含有磷酸肌醇3-激酶抑制剂的药物组合物
文献发布时间:2024-01-17 01:27:33
本申请是以CN申请号为202210492783.4,申请日为2022年5月7日的申请为基础,并主张其优先权,该CN申请的公开内容在此作为整体引入本申请中。
技术领域
本申请涉及生物医药领域,具体的涉及一种含有磷酸肌醇3-激酶抑制剂的药物组合物。
背景技术
磷酸肌醇3-激酶(Phosphoinositide 3-kinase,PI3K)信号转导途径是人类癌症中最高度突变的系统之一。PI3K是细胞内脂质激酶的独特且保守的家族的成员,其使磷脂酰肌醇或磷酸肌醇上的3’-OH基团磷酸化。PI3K家族包括15种激酶,其具有不同的底物特异性、表达模式和调节模式。I类PI3K(p110α、p110β、p110δ和p110γ)通常由酪氨酸激酶或G-蛋白偶联受体活化以产生(3,4,5)-三磷酸磷脂酰肌醇(PIP3),其接合下游效应子,如AKT/PDK1途径中的那些,mTOR、Tec家族激酶和Rho家族GTP酶。II类和III类PI3K通过合成3-二磷酸磷脂酰肌醇(PI(3)P)和(3,4)-二磷酸磷脂酰肌醇(PI(3,4)P2),在细胞内运输中起关键作用。PI3K是控制细胞生长(mTORC1)或监测基因组完整性(ATM、ATR、DNA-PK和hSmg-1)的蛋白激酶。
存在四种I类PI3K的哺乳动物亚型:PI3K-α、β、δ(Ia类PI3K)和PI3K-γ(Ib类PI3K)。这些酶催化PIP3的产生,导致对细胞存活、分化和功能重要的下游效应途径的活化。PI3K-α和PI3K-β广泛表达,是来自细胞表面受体的信号转导的重要介质。PI3K-α是最常见于癌症中的突变的亚型,并且在胰岛素信号转导和葡萄糖稳态中起作用(Knight等人Cell(2006)125(4):733–47;Vanhaesebroeck等人Current Topic Microbiol.Immunol.(2010)347:1–19)。PI3K-β在磷酸酶和张力蛋白同源物(PTEN)缺失的癌症中被活化。两种亚型都是研发中的小分子疗法的癌症靶标。
PI3K-δ和-γ优先在白细胞中表达,并且在白细胞功能中是重要的。这些亚型也有助于血液学恶性肿瘤的发展和维持(Vanhaesebroeck等人Current TopicMicrobiol.Immunol.(2010)347:1–19;Clayton等人J Exp Med.(2002)196(6):753–63;Fung-Leung Cell Signal.(2011)23(4):603–8;Okkenhaug等人Science(2002)297(5583):1031–34)。PI3K-δ通过与PI3K调节亚基(p85)的Sarc同源2(SH2)结构域的相互作用或通过与RAS的直接相互作用被细胞受体(例如,受体酪氨酸激酶)活化。
Ibrutinib(依鲁替尼)是首个被FDA批准的布鲁顿酪氨酸激酶(BTK)抑制剂,对B细胞淋巴瘤,包括MCL和CLL等有良好疗效。
发明内容
本发明所述的药物组合物或者组合治疗适用于治疗或延迟受试对象中B细胞增殖性紊乱的进展,例如血液学恶性肿瘤。
一方面,本发明提供了一种技术方案I:
一种药物组合物,其特征在于,包括:i)至少一种有效量的磷酸肌醇3-激酶(PI3K)抑制剂、ii)至少一种有效量的Bruton酪氨酸激酶(BTK)抑制剂以及iii)来那度胺、泊马度胺、沙利度胺中的任意一种或其组合,优选为来那度胺,其中,所述的药物组合物可以处于同一和/或分开的剂型中。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如,所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的Bruton酪氨酸激酶(BTK)抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的来那度胺、泊马度胺、沙利度胺中的任意一种或其组合(优选为来那度胺)可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案I的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的磷酸肌醇3-激酶(PI3K)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案I的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的Bruton酪氨酸激酶(BTK)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、
