/87DNA转染的细胞产生的发光的比较
用总计0.2、0.02、0.002或0.0002μg pF4A Ag FRB-NLpoly5P和pF4A Ag FKBP-NLpep80/NLpep87,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到2μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图115说明可用低至2.5pg的DNA水平实现如实施例69和71中测定的高于本底的发光和雷帕霉素诱导。
实施例71
在没有结合伴侣时经不同量的FRB-NLpoly5P或FKBP-NLpep80/87
DNA转染的细胞产生的发光的比较
用总计0.2、0.02、0.002或0.0002μg pF4A Ag FRB-NLpoly5P或pF4A Ag FKBP-NLpep80/NLpep87,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到2μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图116说明当使用较少DNA时,单独组分产生的发光无明显变化。
实施例72
在用雷帕霉素处理不同长度时间之后经不同量的FRB-NLpoly5P和FKBP-NLpep80或FKBP-NLpep87 DNA转染的细胞产生的发光的比较
用总计2、0.2、0.02或0.002μg pF4A Ag FRB-NLpoly5P和pF4AAg FKBP-NLpep80或FKBP-NLpep87,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HEK293T细胞(400,00)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到2μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板上并且再孵育24小时。用PBS洗涤细胞,然后在有或无20nM雷帕霉素的无酚红OptiMEMI中孵育5/15/30/60/120分钟。直接向每个孔添加于OptiMEM中有或无20nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图117和118说明随时间推移用较少DNA的发光减少并且雷帕霉素诱导增加。
实施例73
表达FRB-NLpoly5P或FRB-NLpoly5A2与FKBP-NLpep80/87/95/96/97的不同组合的细胞产生的发光的比较
在该实施例中,以两天和三天形式进行测定。对于2天测定而言,在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly5P或FRB-NLpoly5A2和pF4A Ag FKBP-NLpep80/87/95/96/97,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染20,000个HEK293T细胞。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEMI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。
对于3天测定而言,用总计0.002μg pF4A Ag FRB-NLpoly5P和pF4A Ag FKBP-NLpep80/87/95/96/97,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染400,000个HEK293T细胞。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板上并且再孵育24小时。用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEMI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图119和120说明了在2天和3天测定中相似水平的发光。用NLpoly5A2进行的测定显示出相对于NLpoly5P的较佳的雷帕霉素诱导,并且用NLpoly5A2和NLpep96进行的测定显示出所有试验组合中的最强雷帕霉素诱导。
实施例73
表达FRB-NLpoly5A2或FRB-NLpoly11S与FKBP-NLpep101/104/105/106/107/108/109/110的不同组合的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly5A2/11S和pF4A AgFKBP-NLpep101/104/105/106/107/108/109/110,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于Opt iMEMI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图121说明,在试验组合中,具有NLpep101的NLpoly11S显示出最强雷帕霉素诱导和最强雷帕霉素-特异性发光信号之一。
实施例74
用FRB-NLpoly5A2或FRB-NLpoly11S与FKBP-NLpep87/96/98/99/100/101/102/103的不同组合转染的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly5A2/11S和pF4A AgFKBP-NLpep87/96/98/99/100/101/102/103,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiME MI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图122说明NLpoly11S和NLpep101组合在保持高水平特定发光的同时产生最高诱导。
实施例75
经不同水平的FRB-NLpoly11S和FKBP-NLpep87/101/102/107DNA
转染的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.01、0.1、1或10ng pF4A Ag FRB-NLpoly11S和pF4A Ag FKBP-NLpep87/101/102/107,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEMI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图123说明在所有试验DNA水平下,在未处理的样品中具有NLpep101的NLpoly11S产生总体最低的发光,并且所述组合在经雷帕霉素处理的样品中维持相对较高的发光水平。
实施例76
经不同水平的FRB-NLpoly5A2和FKBP-NLpep87/101/102/107DNA
转染的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.01、0.1、1或10ng pF4A Ag FRB-NLpoly5A2和pF4A Ag FKBP-NLpep87/101/102/107,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEMI中有或无50nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图124说明NLpoly5A2在未处理的样品中产生比实施例75中所示的NLpoly11S更高的发光。
实施例77
显示了表达FRB-NLpoly5P和FKBP-NLpep80/87DNA的细胞的发光的雷帕霉素剂量响应曲线
用总计0.001μg pF4A Ag FRB-NLpoly5P和0.001μg pF4A Ag FKBP-NLpep80/NLpep87,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(400,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将10,000个细胞重新涂铺于不透明的96孔测定板中并且再孵育24小时。用PBS洗涤细胞,然后在有0至500nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有0至500nM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMaxMulti上以0.5秒积分时间测量发光。用用于Windows的GraphPad Prism 5.00版计算Kd。图125说明了发光的雷帕霉素特异性增加。
实施例78
显示了表达FRB-NLpoly5A2和FKBP-NLpep87/101DNA的细胞的发光的雷帕霉素剂量响应曲线
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly5A2/11S和pF4A AgFKBP-NLpep87/101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有0至1μM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEMI中有0至1μM雷帕霉素的10μM腔肠素类似物底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图126说明了以NLpoly5A2/NLpep101和NLpoly11S/NLpep101组合对雷帕霉素的S形剂量响应。虽然用雷帕霉素与NLpep87的组合显示发光增加,但是收集的数据点与S形曲线偏离更多。
实施例79
表达FRB-11S和FKBP-101并且经底物PBI-4377或腔肠素类似物处理的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.1/1/10ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有0或50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEMI中有0至50nM雷帕霉素的10μM腔肠素类似物或PBI-4377底物(细胞上的最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图127说明了与腔肠素类似物底物相比,用PBI-4377底物的发光和诱导倍数减小。
实施例80
在存在或没有雷帕霉素时进行的表达FRB-NLpoly11S/5A2和
FKBP-NLpep87/101的细胞的时程
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly11S/5A2和pF4A AgFKBP-NLpep87/101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后手动或经由仪器注射添加有0或50nM雷帕霉素和10μM腔肠素类似物的无酚红OptiMEMI。立即在GloMax Multi上以0.5秒积分时间测量发光。图128和129说明,在所有试验组合中,具有NLpep101的NLpoly11S在0时具有最低发光,更快地达到发光稳定水平并且具有最大的动态范围。
实施例81
如在两台不同仪器上测量的FRB-NLpoly11S和FKBP-NLpep101产生的发光
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或50nM雷帕霉素的无酚红OptiMEMI 20分钟。添加于OptiMEMI中有0或50nM雷帕霉素的10μM腔肠素类似物(细胞上的最终浓度)并且再孵育5分钟。立即在0.5秒积分时间的GloMax Multi上和在具有450nM带通滤波器的Varioskan Flash上测量发光。图130说明可在不同仪器上测量FRB-NLpoly11S和FKBP-NLpep101的雷帕霉素特异性诱导。
实施例82
