一种靶向Nur77的吲哚衍生物及其应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于肝纤维化治疗药物技术领域,具体涉及一种靶向Nur77的吲哚衍生物及其应用。
背景技术
近年来肝纤维化和肝硬化发病率迅速增加,然而,当前尚无获批上市的专门治疗肝纤维化的药物。临床上对肝纤维化最为行之有效的治疗手段是肝移植,由于患者易引起免疫排斥反应及其高额的医疗费用,造成肝纤维化治疗效果不理想。肝星状细胞是肝纤维化的效应细胞,肝星状细胞活化是肝纤维化形成的开端。目前肝纤维化病理机制多关注TGF-β等影响肝星状细胞活性的细胞信号通路,药物靶标也多为其信号通路的活性分子。曾有较多针对这些靶点设计小分子或大分子药物,经过临床前实验或新药临床试验,大都未获得理想结果。因此,对寻找肝纤维化新的治疗分子靶点,阐明其激活肝纤维化新作用机制,对开发靶向抗肝星状细胞活化药物具有重要临床价值。
Nur77,又被称为NR4A1或TR3,属于孤儿核受体亚家族4A(NR4A)的成员。研究表明Nur77在肝纤维化中起重要作用。在TGF-β通路中,Nur77可与SMAD7、AXIN2发生相互作用,并通过AXIN2-RNF12/ARKADIA依赖的方式诱导SMAD7的降解,从而激活TGF-β/SMAD信号通路,持续活化的TGF-β信号则会通过AKT磷酸化和HDAC表观沉默修饰方式使Nur77蛋白功能失活,引起纤维化疾病。综上结果表明,NR4A1是治疗肝纤维化的潜在的重要靶标。然而Nur77如何调控肝星状细胞激活的研究目前未见其报道。本实施例的研究发现在肝纤维化小鼠中,Nur77被诱导,而Nur77的敲除会导致TGF-β信号传导的持续激活,加重了肝纤维化的发生,而Nur77激动剂可以克服这一缺陷,重新平衡TGF-β信号传导并抑制肝纤维化。这提示靶向Nur77的激动剂可作为抗肝纤维化治疗药物开发的方向。因此,设计合成靶向核受体Nur77小分子化合物,并阐明Nur77介导抗肝星状细胞激活新机制,为抗肝纤维化临床治疗和抗肝纤维化药物研发提供理论依据,具有重大研究意义和临床价值。
吲哚类化合物是众多具有生理活性的结构单元之一,因其结构柔性大,可修饰度高,对人体毒性低,抗癌活性较好而备受关注。许多天然的和人工合成的吲哚衍生物具有良好抗纤维化、抗肿瘤活性,包括2-吲哚啉酮衍生物(PMID)抑制二氧化硅(SiO2)小鼠肺纤维化进展,吲哚衍生物尼达尼布(Nintedanib)已成为肺纤维化一线药物,同时吲哚衍生物C-DIM5作为Nur77新型激动剂,通过诱导Nur77的表达,介导肿瘤细胞凋亡蛋白(TRAIL)的表达,从而促进肿瘤细胞凋亡和体内抑制肿瘤生长。这表明吲哚衍生物可开发为靶向Nur77的治疗纤维化和癌症的药物,但目前在以Nur77为靶点的抗肝纤维化药物至今未见其报道。
发明内容
本发明目的在于提供一种靶向Nur77的吲哚衍生物。
本发明的另一目的在于提供上述吲哚衍生物的应用。
本发明的技术方案如下:
一种靶向Nur77的吲哚衍生物,其结构式为
在本发明的一个优选实施方案中,其为1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基脲和1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基硫脲中的至少一种。
上述吲哚衍生物在制备用于治疗肝纤维化疾病的组合物中的应用,该组合物的有效成分包括所述吲哚衍生物或其在药学上可接受的盐、酯或水合物。