15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案I的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的来那度胺、泊马度胺、沙利度胺中的任意一种或其组合(优选为来那度胺),例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂选自PI3Kα抑制剂、PI3Kβ抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂、泛-PI3K抑制剂或PI3Kδ/γ双重抑制剂,优选为PI3Kδ/γ双重抑制剂。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的PI3K抑制剂选自LY294002、hibiscone C、Idelalisib、Copanlisib、Duvelisib、Alpelisib、Taselisib、Perifosine、Buparlisib、Umbralisib、PX-866、Dactolisib、CUDC-907、Voxtalisib、CUDC-907、ME-401、IPI-549、SF1126、Tenalisib、INK1117、pictilisib、XL147、Palomid 529、GSK1059615、ZSTK474、PWT33597、IC87114、TG100–115、CAL263、RP6503、PI-103、GNE-477或AEZS-136中的任意一种或其任意的组合,优选为Idelalisib、Copanlisib、Duvelisib、Alpelisib或者Tenalisib中的任意一种或其任意的组合。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的PI3K抑制剂选自Tenalisib、其代谢产物或其药学上可接受的盐中的任意一种或其组合。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的Bruton酪氨酸激酶(BTK)抑制剂选自依鲁替尼、阿卡替尼、奥布替尼、泽布替尼、CT-1530、DTRMWXHS-12、司布替尼、维卡布替尼、依武替尼、替拉布替尼、芬尼布替尼、泊塞替尼、BMS-986142、ARQ-531、LOU-064、PRN-1008、ABBV-599、AC-058、ARQ-531、BIIB-068、BMS-986195、HWH-486、PRN-2246、TAK-020、GDC-0834、BMX-IN-1、RN486、SNS-062、LFM-A13中的任意一种或其组合,优选为依鲁替尼、阿卡替尼、泽布替尼中的任意一种或其组合。
在本发明的技术方案I的优选技术方案中,其特征在于,所述的Bruton酪氨酸激酶(BTK)抑制剂选自依鲁替尼、阿卡替尼、奥布替尼、泽布替尼中的任意一种或其任意的组合。
除此之外,本发明还提供了一种药盒,其包含本发明的上述技术方案I或其优选技术方案中的任意一项所述的药物组合物,以及药品使用说明书。
另外的,本发明还提供了一种技术方案II:
一种在对有需要的对象治疗与过度B细胞增殖有关的疾病或紊乱的方法,其特征在于,包括向所述的对象施用本发明的上述技术方案I或其优选技术方案中的任意一项药物组合物,其中,所述的药物组合物可以同时、或者先后施用。
在本发明的技术方案II的优选技术方案中,其特征在于,所述的与过度B细胞增殖有关的疾病或紊乱为癌症。
在本发明的技术方案II的优选技术方案中,其特征在于,所述的癌症为血液学恶性肿瘤。
在本发明的技术方案II的优选技术方案中,其特征在于,所述的血液学恶性肿瘤为淋巴瘤、白血病或骨髓瘤。
在本发明的技术方案II的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自急性淋巴细胞白血病(ALL)、急性髓系白血病(AML)、慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、多发性骨髓瘤(MM)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、Waldenstrom巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)、边缘区淋巴瘤(MZL)、Burkitt淋巴瘤、毛细胞白血病(HCL)和Richter转化。
在本发明的技术方案II的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、弥漫性大B细胞淋巴瘤(DLBCL)和边缘区淋巴瘤(MZL)。
另外的,本发明还提供了一种技术方案III:
一种药物组合物,其特征在于,包括:i)至少一种有效量的磷酸肌醇3-激酶(PI3K)抑制剂、ii)DNA甲基化试剂(例如,阿扎胞苷),其中所述的药物组合物可以处于同一和/或分开的剂型中。