显示表达FRB-NLpoly11S和FKBP-NLpep101的细胞在用雷帕霉素处理后的不同时间发光的图像
用1μg pF4 Ag FRB-NLpoly11S和1μg pF4 Ag FKBP-NLpep101,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HeLa细胞(500,000)。在35mm玻璃底培养皿(MatTek#p35gc-1.5-14-C)中转染细胞。转染后24小时,用PBS洗涤细胞,然后用10μM于OptiMEM中的腔肠素类似物孵育5分钟。向细胞内添加50nM于OptiMEMI中的雷帕霉素并且用LV200以10秒间隔,总计20分钟,获取发光图像。仪器在37
实施例83
表达FRB-NLpoly11S和FKBP-NLpep101的单个细胞在用雷帕霉素处理后的不同时间产生的信号的定量
用1μg pF4 Ag FRB-NLpoly11S和1μg pF4 Ag FKBP-NLpep101,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HeLa细胞(500,000)。在35mm玻璃底培养皿(MatTek#p35gc-1.5-14-C)中转染细胞。转染后24小时,用PBS洗涤细胞,然后用10μM于OptiMEM中的腔肠素类似物孵育5分钟。向细胞添加50nM于OptiMEMI中的雷帕霉素并且用LV200以10秒间隔,总计20分钟,获取发光图像。仪器在37℃下,物镜为60X,增益为200并且暴露为600ms。在整个时期内用Image J软件分析视野内每个细胞的信号强度。图132说明单个细胞产生的信号可测量并且每个细胞信号的增加与图128和129中所示在96孔板测定中观察到的增加相应。
实施例84
在表达FRB-NLpoly11S和FKBP-NLpep101的不同细胞系中发光的比较
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T、HeLa或U2-OS细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或50nM雷帕霉素的无酚红OptiMEMI 20分钟。添加于OptiMEMI中有0或50nM雷帕霉素的10μM腔肠素类似物(细胞上的最终浓度)并且再孵育5分钟。立即在GloMax Multi上以0.5秒积分时间测量发光。图133说明了在经FRB-NLpoly11S和FKBP-NLpep101转染的三种不同细胞系中,在没有和存在雷帕霉素时产生的相似水平的发光。
实施例85
在用雷帕霉素竞争性抑制剂FK506处理后,表达FRB-NLpoly11S和FKBP-NLpep101的细胞产生的发光的比较
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或20nM雷帕霉素的无酚红OptiMEMI 20分钟。按5μM的最终浓度向细胞中添加于OptiMEM中的FK506抑制剂并且孵育3或5小时。向细胞添加于OptiMEM中的腔肠素类似物在细胞上达10μM的最终浓度。立即在GloMax Multi上以0.5秒积分时间测量发光。图134说明了在用竞争性抑制剂FK506处理后雷帕霉素诱导的发光减少。
实施例86
在用雷帕霉素竞争性抑制剂FK506处理后,表达FRB-NLpoly11S和FKBP-NLpep101的细胞产生的发光
在不透明的96孔测定板中用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或20nM雷帕霉素的无酚红OptiMEMI 2.5小时。按于OptiMEM(含10μM)中0、1或10μM的最终浓度经由注射器向细胞添加于OptiMEM中的FK506抑制剂。在设为37℃的Gl oMaxMulti上以0.5秒积分时间每10分钟测量一次发光,测量4小时。图135说明了到200秒,FK506抑制剂可使发光降低接近未处理细胞的水平。
实施例87
存在或没有V2R激动剂AVP时经V2R-NLpoly5A2或V2R-NLpoly11S与NLpep87/101-ARRB2的不同组合转染的细胞产生的发光
在不透明的96孔测定板中用总计0.1、1或10ng pF4A Ag V2R-NLpoly11S和pF4AAg ARRB2-NLpep87/101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或1μM AVP和10μM腔肠素类似物的无酚红OptiMEMI 25分钟。然后在GloMax Multi上以0.5秒积分时间测量发光。图136说明具有NLpep101的V2R-NLpoly11S在发光上产生最大的AVP特异性增加。与NLpep87的组合未对AVP显示出显著响应。
实施例88
显示在用AVP处理后经V2R-NLpoly5A2或V2R-NLpoly11S和
NLpep87/101-ARRB2转染的细胞产生的发光的时程
在不透明的96孔测定板中用总计0.1或1ng pF4A Ag V2R-NLpoly11S或1ng pF4AAg V2R-NLpoly5A2和pF4A Ag ARRB2-NLpep87/101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后手动(图137)或经由仪器注射(图138)添加有0或1μMAVP和10μM腔肠素类似物的无酚红OptiMEMI。然后在室温(图137和138)或37℃(图139)下在GloMax Multi上以0.5秒积分时间每5分钟测量一次发光,测量25分钟。图137和138说明了对于具有NLpep101-ARRB2的V2R-NLpoly11S而言,AVP诱导的发光从开始至600秒时达到最高的时间依赖性增加。与V2R-NLpoly5A2和NLpep87的组合随时间推移在发光上未显示出显著增加。图139说明在37℃下受试的全部NLpoly11S和NLpep101组合显示出在约200秒时达到平衡的AVP诱导发光的时间依赖性增加。
实施例89
在表达V2R-NLpep11S和NLpep101-ARRB2的不同细胞系中发光的比较
在不透明的96孔测定板中用总计1ng pF4A Ag V2R-NLpoly11S和pF4A Ag ARRB2-NLpep87/101,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T、HeLa或U2-OS细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有0或1μM AVP的无酚红OptiMEMI 20分钟。然后添加于OptiMEM中的腔肠素类似物至在细胞上的10μM的最终浓度,并且在GloMax Multi上以0.5秒积分时间测量发光。
图140说明了在表达V2R-NLpoly11S和NLpep101-ARRB2的三种不同细胞系中,在没有和存在AVP时相似的发光水平。
实施例90
在用AVP处理后的不同时间表达V2R-NLpoly11S和
NLpep101-ARRB2的细胞的发光
用1μg pF4 Ag V2R-NLpoly11S和1μg pF4 Ag ARRB2-NLpep101,使用FuGENE HD按1:4的DNA:FuGENE比率反向转染HeLa细胞(500,000)。在35mm玻璃底培养皿(MatTek#p35gc-1.5-14-C)中转染细胞。转染后24小时,用PBS洗涤细胞,然后用10μM于OptiMEM中的腔肠素类似物孵育5分钟。向细胞添加1μM于OptiMEMI中的AVP,并且用LV200以15秒间隔,总计30分钟,获取发光图像。仪器在37℃下,物镜为60X或150X,增益为600,并且暴露1秒或2秒。图141和142说明成像可检测在用AVP处理后单个细胞内的发光增加和斑点的形成的增加。
实施例91
NLpep的解离常数
将NLpoly 5P大肠杆菌澄清裂解物(如先前所述制备)1:1,000稀释于PBS+0.1%Prionex中。于PBS+0.1% Prionex中制备4×浓度的NLpep78-HT(如先前所述制备的大肠杆菌澄清裂解物)。混合20uL NLpoly 5P和20uL NLpep78并且在室温下振荡10分钟。添加40uLNanoGlo/Fz并且在室温下振荡10分钟。在GloMax光度计上以0.5秒积分测量发光。使用Graphpad Prism、单点特异性结合、最佳拟合值测定Kd。图80比较了由NLpep78的1或2个重复单元组成的NLpep的解离常数。
实施例92
NLpoly 5A2和NLpep86之间的亲和力
将NLpoly 5A2裂解物(在转染CHO细胞后如先前所述制备)1:10稀释于PBS+0.1%Prionex中。于PBS+0.1% Prionex中制备4×浓度的NLpep86(合成NLpep)。混合20uLNLpoly和20uL NLpep并且在室温下振荡10分钟。添加40uL NanoGlo/Fz并且在室温下振荡10分钟。在GloMax光度计上以0.5秒积分测量发光。使用Graphpad Prism、单点特异性结合、最佳拟合值测定Kd。图81说明了NLpoly 5A2和NLpep86之间的亲和力。
实施例93
NLpoly变体的发光
单独地将各种NLpoly的单菌落接种到200uL基本培养基中并使其在37℃下在振荡器上生长20小时。将10uL过夜培养物稀释至190uL新鲜基本培养基中并使其在37℃下在振荡器上生长20小时。将10uL这种过夜培养物稀释至190uL自动诱导培养基(先前所述)中并使其在25℃下在振荡器上生长18小时。将10uL表达培养物与40uL无NLpep或有NLpep78-HT(1:3,860稀释)或NLpep79-HT(1:10,000稀释)的测定裂解缓冲液(先前所述)混合。在室温下振荡混合物10分钟,添加50uL NanoGlo+Fz并且在室温下再振荡10分钟。在GloMax光度计上以0.5秒积分测量发光。图82展示了来自于无NLpep或有NLpep78或NLpep79的NLpoly变体的发光。结果显示NLpoly变体11S(12S-51)具有比其它变体改善的发光。
实施例94
具有96种NLpep变体的NLpoly的解离常数以及Vmax值
由新英格兰肽(New England Peptide)以阵列形式合成NLpep(N端通过乙酰化并且C端通过酰胺化封端的肽;以约1mg级合成阵列中的肽)(表6)。在3张单独的板中冻干每种肽。将来自3张肽板中的1张的每个孔溶解于100uL超纯水(nanopure water)中,并且测量A260并用于使用每种肽的消光系数计算浓度。然后根据所述肽的纯度调节浓度,并且添加超纯水以产生750uM的最终浓度。
于PBS+0.1% Prionex中将肽稀释至12.66uM(4X),然后在0.5log步骤中连续稀释7次(总共8个浓度)(稀释3.162倍)。如下,将NLpoly5P、8S、5A2或11S稀释至PBS+0.1%Prionex中:5P 1:2,000;8S1:10,000;11S 1:150,000;5A2 1:1,000。将各25uL NLpep+各25uL NLpoly混合并且在室温下孵育30分钟。添加50uL NanoGlo+100uM Fz并且在室温下孵育30分钟。在GloMax Multi+上以0.5秒积分测量发光。使用Graphpad Prism、单点特异性结合、最佳拟合值测定Kd/Vmax。图83-90说明了具有96种变体NLpep的NLpoly的解离常数和Vmax值。结果指示了NLpep中表现出较低结合亲和力而Vmax无损失的特定突变。
表6.肽阵列1
/>
/>
实施例95
NLpoly变体的溶解性
将单个NLpoly 5A2、12S、11S、12S-75、12S-107或5P-B9菌落接种到5mL LB培养物中并且在37℃下振荡孵育过夜。将过夜培养物1:100稀释至新鲜LB中并且在37℃下振荡孵育3小时。向培养物中添加鼠李糖至0.2%并且在25℃下振荡孵育过夜。将900ul这些过夜培养物与100uL 10×FastBreak裂解缓冲液(Promega Corporation)混合并且在室温下孵育15分钟。从每种培养物中取出75ul等分试样(总)并储存用于分析。在4℃下于台式微量离心机中以14,000×rpm离心来自于每份样品的剩余培养物15分钟。从每份样品取出75ul等分试样的上清液(可溶)并储存用于分析。向储存的等分试样添加25ul的4×SDS缓冲液并且在95℃下孵育5分钟。将各5ul样品上样到4-20%Tris-甘氨酸SDS凝胶上并且在约190V下电泳约50分钟。用SimplyBlue Safe Stain为凝胶染色并且在LAS4000上成像。图91显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶。除5A2外,所有变体均在可溶级分中显示出一定百分比的NLpoly。