在本发明的一个优选实施方案中,所述组合物的有效成分为所述吲哚衍生物或其在药学上可接受的盐、酯或水合物。
在本发明的一个优选实施方案中,所述组合物的剂型为片剂、胶囊剂、丸剂、栓剂、气雾剂、口服液体制剂、颗粒剂、散剂、注射剂、糖浆剂、酒剂、酊剂、露剂或膜剂。
在本发明的一个优选实施方案中,所述吲哚衍生物或其在药学上可接受的盐、酯或水合物的体内剂量为15-30mg/kg。
一种用于治疗肝纤维化疾病的组合物,其特征在于:其有效成分包括上述吲哚衍生物或其在药学上可接受的盐、酯或水合物。
在本发明的一个优选实施方案中,其有效成分为权利要求1或2所述的吲哚衍生物或其在药学上可接受的盐、酯或水合物。
在本发明的一个优选实施方案中,其剂型为片剂、胶囊剂、丸剂、栓剂、气雾剂、口服液体制剂、颗粒剂、散剂、注射剂、糖浆剂、酒剂、酊剂、露剂或膜剂。
在本发明的一个优选实施方案中,所述吲哚衍生物或其在药学上可接受的盐、酯或水合物的体内剂量为15-30mg/kg。
本发明的有益效果是:
1、本发明的吲哚衍生物以孤儿核受体Nur77作为作用靶点,通过诱导孤儿核受体Nur77的表达来抑制肝星状细胞激活,进而能够有效抑制肝纤维化。
2、本发明的吲哚衍生物的制备的反应成本低,产率高,反应过程简单易控制,适用于工业化生产。
附图说明
图1为本发明实施例6的实验结果图。
图2为本发明实施例7和8的实验结果图。
图3为本发明实施例9的实验结果图。
具体实施方式
以下通过具体实施方式结合附图对本发明的技术方案进行进一步的说明和描述。
实施例1:1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-对氟苯基氨基脲(J5)的制备
(1)中间体3-N,N-二甲氨基-1H-吲哚-3-基-2-丙烯-1-酮制备:在干燥的圆底烧瓶中依次加入0.318g(0.002mol)3-乙酰吲哚和5.5mL(0.04mol)N,N-二甲基甲酰胺二甲缩醛(DMF-DMA),再在搅拌状态下升温至110℃回流,保温反应8h,TLC跟踪监测。反应完全后冷却至室温,有黄色晶体析出,抽滤烘干得黄色固体3-N,N二甲氨基-1H-吲哚-3-基-2-丙烯-1-酮。收率75%-80%。
(2)中间体6-(1H-吲哚-3-基)-2-甲基烟酸乙酯的制备:在干燥的圆底烧瓶中依次加入1.07g(5mmol)步骤(1)制得的3-N,N-二甲氨基-1H-吲哚-3-基-2-丙烯-1-酮、0.7mL(5.5mmol)乙酰乙酸乙酯、0.42g(5.5mmol)乙酸铵、15mL冰乙酸,搅拌加热至125℃,回流5h,TLC跟踪监测。反应完全后冷却至室温,用30mL乙酸乙酯萃取3次,有机相用无水硫酸钠干燥后,过滤,粗产物经柱色谱(乙酸乙酯 石油=1∶4)纯化,得黄色固体6-(1H-吲哚-3-基)-2-甲基烟酸乙酯,收率70%-75%;
(3)中间体6-(1H-吲哚-3-基)-2-甲基烟酰肼的制备:在干燥的圆底烧瓶中依次加入2.80g(10mmol)步骤(2)制得的6-(1H-吲哚-3-基)-2-甲基烟酸乙酯、10mL乙醇、0.42g(5.5mmol)乙酸铵、5mL水合肼(100mmol),搅拌加热至78℃,回流5h,TLC跟踪监测。反应完全后冷却至室温,过滤,干燥的6-(1H-吲哚-3-基)-2-甲基烟酰肼,收率70%-75%;