在本发明的技术方案III的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案III的优选技术方案中,其特征在于,所述的DNA甲基化试剂(例如,阿扎胞苷)可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案III的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的磷酸肌醇3-激酶(PI3K)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案III的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的DNA甲基化试剂(例如,阿扎胞苷),例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案III的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂选自PI3Kα抑制剂、PI3Kβ抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂、泛-PI3K抑制剂或PI3Kδ/γ双重抑制剂,优选为PI3Kδ/γ双重抑制剂。
在本发明的技术方案III的优选技术方案中,其特征在于,所述的PI3K抑制剂选自LY294002、hibiscone C、Idelalisib、Copanlisib、Duvelisib、Alpelisib、Taselisib、Perifosine、Buparlisib、Umbralisib、PX-866、Dactolisib、CUDC-907、Voxtalisib、CUDC-907、ME-401、IPI-549、SF1126、Tenalisib、INK1117、pictilisib、XL147、Palomid 529、GSK1059615、ZSTK474、PWT33597、IC87114、TG100–115、CAL263、RP6503、PI-103、GNE-477或AEZS-136中的任意一种或其任意的组合,优选为Idelalisib、Copanlisib、Duvelisib、Alpelisib或者Tenalisib中的任意一种或其任意的组合。
在本发明的技术方案III的优选技术方案中,其特征在于,所述的PI3K抑制剂选自Tenalisib、其代谢产物或其药学上可接受的盐中的任意一种或其组合。
另外的,本发明还提供了一种技术方案IV:
一种药物组合物,其特征在于,包括:i)至少一种有效量的磷酸肌醇3-激酶(PI3K)抑制剂、ii)至少一种有效量的Bcl-2抑制剂,其中所述的药物组合物可以处于同一和/或分开的剂型中。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的Bcl-2抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案IV的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的磷酸肌醇3-激酶(PI3K)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案IV的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的Bcl-2抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂选自PI3Kα抑制剂、PI3Kβ抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂、泛-PI3K抑制剂或PI3Kδ/γ双重抑制剂,优选为PI3Kδ/γ双重抑制剂。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的PI3K抑制剂选自LY294002、hibiscone C、Idelalisib、Copanlisib、Duvelisib、Alpelisib、Taselisib、Perifosine、Buparlisib、Umbralisib、PX-866、Dactolisib、CUDC-907、Voxtalisib、CUDC-907、ME-401、IPI-549、SF1126、Tenalisib、INK1117、pictilisib、XL147、Palomid 529、GSK1059615、ZSTK474、PWT33597、IC87114、TG100–115、CAL263、RP6503、PI-103、GNE-477或AEZS-136中的任意一种或其任意的组合,优选为Idelalisib、Copanlisib、Duvelisib、Alpelisib或者Tenalisib中的任意一种或其任意的组合。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的PI3K抑制剂选自Tenalisib、其代谢产物或其药学上可接受的盐中的任意一种或其组合。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的Bcl-2抑制剂包括navitoclax、venetoclax、A-1155463、A-1331852、ABT-737、obatoclax、TW-37、A-1210477、AT101、HA14-1、BAM7、S44563、sabutoclax、UMI-77、藤黄酸、maritoclax、MIM1、甲泼尼龙、iMAC2、Bax抑制剂肽V5、Bax抑制剂肽P5、Bax通道阻滞剂和ARRY 520三氟乙酸盐中的任意一种或其组合。