实施例96
NLpoly变体的溶解性和解离常数
将单个NLpoly菌落(在图92中列出)接种到5mL LB培养物中并且在37℃下振荡孵育过夜。将过夜培养物1:100稀释至新鲜LB中并且在37℃下振荡孵育3小时。向培养物中添加鼠李糖至0.2%并且在25℃下振荡孵育过夜。将900ul这些过夜培养物与100uL 10×FastBreak裂解缓冲液(Promega Corporation)混合并且在室温下孵育15分钟。从每种培养物中取出75ul等分试样(总)并储存用于分析。在4℃下于台式微量离心机中以14,000×rpm离心来自于每份样品的剩余培养物15分钟。从每份样品取出75ul等分试样的上清液(可溶)并储存用于分析。向储存的等分试样中添加25ul的4×SDS缓冲液并且在95℃下孵育5分钟。将各5ul样品上样到4-20% Tris-甘氨酸SDS凝胶上并且在约190V下电泳约50分钟。用SimplyBlue Safe Stain为凝胶染色并且在LAS4000上成像。图92显示了对于NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶以及含有相同变体的解离常数的表。
实施例97
具有NLpep79的NLpoly 5P和11S的底物特异性
如先前所述为NLpoly 5P或11S制备大肠杆菌澄清裂解物。然后在10倍步骤中将NLpoly裂解物连续稀释到PBS+0.1% Prionex中。混合25uL NLpoly和25uL合成NLpep79(400nM,4×)并且在室温下孵育10分钟。添加50uL NanoGlo+100μM Fz,在室温下孵育10分钟,在GloMax Multi+上以0.5秒积分测量发光。图93显示了具有NLpep79的5P和11S的底物特异性并且证明11S对腔肠素类似物具有比5P更高的特异性。
实施例98
来自于演化各个步骤的NLpoly变体的溶解性
将单个NLpoly WT、5A2、5P、8S或11S菌落接种到5mL LB培养物中并且在37℃下振荡孵育过夜。将过夜培养物1:100稀释至新鲜LB中并且在37℃下振荡孵育3小时。向培养物中添加鼠李糖至0.2%并且在25℃下振荡孵育过夜。将900ul这些过夜培养物与100uL10×FastBreak裂解缓冲液(Promega Corporation)混合并且在室温下孵育15分钟。从每种培养物中取出75ul等分试样(总)并储存用于分析。在4℃下于台式微量离心机中以14,000×rpm离心来自于每份样品的剩余培养物15分钟。从每份样品取出75ul等分试样的上清液(可溶)并储存用于分析。向储存的等分试样中添加25ul的4×SDS缓冲液并且在95℃下孵育5分钟。将各5ul样品上样到4-20% Tris-甘氨酸SDS凝胶上并且在约190V下电泳约50分钟。用SimplyBlue Safe Stain为凝胶染色并且在LAS4000上成像。图104显示了对于来自演化过程各个步骤的NLpoly变体而言,总裂解物和相同裂解物的可溶级分的蛋白质凝胶。这些结果证明在演化过程中NLpoly的溶解性明显增加。
实施例99
蛋白质的化学标记
本发明的不发光肽(NLpep)可用于化学标记蛋白质。本发明的NLpep可合成为含有反应性基团,例如生物素、琥珀酰亚胺酯、马来酰亚胺等,并且连接(例如,缀合、链接、标记等)到蛋白质,例如抗体上。然后NLpep标记的蛋白,例如NLpep-抗体,可用于各种应用,例如ELISA中。将通过添加本发明的NLpoly和
实施例100
使用HaloTag-NLpep通过蛋白质印迹检测翻译后蛋白质修饰
可通过AMP化或ADP-核糖基化翻译后修饰几种蛋白质。在AMP化中,使用ATP作为供体分子通过磷酸二酯键向靶蛋白添加AMP。类似地,在ADP-核糖基化中,使用NAD+作为供体分子通过磷酸二酯键向靶蛋白添加ADP-核糖部分。已经显示ATP和NAD+的N6位置可用于标记接头,而不影响翻译后事件。如果使用经N6修饰的氯烷烃-ATP或-NAD+进行单磷酸腺苷化或ADP-核糖基化反应,则靶蛋白将被修饰为含有氯烷烃-ATP或-NAD+。
经N6修饰的ATP/NAD已经与点击化学组合用于开发基于胶内荧光的检测系统。通过蛋白质印迹技术检测这些翻译后修饰需要常常无特异性或不可用的抗体。替代方法可以结合
使用细胞裂解物或纯化蛋白,用氯烷烃-ATP(对于AMP化而言)或氯烷烃-NAD+(对于ADP-核糖基化而言)翻译后修饰靶蛋白后,可通过SDS-PAGE分析样品并转移到PVDF膜上。封闭后,可将印迹与HaloTag-NLpep孵育。HaloTag将与经翻译后修饰的蛋白结合。在下一步骤中,可向印迹添加NLpoly和腔肠素类似物以检测生物发光。这种检测方法是用于蛋白质印迹检测的基于化学发光的方法的替代方案。基于化学发光的方法可涉及在下一步骤中孵育HaloTag-蛋白G融合物(作为一抗),在ECL反应后可使用与HRP链接的任何二抗。
实施例101
翻译后修饰测定
蛋白质翻译后修饰(PTM)是生物调节所有方面的中心。PTM通过向蛋白质共价添加官能团增强了蛋白质组的各种功能。这些修饰包括磷酸化、甲基化、乙酰化、糖基化、泛素化、亚硝基化、脂化并且影响正常细胞生物学和发病机制的许多方面。更具体地,组蛋白相关的PTM有重要意义。组蛋白蛋白的表观遗传共价修饰对基因转录调控和细胞活性具有较强的影响。翻译后修饰酶的实例包括但不限于激酶/磷酸酶、甲基转移酶(HMT)/脱甲基酶(HDMT)、乙酰转移酶/组蛋白脱乙酰基酶、糖基转移酶/葡聚糖酶和ADP-核糖基转移酶。在正常生理条件下,PTM酶的调控受严格调控。然而,在病理条件下,这些酶活性可受异常调节,并且受这些酶控制的胞内网络的破坏导致许多疾病,包括癌症和炎症。
本发明的不发光肽(NLpep)和不发光多肽(NLpoly)可用于通过监测向与本发明的NLpep链接的特异性肽底物的共价基团转移(例如磷酰基、乙酰基)的变化,测定PTM酶的活性。将通过肽合成使NLpep链接到PTM酶特异性小肽上并且用作PTM酶的底物。
A)PTM转移酶测定(HAT)
一旦发生PTM酶反应,就可使用氨肽酶降解未修饰肽(NLpep;对照)。因为已知氨肽酶活性会受PTM影响,所以经修饰(乙酰化)肽(NLpep-PTM酶底物)将以极低速率降解或完全不降解。一旦氨肽酶反应完成,则随含有腔肠素类似物的
将在最佳酶反应条件下使用与本发明的NLpep链接的组蛋白肽底物和乙酰辅酶A(CoA)或SAM作为乙酰基或甲基供体进行所述反应。将添加含有氨肽酶或氨肽酶混合物的缓冲液以特异性降解所有未修饰底物。将添加含本发明的NLpoly和氨肽酶抑制剂的缓冲液。将添加
B)PTM水解酶测定(HDMT)
与A)相似的概念可用于组蛋白脱甲基酶(HDMT)。然而,替代氨肽酶,可用PTM特异性抗体产生活性干扰。可通过肽合成使本发明的NLpep链接到小的甲基化肽上并且用作水解酶的底物。一旦水解酶反应已经完成,就可向反应物中添加抗甲基抗体。这种抗体将与甲基化肽(对照)特异性结合。HDMT生成的肽产物将不与抗体结合。然后,可添加本发明的NLpoly。如果抗体干扰NLpep和NLpoly的相互作用,则不会产生发光。如果存在通过脱甲基酶的PTM水解,则NLpep和NLpoly将相互作用,并且将产生发光。图198中对于普通水解酶概念和图146中对于H3K4/9脱甲基酶而言,例证了这种概念。
除将是信号测定损失,而不是信号增益外,未修饰底物的氨肽酶降解的概念也可用于水解酶测定。将在最佳酶反应条件下使用与本发明的NLpep链接的经修饰(甲基化或乙酰化)组蛋白肽底物进行所述反应。将添加含有能够识别甲基或乙酰基的抗体的缓冲液。将添加含有本发明的NLpoly的缓冲液。NLpoly将和未与抗体结合的NLpep相互作用。将添加
在这些概念的另一种形式中,肽-NLpep上的PTM转移或水解就足以减少或增强NLpep与NLpoly的相互作用并且因此减少或增加发光信号而无需氨肽酶或抗体。
使用以下NLpep-SRC底物肽,使用本发明的方法测定代表性转移酶,酪氨酸激酶SRC:YIYGAFKRRGGVTGWRLCERILA。在10μl反应缓冲液A(40mM Tris 7.5、20mM MgCl2和0.1mg/ml BSA)中,在150μM ATP和2.5μM NLpep-Src的存在下滴定SRC酶并且在23℃下孵育1小时。孵育后,添加10μl的氨肽酶M(APM)试剂(40mM Tris 7.5、0.1mg/ml BSA和50mUAPM),在定轨振荡器上混合2分钟,然后在37℃下孵育2小时。向样品中,添加30μl的NLpoly试剂,并且在室温下孵育样品。NLpoly试剂含有NLpoly片段和氨肽酶抑制剂。30分钟后,添加50μl
实施例102
哺乳动物细胞、细胞裂解物或临床样品中目标特异性RNA(非编码RNA或mRNA)的检测
本发明的不发光肽(NLpep)和不发光多肽(NLpoly)可拴系到具有工程化序列特异性的RNA结合结构域(RBD)。可通过改变赋予RBD碱基特异性的独特氨基酸精确地改变RBD的特异性。一种此类RBD的实例为人pumilio结构域(本文称为PUM)。PUM的RNA识别代码已经非常确实。PUM由8个串联重复序列组成(每个重复序列由折叠成由α螺旋组成的紧凑结构域的34个氨基酸组成)。来自于每个重复序列中心的保守性氨基酸与RNA识别序列(由8个碱基组成)内的单独碱基产生特异性接触。可通过改变RNA识别序列内碱基识别中所涉及的保守性氨基酸(通过定点诱变)精确地改变PUM的序列特异性。对于细胞内特异性RNA的检测而言,可使对靶RNA具有自定义序列特异性的PUM结构域(PUM1和PUM2)拴系到本发明的NLpep和NLpoly上(例如,经由基因工程作为遗传融合蛋白)并且可在哺乳动物细胞内表达。将PUM1和PUM2设计为识别靶RNA内相互靠近(经实验测定,仅间隔几个碱基对)的8核苷酸序列。通过引入将PUM与不发光肽和不发光多肽隔开的柔性接头(经实验测定接头的序列和长度)保证PUM1和PUM2与其靶序列的最佳相互作用。PUM1和PUM2与其靶序列的结合将使NLpep和NLpoly在导致能够在我们的特定测定条件下产生生物发光信号的功能性复合物形成的方向上紧密靠近。在没有RNA靶标时由于构成复合物的NLpep和NLpoly对的相互作用不稳定,将不会产生功能性生物发光复合物。
类似策略也可用于检测体外临床样品中的RNA。可表达具有自定义RNA特异性的NLpep-PUM融合蛋白并从适合的蛋白质表达系统(例如大肠杆菌或哺乳动物蛋白质表达系统)纯化。可连同适合底物和测定组分一起向生物样品中添加纯化组分以产生生物发光信号。
实施例103
临床样品或哺乳动物细胞裂解物中特异性RNA(非编码RNA或
mRNA)的基于DNA寡聚物的检测
本发明的不发光肽(NLpep)和不发光多肽(NLpoly)可经适合接头(氨基酸或核苷酸)连接到与靶RNA互补的寡核苷酸上。仅在DNA寡聚物与其靶RNA的序列特异性杂交使NLpep和NLpoly在测定条件下呈对于产生生物发光信号而言最佳的理想构象紧密靠近时,发生生物发光复合物的功能性装配。也可通过涉及两种NLpep和第三NLpoly的三组分互补系统实现检测。例如,两种NLpep-DNA缀合物将与靶RNA混合。通过后续添加第三NLpoly实现生物发光复合物的功能性装配。因此,如果在特定测定条件下使用临床样品或细胞裂解物产生可检测信号,则推断在此类样品中存在靶RNA。此类测定用于检测源自传染剂的RNA(病毒RNA)和特异性RNA生物标记(牵涉于许多病状中,例如各种形式的癌症、肝病和心脏病),并且可为许多疾病病状的诊断和预后提供新途径。
实施例104
体内成像
生物技术来源的产品(Biologics),包括抗体、肽和蛋白质,有着作为治疗剂的巨大希望。与小分子药物不同,生物制剂是具有二级和三级结构的大分子并且通常含有翻译后修饰。生物制剂的内化、胞内运输、生物分布、药代动力学和药效动力学(PK/PD)、免疫原性等与小分子药物明显不同,并且需要在体内“追踪”这些抗体的新工具。用酶报道分子(HRP、荧光素酶等)或小荧光标签的常规化学标记可明显改变生物制剂的治疗价值并且对于使用生物制剂的体内成像而言并不理想。用于基于PET的成像的放射性同位素标记也不方便。