(4)1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-对氟苯基氨基脲(J5):在干燥的反应瓶中,先依次加入步骤(3)制得的6-(1H-吲哚-3-基)-2-甲基烟酰肼(0.266g,1mmol),乙醇(5mL),对氟苯基异氰酸酯(0.137g,1mmol)再在搅拌状态升温70-80℃,保温反应8-10h。TLC检测反应已完全,停止反应。冷却,过滤,甲苯洗涤,干燥。得白色固体产物1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基硫脲0.32g,收率77%;黄色固体,
实施例2:1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-对甲氧基苯基氨基脲(J9)的制备
步骤(1)至(3)同实施例1
(4)在干燥的反应瓶中,先依次加入步骤(3)制得的6-(1H-吲哚-3-基)-2-甲基烟酰肼(0.266g,1mmol),乙醇(5mL),对甲氧基苯基异氰酸酯(0.149g,lmmol)再在搅拌状态升温70-80℃,保温反应8-10h。TLC检测反应已完全,停止反应。冷却,过滤,甲苯洗涤,干燥。得白色固体产物1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基硫脲0.32g,收率77%;白色固体,
实施例3:1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻氯苯基氨基脲(J14)的制备
步骤(1)至(3)同实施例1
(4)在干燥的反应瓶中,先依次加入步骤(3)制得的6-(1H-吲哚-3-基)-2-甲基烟酰肼(0.266g,1mmol),乙醇(5mL),邻氯苯基异氰酸酯(0.153g,1mmol)再在搅拌状态升温70-80℃,保温反应8-10h。TLC检测反应已完全,停止反应。冷却,过滤,甲苯洗涤,干燥。得白色固体产物1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基硫脲0.32g,收率77%;白色固体,
实施例4:1-(2-(1H-吲哚-3-基).6.甲基烟酰)-4-邻甲基苯基氨基硫脲(J20)的制备
步骤(1)至(3)同实施例1
(4)在干燥的反应瓶中,先依次加入步骤(3)制得的6-(1H-吲哚-3-基)-2-甲基烟酰肼(0.266g,1mmol),乙醇(5mL),邻甲苯基硫代异氰酸酯(0.149g,1mmol)再在搅拌状态升温70-80℃,保温反应8-10h。TLC检测反应已完全,停止反应。冷却,过滤,甲苯洗涤,干燥。得白色固体产物1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基硫脲0.32g,收率77%;
实施例5:1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基脲(J25)的制备
步骤(1)至(3)同实施例1。
(4)在干燥的反应瓶中,先依次加入步骤(3)制得的6-(1H-吲哚-3-基)-2-甲基烟酰肼(0.266g,1mmol),乙醇(5mL),邻甲基苯异氰酸酯(0.133g,1mmol)再在搅拌状态升温70-80℃,保温反应8-10h。TLC检测反应已完全,停止反应。冷却,过滤,甲苯洗涤,干燥。得白色固体产物1-(2-(1H-吲哚-3-基)-6-甲基烟酰)-4-邻甲基苯基氨基脲(J25)0.30g,收率77%;
实施例6:孤儿核受体Nur77可作为抗肝纤维化的药靶