在本发明的技术方案IV的优选技术方案中,其特征在于,所述的Bcl-2抑制剂选自venetoclax、navitoclax、obatoclax、sabutoclax或者maritoclax中的任意一种或其组合。
另外的,本发明还提供了一种技术方案V:
一种药物组合物,其特征在于,包括:i)至少一种有效量的磷酸肌醇3-激酶(PI3K)抑制剂、ii)至少一种有效量的DNA甲基化试剂(例如,阿扎胞苷)以及iii)至少一种有效量的Bcl2抑制剂,其中所述的药物组合物可以处于同一和/或分开的剂型中。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如,所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的DNA甲基化试剂(例如,阿扎胞苷)可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的来Bcl-2抑制剂可以配制成经口服、口含、或者经消化道外途径的剂型,例如所述的口服制剂可以是片剂、胶囊、粉末、丸剂、颗粒、悬浮液、溶液、溶液预浓缩剂、乳液或者乳液预浓缩剂,优选为片剂或者胶囊;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案V的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的磷酸肌醇3-激酶(PI3K)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案V的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的DNA甲基化试剂(例如,阿扎胞苷),例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案V的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的Bcl-2抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂选自PI3Kα抑制剂、PI3Kβ抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂、泛-PI3K抑制剂或PI3Kδ/γ双重抑制剂,优选为PI3Kδ/γ双重抑制剂。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的PI3K抑制剂选自LY294002、hibiscone C、Idelalisib、Copanlisib、Duvelisib、Alpelisib、Taselisib、Perifosine、Buparlisib、Umbralisib、PX-866、Dactolisib、CUDC-907、Voxtalisib、CUDC-907、ME-401、IPI-549、SF1126、Tenalisib、INK1117、pictilisib、XL147、Palomid 529、GSK1059615、ZSTK474、PWT33597、IC87114、TG100–115、CAL263、RP6503、PI-103、GNE-477或AEZS-136中的任意一种或其任意的组合,优选为Idelalisib、Copanlisib、Duvelisib、Alpelisib或者Tenalisib中的任意一种或其任意的组合。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的PI3K抑制剂选自Tenalisib、其代谢产物或其药学上可接受的盐中的任意一种或其组合。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的Bcl-2抑制剂包括navitoclax、venetoclax、A-1155463、A-1331852、ABT-737、obatoclax、TW-37、A-1210477、AT101、HA14-1、BAM7、S44563、sabutoclax、UMI-77、藤黄酸、maritoclax、MIM1、甲泼尼龙、iMAC2、Bax抑制剂肽V5、Bax抑制剂肽P5、Bax通道阻滞剂和ARRY 520三氟乙酸盐中的任意一种或其组合。
在本发明的技术方案V的优选技术方案中,其特征在于,所述的Bcl-2抑制剂选自venetoclax、navitoclax、obatoclax、sabutoclax或者maritoclax中的任意一种或其组合。
除此之外,本发明还提供了一种药盒,其包含本发明的技术方案III、IV、V或其优选技术方案中的任意一项所述的药物组合物,以及药品使用说明书。
除此之外,本发明还提供了一种技术方案VI:
一种在对有需要的对象治疗与过度B细胞增殖有关的疾病或紊乱的方法,其特征在于,包括向所述的对象本发明的技术方案III、IV、V或其优选技术方案中的任意一项所述的药物组合物。
在本发明的技术方案VI的优选技术方案中,其特征在于,所述的与过度B细胞增殖有关的疾病或紊乱为癌症。
在本发明的技术方案VI的优选技术方案中,其特征在于,所述的癌症为血液学恶性肿瘤。
在本发明的技术方案VI的优选技术方案中,其特征在于,所述的血液学恶性肿瘤为淋巴瘤、白血病或骨髓瘤。
在本发明的技术方案VI的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自急性淋巴细胞白血病(ALL)、急性髓系白血病(AML)、慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、多发性骨髓瘤(MM)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、Waldenstrom巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)、边缘区淋巴瘤(MZL)、Burkitt淋巴瘤、毛细胞白血病(HCL)和Richter转化。
在本发明的技术方案VI的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、弥漫性大B细胞淋巴瘤(DLBCL)和边缘区淋巴瘤(MZL)。
除此之外,本发明还提供了一种技术方案VII:
一种药物组合物,其特征在于,包括:i)至少一种有效量的磷酸肌醇3-激酶(PI3K)抑制剂、ii)至少一种有效量的化疗试剂,其中所述的药物组合物可以处于同一和/或分开的剂型中。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的磷酸肌醇3-激酶(PI3K)抑制剂选自PI3Kα抑制剂、PI3Kβ抑制剂、PI3Kδ抑制剂、PI3Kγ抑制剂、泛-PI3K抑制剂或PI3Kδ/γ双重抑制剂,优选为PI3Kδ/γ双重抑制剂。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的PI3K抑制剂选自LY294002、hibiscone C、Idelalisib、Copanlisib、Duvelisib、Alpelisib、Taselisib、Perifosine、Buparlisib、Umbralisib、PX-866、Dactolisib、CUDC-907、Voxtalisib、CUDC-907、ME-401、IPI-549、SF1126、Tenalisib、INK1117、pictilisib、XL147、Palomid 529、GSK1059615、ZSTK474、PWT33597、IC87114、TG100–115、CAL263、RP6503、PI-103、GNE-477或AEZS-136中的任意一种或其任意的组合,优选为Idelalisib、Copanlisib、Duvelisib、Alpelisib或者Tenalisib中的任意一种或其任意的组合。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的PI3K抑制剂选自Tenalisib、其代谢产物或其药学上可接受的盐中的任意一种或其组合。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的化疗试剂选自环磷酰胺、多柔比星、紫杉醇、泼尼松龙的组合。
在本发明的技术方案VII的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的磷酸肌醇3-激酶(PI3K)抑制剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案VII的优选技术方案中,其特征在于,每单位剂型中包含1-1000mg的化疗试剂,例如1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、30、35、40、45、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000mg或以上任意两个数值之间的值。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的环磷酰胺、多柔比星、紫杉醇、泼尼松龙的质量比例为10-50:1-5:0.1-1:0.1-0.5。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的环磷酰胺、多柔比星、紫杉醇、泼尼松龙的质量比例40:3:0.5:0.2。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的环磷酰胺可以配制成经消化道外途径的剂型;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的多柔比星可以配制成经消化道外途径的剂型;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