本文所述的NLpoly和NLpep为生物制剂的体内成像提供了新的解决方案。NLpep可遗传编码至生物治疗剂内,而无需任何合成步骤。遗传编码允许精确控制每个生物分子的肽的量及其位置,从而将对其治疗价值的任何扰动减到最小。对于成像而言,可向动物注射NLpoly,连同底物,例如腔肠素类似物。如果NLpep-生物制剂和NLpoly相互作用,则将产生发光。可选地,表达NLpoly的转基因动物可用作模型系统。
实施例105
BRET应用
这种概念从根本上测量了集合在一起的三个部分。NLpoly和/或NLpep的两个形成复合物,并且具荧光或生物发光的第三部分提供了能量转移组分。如果形成的复合物具生物发光,则可测量生物发光和能量转移(即,BRET)。如果形成的复合物具荧光,则若第三组分为生物发光分子时可测量能量转移的幅度。
A)该实施例展示了与NLpep连接的荧光染料。可选地,荧光蛋白可与NLpoly或NLpep(由遗传构建体产生)融合,例如融合蛋白。
如先前所述制备NLpoly WT的大肠杆菌澄清裂解物。将40uL NLpoly WT裂解物与10uL的PBI-4730(NLpep1)或PBI-4877(NLpep1-TMR)混合并且在室温下孵育10分钟。添加50uL于50mM HEPESpH 7.4中的100uM腔肠素类似物并且在室温下孵育30分钟。在TECANM1000上在400-700nm内测量发光。
图147说明了从NLPoly/NLPep复合物(供体)到TMR(受体)非常有效的能量转移,及发射光线波长上的相应红移。
B)该实施例展示了在检测中使用BRET,例如检测小分子浓度或酶活性。因为能量转移强烈地依赖于距离,所以往往可将能量转移的幅度与系统的构象关联起来。例如,螯合钙的多肽插入可用于通过调节能量转移测量钙浓度。
可通过如本文所述的系统测量也通过如上引起传感器构象变化或通过从荧光部分上裂解传感器改变距离的酶。当NLpoly和NLpep相互作用时,与荧光部分结合的NLpoly或NLpep产生能量转移。这种的一个实例为已经制成的肽传感器,其中NLpep与荧光TOM染料经由DEVD接头(半胱天冬酶-3裂解位点)缀合。当暴露于NLpoly时,观察到能量转移。当暴露于半胱天冬酶-3时,消除能量转移,但是460nm下的发光保持。
纯化NLpoly 5A2和NL-HT(与HaloTag融合的NanoLuc)。将20uL的8pM NL-HT与20uL的100nM PBI-3781混合(见,例如美国专利申请序列NO.13/682,589,其以引用的方式整体并入本文)并且在室温下孵育10分钟。添加40uL NanoGlo+100uM腔肠素类似物,并且在TECAN M1000上在300-800nm内测量发光。
将20uL的33ng/μL NLpoly 5A2与20uL的约500uM PBI-5074(TOM-NCT-NLpep)混合。添加40uL NanoGlo+100μM腔肠素类似物,并且在TECAN M1000上在300-800nm内测量发光。
图148说明了从NLPoly/NLPep复合物(供体)到TOM-染料(受体)的能量转移,及发射光线波长上的相应红移。
C)三元相互作用
用NLpoly和NLpep的能量转移也可用于测量三分子相互作用。一个实例将为经NLpoly标记的GPCR和带NLpep的在其相互作用时形成生物发光复合物的GPCR相互作用蛋白。这允许测量二元相互作用。如果携带用于能量转移的适当荧光部分的小分子GPCR配体与该系统相互作用,则将发生能量转移。因此,可在相同实验中测量蛋白质-蛋白质二元相互作用和药物-蛋白质-蛋白质三元相互作用。同样,荧光分子仅在与蛋白对相互作用时产生信号,其从与失活蛋白相互作用的配体上去除任何信号(图149)。
实施例106
6-四甲基罗丹明-PEG3-NH
向6-四甲基罗丹明琥珀酰亚胺酯(0.25g,0.5mmol)于DMF(5mL)中的溶液,添加1-Boc-4,7,10-三氧杂十三烷-1,13-二胺(0.15g,0.5mmol),接着添加二异丙基乙胺(0.25mL,1.4mmol)。搅拌16小时后,通过HPLC分析反应物以确认6-四甲基罗丹明琥珀酰亚胺酯完全消耗。反应物浓缩至粉红色膜,将粉红色膜溶于三异丙基硅烷(0.2mL)和三氟乙酸(4mL)的组合中。搅拌粉红色溶液2小时,之后分析HPLC确认原材料完全消耗。反应物浓缩至干以提供呈粉红色膜的粗6-四甲基罗丹明-PEG3-NH2。
H-GVTGWRLCERILA-PEG-TMR(PBI-4877):
通过标准固相肽合成,使用Fmoc技术合成完全受保护的肽Boc-GVTGWRLCERILA-树脂,然后使用二氯乙酸从树脂上裂解以释放呈白色固体的完全受保护的肽。向6-四甲基罗丹明-PEG3-NH2(0.05g,0.08mmol)于DMF(1.5mL)中的溶液,添加这种Boc-GVTGWRLCERILA-OH(0.2g,0.07mmol)、1-羟基氮杂苯并三唑(11mg,0.08mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(15mg,0.08mmol)和二异丙基乙胺(0.28mL,0.16mmol)。搅拌30分钟后,浓缩反应物,并且将所得粗产物在CH
TOM-DEVDGVTGWRLCERILA-OH(PBI-5074):
通过标准固相肽合成,使用Fmoc技术合成完全受保护的肽H-DEVDGVTGWRLCERILA-树脂。虽然仍在树脂上,但是添加了6-TOM(PBI-3739)琥珀酰亚胺酯的溶液并使其与游离N-端反应。然后使肽从树脂上裂解并且使用三氟乙酸(TFA)完全去保护以提供蓝色固体。使用在0.1%水性TFA中的一定梯度的ACN,经反相HPLC纯化这种固体以提供呈蓝色粉末的PBI5074:MS(M+Z/2)计算值:1238.9,实测值:1238.8。
实施例107
NLPep78的合成、N端融合物和C端融合物之间的互补比较
以
将含有NLpoly11S的大肠杆菌裂解物以1:10
结果(图150)比较了NLpoly11S与合成NLPep78和在融合伴侣(HaloTag)的N或C端的NLPep78的结合。未发现结合亲和力明显变化,但是当NLPep78在C端时Bmax减小。
实施例108
NLPep79的合成、N端融合物和C端融合物之间的互补比较
以
将含有NLpoly11S的大肠杆菌裂解物1:10
结果(图151)比较了NLpoly11S与合成NLPep79和在融合伴侣(HaloTag)的N或C端的NLPep79的结合。未发现结合亲和力明显变化,但是当NLPep79在C端时Bmax减小。
实施例109
与PBI-4877(NLPep1-荧光团)相比有NLPep86的NLpoly11S的光谱扫描
于PBS pH 7+0.01% Prionex+1mM DTT中将纯化的NLpoly11S稀释至1nM。于PBSpH 7+0.01% Prionex+1mM DTT中将NLPep86或PBI-4877稀释至40uM。混合25uL NLpoly11S和25uL NLPep86或PBI-4877,然后在环境温度下孵育10分钟。然后添加50uL缓冲液(PBS pH7+0.01% Prionex+1mM DTT)+100uM Fz。在Tecan Infinite M1000上测量发光:300-800nm,每5nm,带宽10nm,增益127,积分0.5秒,z-位置22,000um。
结果证明(图152)NLPep可与小分子例如荧光染料缀合并且保持与NLpoly11S的相互作用以产生发光。还证明了有效的能量转移和改变发射光谱的能力。
实施例110
与PBI-5434(荧光团-NLPep1)相比有NLPep86的NLpoly11S的光谱扫描
于PBS pH 7+0.01% Prionex+1mM DTT中将纯化的NLpoly11S稀释至1nM。于PBSpH 7+0.01% Prionex+1mM DTT中将NLPep86或PBI-5434稀释至40uM。混合25uL NLpoly11S和25uL NLPep86或PBI-5434,然后在环境温度下孵育10分钟。然后添加50uL缓冲液(PBS pH7+0.01% Prionex+1mM DTT)+100uM Fz。在Tecan Infinite M1000上测量发光:300-800nm,每5nm,带宽10nm,增益127,积分0.5秒,z-位置22,000um。
结果证明(图153)NLPep可与小分子例如荧光染料缀合并且保持与11S的相互作用以产生发光。这连同实施例109中用PBI-4877的结果一起,还表明用于缀合的末端和/或接头长度可显著影响能量转移。
实施例111
与PBI-5436(荧光团-NLPep1)相比有NLPep86的NLpoly11S的光谱扫描
于PBS pH 7+0.01% Prionex+1mM DTT中将纯化的NLpoly11S稀释至1nM。于PBSpH 7+0.01% Prionex+1mM DTT中将NLPep86或PBI-5436稀释至40uM。混合25uL NLpoly11S和25uL NLPep86或PBI-5436,然后在环境温度下孵育10分钟。然后添加50uL缓冲液(PBS pH7+0.01% Prionex+1mM DTT)+100uM Fz。在Tecan Infinite M1000上测量发光:300-800nm,每5nm,带宽10nm,增益127,积分0.5秒,z-位置22,000um。
结果证明(图154)NLPep可与小分子例如荧光染料缀合并且保持与11S的相互作用以产生发光。还证明了有效的能量转移和改变发射光谱的能力。
实施例112
11S与各种NLPep在亲和缓冲液中的Km值比较
于PBS pH 7+0.01% Prionex+1mM DTT+0.005% Tergitol(亲和缓冲液)或NanoGlo测定试剂(Promega Corporation)中将纯化的NLpoly11S稀释至40pM。于亲和缓冲液或NanoGlo测定试剂中将NLPep(NLpep86、78、99、101、104、128和114)稀释至400uM(NLPep稀释至1mM)。混合300uL NLpoly11S和300uL的NLPep并且在环境温度下孵育30分钟。然后向白色96孔板的孔内添加50ul。向每个孔添加50ul亲和缓冲液+2×Fz(12.5uM稀释2倍,7次)或50ul NanoGlo+2×Fz(100uM稀释2倍,7次),并且在Glomax Multi+上使用0.5秒积分测量发光。使用GraphPad Prism,Michaelis-Menten测定Km。
结果展示了在亲和缓冲液(图155)或NanoGlo测定缓冲液(图156)中的底物与NLpoly11S和各种NLPep之间的复合物的结合。在指定的NLPep的情况下,测定的Km值不明显波动。
实施例113
在不同浓度的腔肠素类似物下NLPep1与NLpoly11S的结合亲和力
于亲和缓冲液(PBS pH 7+0.01%prionex+1mM DTT+0.005%terg itol)中将纯化的NLpoly156和NLpoly11S稀释至40pM。在亲和缓冲液中对于NLpoly156而言将合成NLPep1(WT)稀释至560uM或对于NLpoly11S而言稀释至80uM,然后连续稀释3倍制成8个浓度。混合350uL NLPep1和350uL NLPoly156或11S,然后在环境温度下孵育30分钟。然后将50uL等分至白色96孔测定板的孔内。向亲和缓冲液中添加Fz至40、20、10、5、2.5和1.25uM,向每个孔添加50uL Fz/亲和缓冲液并且在环境温度下孵育2分钟。在Glomax Multi+上以0.5秒积分测量发光。使用GraphPad Prism和单点特异性结合计算在每个Fz浓度下的Kd。
结果(图157)指示随Fz浓度增加,亲和力(NLPoly/NLPep)的变化。
实施例114
在不同浓度的NLPep1下NLpoly156/NLPep1和NLpoly11S/NLPep1的腔肠素类似物Km值