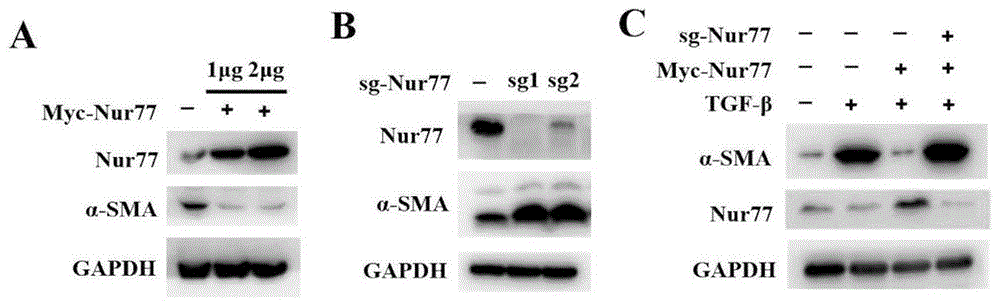
肝星状细胞(HSCs)作为肝纤维化的药效细胞,HSCs活化是肝纤维化形成的开端。在肝星状细胞LX2中,本实施例通过过表达和敲除Nur77,并且通过TGF-β1诱导LX2活化,发现在LX2细胞中缺乏活性Nur77会导致HSCs激活标准物α-SMA蛋白表达,而Nur77的过表达显著抑制HSCs活化,如图1(A-C)所示,这与Palumbo-Zerr K等人报道结果一致,表明Nur77是抗肝纤维化治疗良好靶标。
实施例7:J20和J25对TGF-β1诱导的HSCs活化的药效筛选
(1)在LX2细胞中,本实施例通过建立一个基于稳定转染的CAGA(12)-α-SMA报告基因质粒可用于高通量筛选系统有效抑制肝星状细胞激活的先导化合物。在TGF-β1(5ng/mL)诱导的LX2细胞中,实施例1至5制备的化合物在10μM下作用24h,α-SMA-Luciferase Assay报告基因实验结果表明(表1),J5、J9、J14、J20和J25均有良好的抑制α-SMA转录活性,其中J20和J25抑制活性大于50%,后续以先导物J20和J25展开进一步研究。
表1α-SMA荧光素酶报告基因药物筛选
(2)利用MTT法对化合物进行体外毒性测试。在本实验中,该化合物设置0.625μM、1.25μM、2.5μM、5μM、10μM共5个浓度,加药时间为24h,测定其OD值,根据测得的吸光度值(OD值),并计算IC50值。结果实施例1制得的J20和实施例2制得的J25两个化合物在正常肝细胞中和肝星状细胞LX2中IC50均大于79μM,表明其毒性较小,如表2所示。
表2 MTT法对化合物细胞毒性筛选(IC
(3)采用对肝纤维化标志基因α-SMA建立Lucifrase报告基因药物筛选体系,即α-SMA荧光素酶报告基因药物筛选体系,对细胞毒性较小的化合物J20和J25进行报告基因检测。采用LX2细胞,通过TGF-β1刺激后,加入待选药物J20和J25(10μM)浓度下处理24h后,通过报告基因检测,结果显示化合物J20和J25能显著抑制α-SMA荧光素酶活性,表明J20和J25可以显著抑制肝星状细胞活化标志物α-SMA的转录活性,如图2所示。
实施例8:J20和J25依赖于Nur77的诱导表达抑制肝星状细胞活化
本实施例通过单独使用TGF-β1或联合使用J20或J25处理LX2细胞,研究J20和J25在TGF-β通路激活的条件下Nur77的表达情况。结果表明,J20单独处理可以明显诱导Nut77表达,J25较弱,有趣的是,J20和J25均在细胞加入TGF-β的情况下,大量诱导Nur77的表达,伴随α-SMA表达的抑制,如果Nur77被敲低,J20和J25对于TGF-β诱导α-SMA的拮抗作用均被逆转,表明J20和J25依赖于Nur77的表达,抑制肝星状细胞的活化,结果如图2所示。
实施例9:J20和J25体内抗肝纤维化作用
本实施例通过CCl
综上结果表明,本发明实施例1至5制得的吲哚衍生物都有一定的抗肝纤维化作用,其中J20和J25对肝纤维化有良好的功效,无不良反应,可显著诱导Nur77的表达,从而抑制TGF-β1诱导的体外肝星状细胞(HSCs)激活和减缓CCl4诱导的小鼠肝纤维化,在治疗、缓解或改善肝纤维化疾病方面具有良好的应用前景.
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。