在本发明的技术方案VII的优选技术方案中,其特征在于,所述的紫杉醇可以配制成经消化道外途径的剂型;所述的消化道外途径的剂型可以是静脉内、腹腔内、皮内、皮下、肌肉、颅内、鞘内、经皮渗透、经黏膜给药的剂型,例如可以是溶液制剂或者冻干粉制剂。
除此之外,本发明还提供了一种技术方案VIII:
一种药盒,其包含本发明技术方案VII中的任意一项所述的药物组合物,以及药品使用说明书。
除此之外,本发明还提供了一种技术方案IX:
一种在对有需要的对象治疗与过度B细胞增殖有关的疾病或紊乱的方法,其特征在于,包括向所述的对象施用本发明技术方案VII中的任意一项药物组合物。
在本发明的技术方案IX的优选技术方案中,其特征在于,所述的与过度B细胞增殖有关的疾病或紊乱为癌症。
在本发明的技术方案IX的优选技术方案中,其特征在于,所述的癌症为血液学恶性肿瘤。
在本发明的技术方案IX的优选技术方案中,其特征在于,所述的血液学恶性肿瘤为淋巴瘤、白血病或骨髓瘤。
在本发明的技术方案IX的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自急性淋巴细胞白血病(ALL)、急性髓系白血病(AML)、慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、多发性骨髓瘤(MM)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、Waldenstrom巨球蛋白血症(WM)、弥漫性大B细胞淋巴瘤(DLBCL)、边缘区淋巴瘤(MZL)、Burkitt淋巴瘤、毛细胞白血病(HCL)和Richter转化。
在本发明的技术方案IX的优选技术方案中,其特征在于,所述的血液学恶性肿瘤选自慢性淋巴细胞白血病(CLL)、小淋巴细胞性淋巴瘤(SLL)、非Hodgkin淋巴瘤(NHL)、外套细胞淋巴瘤(MCL)、滤泡性淋巴瘤(FL)、弥漫性大B细胞淋巴瘤(DLBCL)和边缘区淋巴瘤(MZL)。
在本发明的上述技术方案中,所述的Tenalisib药学上可接受的盐选自以下任意一组:
a)Tenalisib的苯磺酸盐的结晶形式,其在X射线衍射图上显示13.28°,20.72°,21.70°,18.80°,18.14°,17.26°,23.81°,23.02°,11.12°,14.00°,23.44°,22.14°(±0.2°)的衍射角2θ处的峰;
b)Tenalisib的己二酸盐的结晶形式,其在X射线衍射图上显示5.09°,12.26°,10.31°,20.41°,27.02°,6.90°,21.67°,14.62°,8.98°,18.92°,13.63°,3.46°,7.26°,15.15°,17.59°,10.67°,(±0.2°)的衍射角2θ处的峰;
c)Tenalisib的富马酸盐的结晶形式,其在X射线衍射图上显示3.30°,6.60°,22.70°,28.92°,12.08°,27.58°,11.70°,20.28°,19.44°,23.54°,29.45°,24.22°,16.10°(±0.2°)的衍射角2θ处的峰;
d)Tenalisib的半富马酸盐的结晶形式1,其在X射线衍射图上显示3.26°,9.64°,21.67°,22.46°,26.75°,16.00°(±0.2°)的衍射角2θ处的峰;
e)Tenalisib的半富马酸盐的结晶形式2,其在X射线衍射图上显示3.36°,6.92°,22.91°,19.65°,16.37°,18.98°,9.13°,27.52°,6.37°,26.32°,25.18°,11.64°,9.84°,10.92°,
15.80°,20.26°,24.13°,20.98°,27.77°,12.12°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的苯磺酸盐的结晶形式还包括在27.25°,28.31°,22.43°,26.47°,34.51°,28.84°,20.32°,16.52°,27.97°,27.251°,26.91°,33.14°,31.07°,24.96°,8.94°,29.31°,29.65°,32.60°,31.32°,38.81°,33.51°,37.77°,37.09°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的苯磺酸盐的结晶形式具有如图8所述的XRPD衍射图。