于亲和缓冲液(PBS pH 7+0.01%prionex+1mM DTT+0.005%terg itol)中将纯化的NLpoly156和NLpoly11S稀释至40pM。在亲和缓冲液中对于NLpoly156而言将合成NLPep1(WT)稀释至560uM或对于NLpoly11S而言稀释至80μM,然后连续稀释3倍制成8个浓度。然后将50uL等分至白色96孔测定板的孔内。向亲和缓冲液中添加Fz至40、20、10、5、2.5和1.25uM,向每个孔添加50uL Fz/亲和缓冲液并且在环境温度下孵育2分钟。在Glomax Multi+上以0.5秒积分测量发光。使用GraphPad Prism和单点特异性结合计算在每个浓度的NLPep1下的Kd。
结果(图158)指示随NLPep1浓度增加,亲和力(NLPoly/NLPep)的变化。
实施例115
NLPoly156/NLPep1、NLPoly11S/NLPep1和
于亲和缓冲液(PBS pH 7+0.01%prionex+1mM DTT+0.005%terg itol)中将纯化的NLpoly156、NLpoly11S或
结果(图159)展示了通过NLPep1激活后NLPoly156或NLPoly11S的最高活性与NanoLuc荧光素酶的最高活性。
实施例116
用不同NLPep滴定NLpoly11S产生的发光值
于PBS pH 7+0.01% Prionex+1mM DTT+0.005% Tergitol(亲和缓冲液)中将纯化的NLPoly11S稀释至40pM。如下,在亲和缓冲液中稀释合成NLPep(NLPep86、78、79、99、101、104、114、128或wt型):NLPep86=60nM、NLPep78=280nM、NLPep79=800nM、NLPep99=4uM、NLPep101=34uM、NLPep104=20uM、NLPep128=4uM、NLPep114=4.48mM和NLPepWT=20uM。混合25uL NLPoly11S和25uL NLPep,然后在环境温度下孵育30分钟。然后向每种混合物添加50ul亲和缓冲液+20uM Fz,并且在GlomaxMulti+上使用0.5秒积分测量发光。使用GraphPad Prism和单点特异性结合测定Bmax和Kd值。
结果(图160)展示了使用NLPoly11S和各种NLPep,约100,000倍的亲和力范围。在高亲和力和低亲和力NLPep之间观察到Bmax的损失最小。
实施例117
NLPoly156、NLPoly11S和
细胞中后的蛋白质印迹
在第1天,制备2ng NLPoly156、NLPoly11S或
在第2天,用无酚红DMEM洗涤细胞,向每个孔添加500uL无酚红DMEM,并且在-70℃下冷冻细胞至少30分钟。然后将细胞解冻,将500uL转移至微量离心管,并且将20uL与80uL的1.25×SDS上样缓冲液混合并且在95℃下孵育5分钟。将10ul上样到具有MES运行缓冲液的10% Bis-Tris NuPAGE凝胶上。使用iBlot将蛋白质转移到PVDF上,并且于甲醇中洗涤所述膜。然后在环境温度下于TBST+5% BSA中封闭所述膜1小时,于TBST中洗涤3次,然后在4℃下用10mL TBST+2uL兔抗Nluc多克隆抗体+2uL兔抗β-肌动蛋白多克隆抗体(Abcam#ab8227)孵育过夜。
在第3天,于TBST中洗涤所述膜3次,在环境温度下与10mL TBST+2uL抗兔HRP缀合抗体孵育1小时,再用TBST洗涤3次并且与12mL ECL蛋白质印迹底物孵育1分钟。用LAS4000Image Quant为化学发光成像。
结果(图161)显示了与全长
实施例118
NLPoly11S/NLPep114亲和力对β-内酰胺酶(SME)和β-内酰胺酶抑制蛋白(BLIP)之间的相互作用的影响的测定及通过11S/114和β-内酰胺酶活性测量的亲和力值之间的比较
蛋白质纯化
用鼠李糖诱导pF1K-信号-6H-SME、pF1K-信号-6H-SME-11S、pF1K-信号-6H-BLIPY50A和pF1K-信号-6H-BLIPy50A-114(用于在大肠杆菌内基于T7启动子表达重组蛋白的Promega Flexi载体;信号指SME或BLIP的天然信号肽)以在25℃下在KRX细胞的周质内表达18-20小时。使细胞沉淀并重新悬浮于B-Per裂解试剂(Pierce;1/50培养体积)中并且在环境温度下孵育15分钟。然后通过添加1.5×体积的20mM Tris pH 8+500mM NaCl稀释裂解物并且在12,000×g下离心10分钟。将上清液转移到干净的管内,添加1mL RQ1 DNA酶(Promega Corporation)并且在12,000×g下再离心10分钟。将上清液在HisTALON柱Clontech)上用25mM Tris pH 8和500mM NaCl上样缓冲液纯化并用25mM Tris pH 8、500mMNaCl和50mM咪唑洗脱。将洗脱蛋白透析至25mM Tris pH 7.5和25mM NaCl中并且在HiTrapQFF柱(GE Healthcare)上用25mM Tris pH 7.5和25mM NaCl上样缓冲液纯化并用25mMTris pH 7.5和125mM NaCl洗脱。调节离子强度至150mM NaCl的最终浓度,并且使用VivaSpin浓缩器浓缩。
测定
于亲和缓冲液(PBS pH 7、0.01%prionex、0.005%tergitol、1mM DTT)中将BLIPY50A和BLIPY50A-114稀释至312.5nM,然后连续稀释1.5倍。于亲和缓冲液中将SME和SME-11S稀释至0.2nM。混合11.11uL SME和88.89uL BLIP,然后在环境温度下孵育2小时。将90uL混合物转移到具有10uL的100uM头孢硝噻吩(Nitrocefin)(于亲和缓冲液中的Calbiochem)的透明96孔板内。将90uL的SME-11S/BLIPY50A-114转移到具有10uL的100uMFz(于亲和缓冲液中)的白色96孔板内。在30分钟内在486nm下每15秒测量吸光度(头孢硝噻吩),并且在30分钟内每2分钟测量发光(Fz)。
对于头孢硝噻吩而言,使用Excel拟合初速度。为初速度对比BLIP浓度绘图。使用E_free=[E]-([E_0]+[I_0]+K_app-√(([E_0]+[I_0]+K_app)^2-(4[E_0][I_0])))/2和K_app=K_i(1+([S])/K_M)拟合Ki。对于Fz而言,使用RLU=(Bmax×[BLIP-114])/([BLIP-114]+K_D)拟合Kd。
结果(图162)比较了作为未融合蛋白的蛋白质相互作用(β-内酰胺酶SME及其抑制剂BLIPY50A)的亲和力与当NLPoly和NLPep与之融合时的亲和力并且证明NLPoly11S和NLPep114之间的亲和力未导致SME/BLIPY50A相互作用的增加的表观亲和力。这还证明了NLPoly11S和NLPep114用于测量蛋白质相互作用的平衡结合常数的用途,并且通过NLPoly11S和NLPep114测量的亲和力与通过靶蛋白(SME)的活性测量的亲和力一致。
实施例119
表达FRB-NLPoly11S与FKBP-NLPep101和111-136的不同组合的细胞产生的发光的比较
向不透明的96孔测定板的孔内,用总计1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep101或111-136质粒DNA,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEM中有或无50nM雷帕霉素的10μM腔肠素类似物底物(最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间读取发光。
图163证明,在试验组合中,具有NLpep114的NLpoly11S显示出最强的雷帕霉素诱导和最强的雷帕霉素特异性发光信号之一。
实施例120
表达FRB-NLpoly11S与FKBP-NLpep114和137-143的不同组合的细胞产生的发光的比较
向不透明的96孔测定板的孔内,用总计1ng pF4A Ag FRB-NLpoly11S和pF4AAgFKBP-NLpep114或137-143质粒DNA,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEM中有或无50nM雷帕霉素的10μM腔肠素类似物底物(最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间读取发光。
图164证明,在试验组合中,具有NLpep114的NLpoly11S显示出最强的雷帕霉素诱导和最强的雷帕霉素特异性发光信号之一。
实施例121
表达FRB-NLpoly11S和FKBP-NLpep78/79/99/101/104/114/128的细胞的雷帕霉素剂量响应曲线
向不透明的96孔测定板的孔内,用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep78/79/99/101/104/128质粒DNA,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将细胞用PBS洗涤,然后在有0至300nM雷帕霉素的无酚红OptiMEMI中孵育2小时。直接向每个孔添加于OptiMEM中有0至300nM雷帕霉素的10μM腔肠素类似物底物(最终浓度)并且在室温下孵育5分钟。然后在GloMax Multi上以0.5秒积分时间读取发光。使用GraphpadPrism将数据拟合为S形曲线并计算EC50值。
图165显示了具有NLpep78/79/99/101/104/114/128的NLpoly11S对雷帕霉素的S形剂量响应。在绘制的组合中,具有NLpep114的NLpoly11S显示出最强的动态范围。
实施例122
表达FRB-NLpoly11S和FKBP-NLpep78/79/99/101/104/114/128的细胞对雷帕霉素竞争性抑制剂FK506的响应
向不透明的96孔测定板的孔内,用总计0.1ng pF4A Ag FRB-NLpoly11S和pF4A AgFKBP-NLpep78/79/99/101/104/128质粒DNA,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后添加有10nM雷帕霉素的无酚红OptiMEMI 2小时。按0至50μM的最终浓度向细胞添加于OptiMEM中的FK506抑制剂并且孵育3小时。向细胞添加于OptiMEM中的腔肠素类似物以在细胞上达10μM的最终浓度。立即在GloMax Multi上以0.5秒积分时间读取发光。使用Graphpad Prism绘制数据,拟合为S形曲线,并计算IC50值。
图166展示了用雷帕霉素竞争性抑制剂FK506,FRB-NLpoly11S和FKBP-78/79/99/101/104/114/128的雷帕霉素诱导信号的剂量依赖性减少。
实施例123
经不同比率的FRB-NLpoly11S和FKBP-NLpep114转染的细胞产生的发光的比较
向不透明的96孔测定板的孔内,用1ng pF4A Ag FRB-NLpoly11S和0.01、0.1、1、10或100ng pF4AAg FKBP-NLpep114质粒DNA,使用FuGENE HD按1:8的DNA:FuGENE比率反向转染HEK293T细胞(20,000)。还用1ng pF4A Ag FKBP-NLpep114和0.01、0.1、1、10或100ngpF4A Ag FRB-NLpoly11S反向转染HEK293T细胞(20,000)。在两种情况下,添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,用PBS洗涤细胞,然后在有或无50nM雷帕霉素的无酚红OptiMEMI中孵育1.5小时。直接向每个孔添加于OptiMEM中有或无50nM雷帕霉素的10μM腔肠素类似物底物(最终浓度)并且在室温下孵育5分钟。然后,在GloMax Multi上以0.5秒积分时间读取发光。
图167证明虽然在试验的所有DNA比率下均观察到显著诱导,但是1:1的DNA比率产生最强的雷帕霉素诱导。
实施例124
在不同方向并以不同接头长度表达FRB/FKBP的NLpoly11S/NLpep114融合物的细胞产生的发光的比较
向96孔板的孔内,用表达pF4Ag NLpoly11S和pF4Ag NLpep114与FRB或FKBP的N和C端融合物的组合的载体转染至HEK293T细胞(20,000)中。在这些构建体中,NLpoly11S/NLpep114与其融合伴侣经一个4、10或15丝氨酸/甘氨酸接头隔开。每个孔按1:8的DNA:FuGENE比率转染0.1ng NLpoly11S和NLpep114 DNA。转染后24小时,用PBS洗涤细胞,然后在于OptiMEMI中有或无50nM雷帕霉素的无酚红OptiMEMI中孵育2小时。然后添加10μM腔肠素类似物底物,并且在室温下孵育5分钟后,使用GloMax Multi以0.5秒积分时间读板。
图168说明了不管融合方向或接头长度如何,RLU的雷帕霉素特异性增加。
实施例125
由FRB-NLpoly11S/FKBP-NLpep114和分离萤火虫互补系统产生的雷帕霉素剂量响应曲线和时程的比较
向6孔板的孔内,用总计20ng pF4A Ag FRB-NLpoly11S和pF4AAg FKBP-NLpep114或750ng pF4AAg N-Fluc(1-398)-FRB和FKBP-C-Fluc(394-544),使用FuGENE HD按1:4的DNA:FuGENE比率转染HEK293T细胞(800,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将20,000个细胞重新涂铺于不透明的96孔测定板的孔内并且再孵育24小时。
对于剂量响应实验(图169A)而言,于无酚红OptiMEMI中用0-1μM雷帕霉素处理表达NLpoly11S/NLpep114的细胞3小时,然后在GloMax Multi上记录发光之前,与10μM腔肠素类似物孵育5分钟。用0-1μM于无酚红中的雷帕霉素孵育表达N-Fluc(1-398)/C-Fluc(394-544)的细胞2小时,接着在GloMax Multi上记录发光之前,在4mM D-萤光素的存在下再孵育1小时。
对于时程实验(图169B)而言,经由GloMax Multi注射器添加于无酚红OptiMEMI中用0或50nM雷帕霉素处理的表达NLpoly11S/NLpep114的细胞,并立即测量发光。于无酚红OptiMEMI中用4mM D-萤光素处理表达N-Flu(1-398)/C-Flu(394-544)的细胞1小时,接着经由注射器添加0或50nM雷帕霉素并且通过GloMax Multi测量发光。使用GraphPad Prism 6软件拟合曲线。
图169A-B证明NLpoly11S/NLpep114和分离萤火虫互补系统以雷帕霉素依赖方式响应,产生S形剂量响应曲线和类似的EC50值。NLpoly11S/NLpep114系统显示出更快的缔合动力学和更高的最大信号。
实施例126
FRB-NLpoly11S/FKBP-NLpep114和分离萤火虫互补系统产生的FK506剂量响应曲线和时程的比较
向6孔板的孔内,用总计20ng pF4A Ag FRB-NLpoly11S和pF4AAg FKBP-NLpep114或750ng pF4AAg N-Fluc(1-398)-FRB和FKBP-C-Fluc(394-544),使用FuGENE HD按1:4的DNA:FuGENE比率转染HEK293T细胞(800,000)。添加pGEM-3Zf(+)DNA以使每次转染中的总DNA达到1μg。转染后24小时,将20,000个细胞重新涂铺于不透明的96孔测定板的孔内并且再孵育24小时。然后于无酚红OptiMEMI中用0或20nM雷帕霉素处理细胞3小时。
对于FK506剂量响应实验(图170A)而言,于无酚红OptiMEMI中用0至100μM FK506抑制剂孵育细胞5小时,用10μM腔肠素类似物处理,然后用GloMax Multi在发光模式下以0.5秒积分读数。对于时程实验(图170B)而言,于含有10μM腔肠素类似物的无酚红OptiMEMI中用10μM FK506处理细胞并且立即用GloMax Multi读取发光。
图170A-B证明NLpoly11S/NLpep114和分离萤火虫互补系统在用FK506抑制剂处理后,在光输出上显示出剂量依赖性减少。NLpoly11S/NLpep114系统的信号损失比分离萤火虫系统开始于更早的时间点,更迅速,并且更完全。
实施例127
显示FKBP-NLpep114和FKBP-Fluc(394-544)的表达水平的蛋白质印迹
用0至30ng的pF4Ag NLpep114-FKBP或pF4Ag FKBP-Fluc(394-544)DNA,使用FugeneHD按1:8的DNA:Fugene比率转染HEK293T细胞(200,000)。转染后48小时,用1×SDS凝胶上样缓冲液收获细胞。在4-10% Tris-HCl SDS-PAGE凝胶上分离样品并转移到PVD F膜上。在5%于TBST的BSA中封闭所述膜1小时,然后用抗FKB P(Abcam#ab2918)孵育过夜。用辣根过氧化物酶缀合的驴抗兔IgG进行二抗孵育1小时,然后使用ECL蛋白质印迹底物(Promega Corpor ation)和Image Quant LAS 4000系统使印迹显影。
图171展示了在相等水平的转染DNA下,FKBP-NLpep114和FKBP-Fluc(394-544)的相似表达水平。
实施例128
IBET-151对NLpoly11S-BRD4和组蛋白H3.3-NLpep114相互作用的剂量-和时间-特异性抑制
向96孔白色测定板的孔内,用10ng的pF4Ag组蛋白H3.3-NLpep114和NLpoly11S-NLpoly11S,使用Fugene HD按1:8的DNA:Fugene比率转染HEK293T细胞(20,000)。
对于剂量响应实验(图172A)而言,在37℃下于无酚红OptiMEMI中用0至10μMIBET-151处理细胞4小时,然后在用GloMax Multi读取发光之前,用10μM腔肠素类似物处理5分钟。
对于时程实验(图172B)而言,用10μM腔肠素类似物预孵细胞5分钟,用0-500nMIBET-151处理,并立即置于GloMax Multi内用于每5分钟测量一次发光。
图172A-B展示了在用BRD4抑制剂IBET-151处理后,在处理3小时内发生的与文献报告一致的发光剂量依赖性下降。
实施例129
响应于GDC0879的RAS/CRAF、BRAF/BRAF和CRAF/BRAF二聚
向96孔测定板的孔内用pF4Ag NLpoly11S-BRAF、NLpoly11S-CRAF、NLpep114-KRAS或NLpep114-BRAF的组合,使用每个孔总计0.1ng DNA和Fugene HD按1:4的比率共转染HEK293T细胞(20,000)。转染后24小时,在无酚红OptiMEMI中用0至10μM的BRAF抑制剂GDC0879处理细胞4小时。添加于无酚红OptiMEMI中的腔肠素类似物底物至10μM,并且立即用设为0.5秒积分时间的GloMax Multi读取发光。
图173展示了响应于BRAF抑制剂GDC0879,RAS/CRAF、BRAF/BRAF和CRAF/BRAF二聚作用的剂量依赖性增加。
实施例130
检查12种合成肽(图180)的结构上互补的三种不同形式的NLpoly11S(即,11S、11S-氨基酸157、11S-氨基酸156和157)的能力。在NanoGlo试剂中将NLpoly的原液制成35nM并且在PBS pH 7.2中将NLpep的原液制成12.5nM。将等体积混合并且在Tecan InfiniteF500读数器(100毫秒积分时间;10分钟时间点)上测量样品的发光(图200)。
实施例131
自发性相互作用的肽NLpep86
于PBS pH 7+0.01% Prionex+1mM DTT+0.005% Tergitol(亲和缓冲液)中将纯化的NLpoly11S稀释至40pM。如下,于亲和缓冲液中稀释合成NLPep(NLPep86、WT、114):NLPep86=60nM、NLPep114=4.48mM和NLPepWT=20uM。混合25uL NLPoly11S和25uL NLPep,然后在环境温度下孵育30分钟。然后向每种混合物中添加50ul亲和缓冲液+20uM Fz,并且在GlomaxMulti+上使用0.5秒积分测量发光。使用GraphPad Prism和单点特异性结合测定Bmax和Kd值。
图174展示了使用NLPoly11S和各种NLPep的约100,000倍亲和力范围。Pep 86是自发性相互作用肽(与LSP 11S)的实例,并且显示了Pep 114作为低亲和力相互作用肽的参考。
实施例132
体内高亲和力肽的滴定
在线性动态范围于
图176展示了广泛的线性范围和检测毫微微摩尔浓度的高亲和力肽标签(NLpep86)的能力。这与最灵敏的蛋白质印迹(WB)+增强化学发光(ECL)试剂盒相匹敌
实施例133
NLpoly和NLpep的蛋白质印迹样实用性
在SDS聚丙烯酰胺凝胶上进行HaloTag(HT7)-NLpep 80(80)或NLpep80-HaloTag(HT7)的滴定。在Typhoon扫描仪上用HaloTag-TMR配体(Promega Corporation)对
图177展示了使用NLpoly11S检测经高亲和力NLPep标记的蛋白质的灵敏度。图177还比较了使用NLPep/NLPoly的检测与使用荧光标记的HaloTag的检测。
实施例134
NLpoly11S试剂的稳定性
将100nM NLpoly11S在NanoGlo测定缓冲液(Promega Corporation)+100uM腔肠素类似物中孵育并且用等量的稀释NLpep86测定。作为对照,使用NanoGlo测定缓冲液+100uM腔肠素类似物测定等体积的稀释
结果(图178)证明NLpoly11S试剂(含Fz)与商用
实施例135
高亲和力NLpep78-HT7融合物的DNA滴定
用来自于与
结果(图179)展示了与实施例131/图27相似的广泛线性范围。除该实施例使用在哺乳动物细胞内重组表达的肽(与HaloTag融合)外,这是与实施例131中所进行的基本上相似的实验。
实施例136
初步结果(阵列肽)
在图183A中,将50nM NLpoly11S与7.5μM NLpep114和37.5μM暗肽(DP)候选物(Q-162、A-162、K-162或E-162)混合。添加
在图183B中,将50nM于测定缓冲液(PBS pH7+0.01% Prionex+1mM DTT+0.005%Tergitol)中的NLpoly11S与7.5μM NLpep114(也于测定缓冲液中)和可变量的暗肽(DP)候选物Q-162或K-162(也于测定缓冲液中)混合。添加
图A表明,如同较少生物发光所示,每种肽候选物(7.5uM)均可抑制NLpoly11S和NLpep114之间的结合。注意这些“暗”肽确实产生了一些发光,因此与完全没有肽相比信号增加。
图B表明用Lys-162和Gln-162肽,抑制为剂量依赖性。
实施例137
高纯度(>95%)暗肽
在图184A中,将5nM NLpoly11S与500nM NLpep114和可变量的暗肽(DP)候选物Q-162或A-162(n=3)混合。添加
在图184B中,将5nM于测定缓冲液中的NLpoly11S与可变量的于测定缓冲液中的暗肽(DP)候选物Q-162或A-162混合(无NLpep114)(n=3)。添加
结果(图184A和B)证实了来自于实施例135的结果,但是因为肽更纯,所以存在更高的置信度。这些结果还表明在试验的暗肽变体中,Ala肽作为抑制剂最有效。
实施例138
暗肽对经环状排列变换的
为确定在经环状排列变换的Nluc(CP Nluc)情况下,“高亲和力/低活性”NLpep(亦称暗肽)是否可与
CP Nluc:NLuc 157-169---33aa-接头---Nluc 1-156
暗肽:VTGWRLCERIL(wt)
1.Gln-162 VSGWQLFKKIS
2.Ala-162 VSGWALFKKIS
将重组CP Nluc制备成大肠杆菌5x浓缩裂解物(T7-启动子;过夜表达)的可溶级分。使用CP Nluc于测定缓冲液(PBS pH 7/0.01%Prionex/1mM DTT/0.005% Tergitol)中的10,000倍稀释液。也在测定缓冲液中,在一定浓度范围内制备合成衍生暗肽。使用30μL的CP NLuc和60μL暗肽设立反应并且通过添加90μL
图185展示了用CP Nluc暗肽的剂量响应。图186展示了用CP Nluc暗肽(56μM肽)的时程。