在本发明的技术方案的优选技术方案中,其特征在于,所述的式(I)结构的化合物的己二酸盐的结晶形式还包括在4.77°,22.83°,28.99°,23.13°,19.96°,16.98°,14.06°,27.84°,18.18°,16.31°,25.48°,24.68°,21.30°,26.51°,30.38°,19.35°,11.75°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的己二酸盐的结晶形式具有如图9所述的XRPD衍射图。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的富马酸盐的结晶形式还包括在15.66°,14.54°,12.98°,24.92°,25.28°,13.32°,9.82°,18.96°,23.54°,29.45°,10.80°,17.18°,35.85°,21.43°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的富马酸盐的结晶形式具有如图10所述的XRPD衍射图。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的半富马酸盐的结晶形式1还包括在11.96°,27.53°,13.86°,10.10°,10.50°,14.30°,12.80°,10.98°,18.44°,17.54°,22.17°,17.86°,20.90°,14.90°,24.71°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的半富马酸盐的结晶形式1具有如图11所述的XRPD衍射图。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的半富马酸盐的结晶形式2还包括在22.53°,12.67°,24.45°,22.03°,17.75°,17.52°,23.61°,29.08°,13.12°,13.74°,17.18°(±0.2°)的衍射角2θ处的峰。
在本发明的技术方案的优选技术方案中,其特征在于,所述的Tenalisib的化合物的半富马酸盐的结晶形式2具有如图12所述的XRPD衍射图。
一般而言,将本发明的药物组合物组合使用会起到下述作用:
(1)与单独施用任一种药剂相比,在减少肿瘤生长和/或转移或甚至消除肿瘤和/或转移方面产生更好的功效,
(2)使化学治疗剂施用量减少,
(3)与用单药剂化学疗法和某些其它联合疗法相比,为患者提供具有良好耐受性(具有较少的有害药理学并发症)的化学治疗,
(4)提供在哺乳动物,特别是人类中更广谱的适于不同癌症类型的治疗,
(5)使治疗的患者具有更高的响应率,
(6)与标准化学疗法治疗相比,为治疗的患者提供更长的存活时间,
(7)为肿瘤进展提供更长的时间,和/或
(8)与其中其它癌症药剂组合产品产生拮抗作用的已知情况相比,产生至少与单独使用的那些药剂同样良好的功效和耐受性结果。
术语定义
本申请中,“磷酸肌醇3-激酶(PI3K)抑制剂”或“PI3K抑制剂”通常是指任何PI3K的抑制剂。PI3K是细胞内脂质激酶的独特且保守的家族的成员,其使磷脂酰肌醇或磷酸肌醇上的3’-OH基团磷酸化。PI3K家族包括具有不同底物特异性、表达模式和调节模式的激酶(参见,例如,Katso等人,2001,Annu.Rev.Cell Dev.Biol.17,615-675;Foster,F.M.等人,2003,J Cell Sci116,3037-3040)。I类PI3K(例如,p110α、p110β、p110γ和p110δ通常由酪氨酸激酶或G-蛋白偶联受体活化以产生PIP3,PIP3结合下游介质,如Akt/PDK1途径中的那些、mTOR、Tec家族激酶和Rho家族GTP酶。II类PI3K(例如,PI3K-C2α、PI3K-C2β、PI3K-C2γ)和III类PI3K(例如,Vps34)通过合成PI(3)P和PI(3,4)P2在细胞内运输中起关键作用。本申请公开了具体的示例性PI3K抑制剂。例如,PI3K抑制剂抑制PI3K-α、PI3K-β、PI3K-γ和PI3K-δ亚型或其组合。
例如,PI3K抑制剂可以是PI3Kδ/γ双重抑制剂Tenalisib,(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮(本发明中以CN401代码表示),其结构式如下:
或其代谢产物(本发明以IN0385表示),其结构式如下:
或者二者的组合物。
术语“P13K-δ选择性抑制剂”(还称为P13K-δ抑制剂)是指这样的化合物,与P13K家族(α、β和γ)的其他同种型相比,其更高效地选择性地抑制P13K-δ同种型的活性。
术语“Bruton酪氨酸激酶”(还称为“BTK”,无丙种球蛋白酪氨酸激酶(ATK),或者B细胞祖细胞激酶(BPK))是指在B细胞抗原受体(BCR)信号传递途径中非受体酪氨酸激酶。BTK(蛋白质酪氨酸激酶的Tec家族成员)在发育的多个阶段主要在B淋巴细胞中表达(除了最终分化的浆细胞)。BTK为信号传导蛋白质,其调节正常的B细胞发育、分化和发挥功能,并且还涉及成熟B细胞淋巴组织增生性紊乱的起始、生存和进展,例如B细胞恶性肿瘤。Akinleye,A.等人,J.Hematol.Oncol.6:59(2013)。如本文所用,BTK得自智人,如美国专利No.6,326,469(Gen Bank Acc.No.NP_000052)所公开。
“BTK的抑制剂”或“BTK抑制剂”是指靶向BTK并抑制BTK酪氨酸磷酸化和/或B细胞活化,和/或以其他方式抑制或降低或消除BTK蛋白质的生物学活性的小分子。“不可逆的BTK抑制剂”是指在与BTK接触时,导致与BTK的氨基酸残基形成新的共价键的分子。BTK的可逆和不可逆的抑制剂都可以用于本发明的方法和试剂盒中。例如,BTK抑制剂可以为泽布替尼(zanubrutinib)及其药学上可接受的盐。