结果表明两种暗肽,特别是Ala162形式,能够显著地抑制CP Nluc产生发光(Ala162>2log;Gln162>1log)。这表明CP Nluc方法对反向互补具有实用性。
实施例139
细胞内的暗肽
在该实施例中,使用以下构建体:
-四种暗肽载体:pF4Ag+FKBP-暗肽Ala-162、Leu-162、Gln-162和Val-162
-两种非暗肽载体:pFc5K2 FKBP-NLpep114(低亲和力肽)和pFc5K2 FKBP-NLpep80(高亲和力肽)
-一种NLpoly载体:pFc5K2 FRB-NLpoly11S
所有构建体均携带有用于哺乳动物细胞表达的CMV启动子。所有融合构建体均含有10aa Gly-Ser柔性接头。
在OptiMem中制备暗肽构建体Ala-162(A)、Leu-162(L)、Gln-162(Q)和Val-162(V)的连续稀释液并且另外含有载体DNA(pGEM-3Z)。
对于仅含NLpoly11S的转染而言,将20ul稀释暗肽与20ul NLpoly11S、60ulOptiMem和8uL Fugene混合。对于含有NLpoly11S和NLpep114或NLpep80的转染而言,将20ul稀释暗肽与20ul NLpoly11S(10ng/μl)、20ul NLpep114或NLpep80(10ng/ul)、40ulOptiMem和8ul Fugene混合。全部在室温下孵育15分钟。向两块96孔板(一块+雷帕霉素,一块无雷帕霉素)的孔内一式三份添加各5ul的转染物。然后向孔内添加100ul于DMEM+10%FBS中的HEK293T(200,000个细胞/ml),并且将转染细胞在37℃下孵育过夜
然后从细胞中去除培养基,并且用200μl DPBS洗涤细胞。添加50μl的50nM雷帕霉素,并且在37℃下孵育细胞1小时。稀释20μl于5ml无酚红OptiMEMI+50nM雷帕霉素中的5mM腔肠素类似物,将50μl直接添加到细胞中并且在GloMax Multi+中孵育5分钟。在GloMax上测量发光。
图187证明暗肽,在与FKBP融合时,可减少NLpoly11S(即,FRB-NLpoly11S)的本底信号。合起来看,图188-190证明暗肽,在与FKBP融合时,可1)与全长NanoLuc(即,FRB-NanoLuc或NanoLuc-FRB)的折叠竞争,并且2)与低和高亲和力肽(还有FKBP融合物)竞争以与NLpoly11S(即FRB-NLpoly11S)结合,并因此减少在活细胞内产生和检测到的总发光。
实施例140
病毒学应用
除使得能够测量病毒滴度外,自发性相互作用NLpep还使得能够研究病毒(例如,流感)的重排。病毒的重排指由双重感染形成新的“杂合”病毒,例如H1N1、H5N1、H3N2(H为血凝素;N为神经氨酸酶);鸟、人、猪、鸡(在猪中最常见)
由于其分段性质,流感基因组在受一种以上病毒感染的宿主细胞内可容易地扰乱。当细胞受来自于不同物种的流感病毒感染时,重排可产生含有来自于通常感染鸟类的菌株的基因和来自于通常感染人类的菌株的基因的子代病毒,导致产生在大多数宿主中从未见过的新菌株。而且,因为已经表征了至少16种不同亚型和9种不同的神经氨酸酶亚型,所以许多不同的衣壳蛋白组合是可能的。在这些亚型中,血凝素的三种亚型(H1、H2和H3)和神经氨酸酶的两种亚型(N1和N2)已经在人类群体中引起持续流行。鸟类是所有流感A亚型的宿主并且是从中向人类引入新的HA亚型的贮库(Palese,2004)。
本系统用于检测重排的应用是使自发性相互作用NLpep的两个组分进入不同病毒粒子,或细胞内的大组分和病毒内的小组分中,并且通过发光检测两种元件的存在(例如,存在于细胞中)。
实施例141
验证自发性相互作用NLpep86作为受蛋白酶体降解的蛋白质的表位标签的用途
在本发明的实施方案开发期间进行实验以验证NLpep86作为标签监测受蛋白酶体降解的蛋白质的表达水平的用途。为此,使Nlpep86与萤火虫荧光素酶变体融合,所述萤火虫荧光素酶变体还与一个或多个PEST、CL1或泛素序列(pBC21、22、24-29)融合。预计这些构建体中的每一个均会在从突变CMV启动子(d1CMV)表达后经历蛋白酶体介导的不同程度的更新。
构建体pBC21、22、24-29和表达未标记萤火虫荧光素酶或与PEST序列(ATG082和ATG083)融合的未标记萤火虫荧光素酶的对照构建体瞬时转染至按10,000个细胞/孔使用100μL的DMEM+10%FBS涂铺于96孔板中的HELA细胞中。第二天,每孔添加10μL的转染混合物(920ul OptiMEM I+各5ug构建体+15ul Fugene HD)并且使细胞在含5% CO2的37℃孵育箱内孵育48小时。通过检测萤火虫荧光素酶活性或添加含有NLpoly11S(添加到
BC21 MVSGWRLFKKIS-GGSGGGGSGG-Fluc(高亲和力)
BC22 MVSGWRLFKKIS-GGSGGGGSGG-FlucP(高亲和力)
BC24 pFC15A/MVSGWRLFKKIS-GGSGGGGSGG-Fluc-CL1
BC25 MVSGWRLFKKIS-GGSGGGGSGG-Fluc-PEST12opt(高亲和力)
BC26 MVSGWRLFKKIS-GGSGGGGSGG-Fluc-CP(高亲和力)
BC27 UBQ G76V-VGKLGRQDP-Fluc(EDAKNIKK..)-GGSGGG GSGG-VSGWRLFKKIS(高亲和力)
BC28 UBQ-RGKLGRQDP-Fluc(EDAKNIKK..)-GGSGGGGSGG-VSGWRLFKKIS(高亲和力)
BC29 UBQ-LGKLGRQDP-Fluc(EDAKNIKK..)-GGSGGGGSGG-VSGWRLFKKIS(高亲和力)
ATG083 D1 FlucP;pF4Ag CMV Luc2-PEST
ATG082 D1Fluc;pF4Ag CMV Luc2
孵育48小时后,向每个孔添加100uL
图191中的结果证明来自于Fluc和NLpep86的信号似乎就相对亮度而论相互反映并且具有相似的RLU。BC21、BC25和BC29是BC系列中最亮的构建体,其中BC21似乎在该实验中最亮,由工程化去稳定作用预计,BC24、26和27最不亮。
实施例142
该实施例证明已知互补肽可用作相同或不同互补NLpep和NLpoly11S(例如,NLpep78(2X))之间的接头。
用含有20μL的NLpep78-HaloTag(HT)或NLpep78(2x)-HT DNA、80μL的无酚Opimex和8μL的FugeneHD的混合物转染HEK293T细胞(20,000)。使细胞在37℃下生长过夜并且在24小时时使用含有33nM纯化NLpoly11S的
结果(图192)证明可使用串联结合肽并且其可足够作为接头。
实施例143
HEK293T裂解物中野生型刺虾荧光素酶残基1-156、NLpoly11S和NanoLuc的比活性比较
将每种克隆插入pFN21A
为定量每份样品的蛋白质表达水平,如下用
为了比较Nluc(全长酶)与wt刺虾1-156和与wt刺虾157-169肽组合的NLpoly11S(双组分蛋白)的比活性,对所有样品进行底物滴定,但是对于双组分样品而言,在多个肽浓度下进行底物滴定。使用这种形式,可能计算Nluc的Vmax值及NLpoly11S和wt刺虾1-156的Vmax和Bmax值。使用这种形式进行三次独立实验并且将Vmax和Bmax值归一化到NanoLuc的Vmax。将相对比活性(根据Vmax和Bmax的平均值计算)归一化到NanoLuc。
实施例144
NLpoly和NLpep对FlucP细胞内半衰期的影响
为确定向Luc2-PEST附加NLpoly11S或NLpep114是否改变放线菌酮(CHX)处理后通过信号衰减测量的细胞内半衰期。
第1天:在6孔板内涂铺Hela细胞。向两张6孔板内涂铺3ml细胞(200,000个/ml)。生长过夜。DMEM+10%FBS。
将含有FlucP、wt157-169 FlucP、NLpoly11S、NLpep114 FlucP和pBC22(全部pF4AgD1-CMV)的构建体转染至Hela细胞内。简言之,将33ul的DNA(3.3ug)添加到122ul的OptiMem中,混合并添加9.9ul的
孵育后,去除生长培养基并更换为0.4mM放线菌酮(CHX)或对照(DMSO)。在每个时间点,添加ONE-Glo
图193证明试验的NLpoly或NLpep组分都不干扰报告酶(FlucP)的细胞内正常降解。
实施例145
细胞外蛋白酶活性测定
在一些实施方案中,本发明提供了细胞外蛋白酶(例如半胱天冬酶)活性的测定。提供了猝灭肽(例如,诸如NLpep86的高亲和力肽),其仅可在由蛋白酶(例如半胱天冬酶)去除猝灭部分后,被NLpoly,例如NLpoly11S接近并重新折叠成活性荧光素酶(图194)。将NLpoly11S和腔肠素类似物作为试剂引入测定,然后测量样品的生物发光。
实施例146
中间连接的前体基团(异构肽和糖基化氨基酸)
提供了通过使用ProNLpep测量酶活性的测定。ProNLpep的这种构型是有一个内部氨基酸与防止肽与NLpoly互补的基团缀合的NLpep。当这种ProNLpep遇到去除封端基团的酶(例如,在WEHD的情况下为半胱天冬酶1或在丝氨酸糖苷的情况下为糖苷酶)时,NLpep与NLpoly互补的能力恢复(图195)。在腔肠素类似物的存在下,这样导致产生与目标酶活性成比例的光线。因为每种酶裂解导致形成荧光素酶,所以预计该系统测定小的酶浓度的灵敏度较高。
实施例147
接头评价
提供了测量物质从抗体释放的测定。NLpep以防止其与NLpoly缔合形成荧光素酶的这种方式连接到抗体、蛋白质、肽或转运蛋白识别部分。在刺激,例如细胞内化后,抗体、蛋白质、肽或转运蛋白识别部分和NLpep之间的接头由于细胞内还原电位而裂解,并且释放NLpep(图196)。NLpep现可与NLpoly互补形成荧光素酶,并且产生的光线将与接头的裂解成比例。这样提供了测量化合物从抗体的释放的系统,其为从抗体药物缀合物的细胞毒性药物递送的替代物。可通过本领域中已知的任何方式例如通过细胞内蛋白酶或pH敏感性裂解接头。同样,因为通过每次裂解产生荧光素酶,所以预计这是测定裂解的灵敏方法。
实施例148
使用抗体靶向并破坏病变细胞已经显示出重大的治疗前景并且通过称为抗体依赖细胞介导的细胞毒作用(ADCC)的过程发生。存在许多监测ADCC活性的方式,包括不同细胞类型交联或使用在效应细胞内表达的特异性荧光素酶报道分子监测基因转录。ADCC作用机制的潜在替代读数可以是在监测在治疗性抗体与细胞表面上呈递的其靶抗原或受体结合后诱导或破坏的特异性蛋白质:蛋白质相互作用中。在一些实施方案中,使用本发明的系统监测特异性蛋白质:蛋白质相互作用,其提供了其它方法需要的在分钟对比小时的时间范围内的读数。
实施例149
免疫测定
本发明的实施方案用于例如图201中所描绘的均相免疫测定,其中NLpep和NLpoly与结合部分(例如,A和B)融合。结合部分A和B可包含许多不同组分,从而构成几种不同形式的免疫测定,其可用作靶标特异性测定或用作免疫测定中的更通用的试剂。结合部分将仅在靶标的存在下紧密靠近,从而使NLpep和NLpoly紧密靠近,导致在添加底物后产生发光。表7列出了结合部分的实例(Mie等The Analyst.2012年3月7日;137(5):1085-9.;
Stains等ACS chemical biology.2010年10月15日;5(10):943-52.;Ueda等Journal of immunological methods.2003年8月;279(1-2):209-18.Ueda等Naturebiotechnology.1996年12月;14(13):1714-8.;Lim等Analytical chemistry.2007年8月15日;79(16):6193-200.;Komiya等Analytical biochemistry.2004年4月15日;327(2):241-6.;Shirasu等Analytical sciences:the international journal of the JapanSociety for Analytical Chemistry.2009年9月;25(9):1095-100.;其以引用的方式整体并入本文)。
表7.
在结合部分由蛋白质A、蛋白质G或蛋白质A或G的结构域组成的一些实施方案中,免疫测定系统利用NLpoly和NLpep融合物以在添加含靶标的样品之前与抗体复合。抗体自然地与蛋白质A和G非共价结合。在复合物形成步骤中引入在抗体和融合物之间的共价缀合引入。NLpep/NLpoly-蛋白质A/G/结构域融合物结合部分可以各种形式与抗体复合,例如:
●单独地与靶向两种不同蛋白质的两种不同特异性抗体复合以确定复合物中是否存在蛋白质;
●和单靶标特异性多克隆抗体复合在一起;
●和二抗(例如,兔抗小鼠IgG)复合在一起以与经一抗(例如,靶标特异性小鼠IgG)预孵的样品结合;和
●单独地与靶向相同靶蛋白上的两个不同表位的两种抗体复合。
如表7所述,在一些实施方案中,结合部分是靶标特异性抗体、靶标特异性抗体的结构域、结合靶配体的受体结构域,或抗体、抗体结构域和靶受体结构域的组合。
在一些实施方案中,在包括但不限于血液、血浆、尿液、血清、细胞裂解物、细胞(初生细胞或细胞系)、细胞培养物上清液、脑脊髓液、支气管肺泡灌洗液、组织活检样品、化学化合物等的样品中监测靶标。
方法描述了对靶标的分析,靶标包括但不限于:蛋白质、小分子和化合物、半抗原、肽、激素、异二聚蛋白质-蛋白质相互作用、细胞表面抗原、受体和配体之间的相互作用、复合物中的蛋白质、病毒和病毒组分、细菌、毒素、合成和天然药物、类固醇、儿茶酚胺、类二十烷酸、蛋白质磷酸化事件等。
应用包括但不限于检测或定量靶标以进行临床疾病监测、诊断、治疗性药物监测、生物学研究、制药、化合物检测及在食品/饮料/香水行业中的监测,病毒进化枝鉴定等。
另外的应用包括高通量筛选能够破坏靶标与其受体的相互作用,从而导致信号测定损失的分子。提出了几种在免疫测定中使用NLpep/NLpoly的几种形式。在一些实施方案中,这些均相进行并且作为试剂盒、作为诊断和研究试剂盒单独组分或作为可为单独测定定制的独立试剂供给。
在其它实施方案中,利用NLpep/NLpoly的均相免疫测定利用HitHunter或CEDIA技术的变型(Yang等Analytical biochemistry.2005年1月1日;336(1):102-7.;Golla和Seethala.Journal of biomolecular screening.2002年12月;7(6):515-25.;其以引用的方式整体并入本文)。在此类测定中,组分包括:靶标特异性抗体、NLpoly、NLpep-重组靶标融合物和底物。当NLpep未与靶标特异性抗体结合时,NLpoly和NLpep-重组靶标融合物形成发光复合物。向测定组分中添加试样后,发光的量与试样中的靶标浓度成正比,因为试样中存在的靶标将与抗体上的NLpep-重组靶标融合物竞争(例如,信号增益表明靶标存在)。
实施例150
自发互补的NLpoly11S的示例性构型
NLpoly11S的各种构型均可用于自发性互补测定或系统。此类构型可包括:C端缺失(例如,减少本底发光)、N和/或C端附属部分(例如,根据其是否会通过His或HaloTag纯化)等。例如,当其N端标签为SDNIAI时,HaloTag留下附属部分。示例性构型包括:SDNAIA-11S(HaloTag纯化);SDN-11S;在C端有单个缺失的SDNAIA-11S;在C端有单个缺失的SDN-11S;在C端有两个缺失的SDNAIA-11S;在C端有两个缺失的SDN-11S;在C端有三个缺失的SDNAIA-11S;在C端有三个缺失的SDN-11S;6His-AIA-11S;6His-11S;在C端有单个缺失的6His-AIA-11S;在C端有单个缺失的6His-11S;在C端有两个缺失的6His-AIA-11S;在C端有两个缺失的6His-11S;在C端有三个缺失的6His-AIA-11S;在C端有三个缺失的6His-11S;11S-6His;减去C端11S残基的11S-6His;减去C端最后两个11S残基的11S-6His;减去C端最后三个11S残基的11S-6His;减去C端11S残基的11S-HT7;减去C端最后两个11S残基的11S-HT7;减去C端最后三个11S残基的11S-HT7;6His-HT7-AIA-11S;6His-HT7-11S;6His-AIA-HT7-11S(有单个、两个、三个11S C端缺失);6His-HT7-11S(有单个、两个、三个11S C端缺失);11S-HT7-6His;11S-HT7-6His(有单个、两个、三个11S C端缺失);和三元11S。
实施例151
用于二元互补研究的蛋白质相互作用
本文所述的二元互补系统已经用于分析各种各样的蛋白质相互作用(见表8)。
表8.用于二元互补研究的蛋白质相互作用
实施例152
具有108种NLpep变体的NLpoly(阵列#2)的解离常数和Bmax值
由New England Peptide以阵列形式合成NLpep(N端通过乙酰化并且C端通过酰胺化封端的肽;以约2mg级合成阵列中的肽)(表9)。在2张单独的板中冻干每种肽。将其中一张肽板的每个孔溶解于100uL超纯水中,并且测量A260并用于使用每种肽的消光系数计算浓度。然后根据所述肽的纯度调节浓度,并且添加超纯水以产生800uM的最终浓度。
表9.肽阵列2序列
/>
/>
/>
于PBS+0.1% Prionex中将肽稀释至400uM(4×),然后在0.5log步骤中(稀释3.162倍)连续稀释7次(总共8个浓度)。将NLpoly 11S1:10^6稀释至PBS+0.1% Prionex中。将各25uL NLpep+25uL NLpoly11S混合并且在室温下孵育30分钟。添加50uL NanoGlo+100uM Fz并且在室温下孵育30分钟。在GloMax Multi+上以0.5秒积分测量发光。使用Graphpad Prism、单点特异性结合、最佳拟合值测定Kd/Bmax。表10显示了NLpoly 11S与所示NLPep的解离常数和Bmax值。结果表明了突变对与NLpoly 11S的结合和复合物产生发光的能力的影响。
表10
/>
/>
实施例153
用于减少来自于NLpoly11S的本底信号的暗肽和猝灭肽
将纯化的NLpoly11S样品稀释至NanoGlo试剂中以产生2uM的最终浓度。Pep86是高亲和力发光肽并且用于为NLpoly11S诱导最大信号。对于工作溶液而言,于PBS(pH 7.2)中制备1nM的Pep86。于PBS pH 7.2或150mM NH4HCO3中将暗肽和猝灭肽(图180)溶解至1mM(或更低)并且等体积添加到NanoGlo/NLpoly11S中,然后在Tecan Infinite F500读数器上使用5分钟时间点为样品读数。
图202A显示GWALFKK和Dabcyl-GWALFKK在没有任何其它发光肽时减少了NLpoly11S产生的本底发光。图202B显示Pep86即使在GWALFKK和Dabcyl-GWALFKK的存在下也能够诱导发光。
图203A显示VTGWALFEEIL(Trp 11mer)和VTGYALFEEIL(Tyr 11mer)诱导高于本底(单独的NLpoly11S;无肽对照)的发光,但是各自的N端Dabcyl形式提供了对这种信号的明显猝灭。图203B显示Pep86即使在Trp 11mer和Tyr 11mer的Dabcyl形式的存在下,也能够诱导发光。
以上说明书中提到的所有出版物和专利均以引用的方式并入本文。在不背离本发明的范围和精神的前提下,本发明的所述方法和系统的各种修改和变化将对本领域中的技术人员显而易见。虽然已连同具体实施方案一起描述了本发明,但是应理解要求保护的本发明不得过度地限于此类具体实施方案。实际上,旨在使相关领域中技术人员显而易见的进行本发明的所述模式的各种修改在本发明的范围之内。
- 通过结构互补激活生物发光
- 一种基于转录激活子样效应因子的生物发光检测探针及其构建方法及应用