如本文所用,术语“协同作用”是指通过化合物的组合给予而产生的高于加和(greater-than-additive)的治疗作用,其中使用所述的组合而获得的治疗作用超过了以其他方式由单独给予单独的化合物而得到的加和的作用。本发明的实施方案包括在血液学癌症的治疗中产生协同作用的方法,其中所述的作用比相应的加和作用高至少5%,至少10%,至少20%,至少30%,至少40%,至少50%,至少60%,至少70%,至少80%,至少90%,至少100%,至少200%,至少500%或至少1000%。
如本文所用,“治疗协同作用”是指如本文所述,试剂的组合给予会产生比单独使用时和/或两种试剂组合时的加和作用更高的治疗作用。
如本发明公开和权利要求书所用,单数形式“一”和“一种”包括复数形式,除非内容中另外清楚地表明。
应该理解的是,本发明中使用语言“包含”和“包括”来描述实施方案时,还提供以“由……组成”和/或“基本上由……组成”所描述的其他类似的实施方案。
本发明中,术语“药学上可接受的盐”指的是以酸或碱形式存在的盐,其非受限的实例为(a)酸性加成盐,无机酸(例如,盐酸、氢溴酸、硫酸、磷酸、硝酸等)和有机酸的盐,有机酸例如醋酸、草酸(oxalicacid)、酒石酸(tartaric acid)、琥珀酸(succinic acid)、苹果酸(malic acid)、抗坏血酸、苯甲酸(benzoic acid)、鞣酸(tannic acid)、扑酸(pamoicacid)、海藻酸(alginic acid)、聚麸胺酸(poly glutamicacid)及水杨酸等;(b)碱性加成盐(base addition salts),形成有金属阳离子,如锌、钙、钠、钾等。
可以将本发明的组合疗法中的各种药物以被健康护理提供者认为适合患者的任意次序单独地施用给患者。它们还可以同时施用;或以杂合(hybrid)方式施用,也就是说,例如,同时施用各种药物中的两种,并与第三种药物分开。还可以将所述组合疗法中的各种药物共配制或提供在药物试剂盒中。本发明还包括前述活性成分的组合,其用于治疗疾病,包括、但不限于癌症诸如淋巴样恶性肿瘤(例如,上面列举的B-细胞恶性肿瘤)和免疫障碍诸如自身免疫疾病和炎症。在本发明中进一步包括前述活性成分的组合在制备用于治疗这些疾病的药物中的用途。可以使用常用于药物制备的载体、赋形剂和其它添加剂来制备含有本发明的活性成分或其药学上可接受的盐的药物组合物。
附图说明
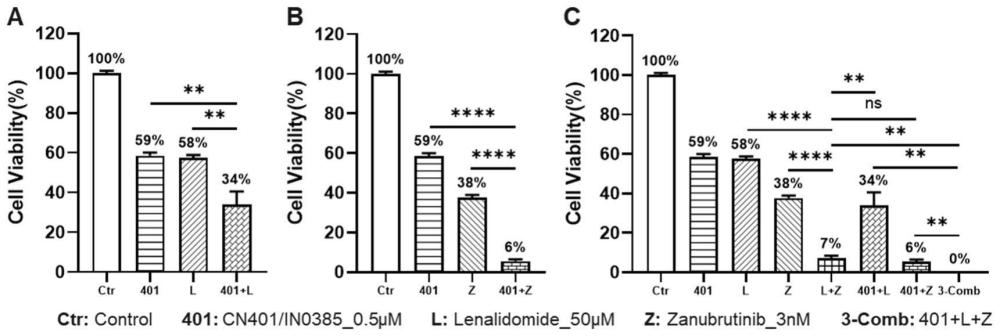
图1CN401/IN0385(0.5μM)与来那度胺(50μM)和泽布替尼(3nM)在人弥漫性大B细胞淋巴瘤细胞系TMD8中的联合用药结果。
图2CN401/IN0385(5μM)与来那度胺(50μM)和泽布替尼(1μM)在人弥漫性大B细胞淋巴瘤细胞系Dohh2中的联合用药结果。
图3CN401/IN0385(5μM)与来那度胺(50μM)和泽布替尼(20μM)在人弥漫性大B细胞淋巴瘤细胞系OCI-ly3中的联合用药结果。
图4CN401/IN0385(5μM)与维奈托克(5nM)和阿扎胞苷(1.2μM)在人急性髓系白血病细胞系MV4-11中的联合用药结果。
图5不同PI3K抑制剂(10μM)与维奈托克(5nM)和阿扎胞苷(1.2μM)在人急性髓系白血病细胞系MV4-11中的联合用药结果。
图6CN401(Tenalisib)(150mg/kg)与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)在Hut78人T细胞淋巴瘤动物皮下模型中联合用药结果。
图7CN401(Tenalisib)(150mg/kg)与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)在HH人T细胞淋巴瘤动物皮下模型中联合用药结果。
图8(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮苯磺酸盐结晶的XRPD衍射图。
图9(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮己二酸盐结晶的XRPD衍射图。
图10(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮富马酸盐结晶的XRPD衍射图。
图11(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮半富马酸盐结晶形式1的XRPD衍射图。
图12(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮半富马酸盐结晶形式2的XRPD衍射图。
图13CN401的5种不同改造盐型及其在体外对人急性髓系白血病细胞系MV4-11生长的抑制曲线。
具体实施方式
本发明以下实施例中所用的术语“CN401/IN0385”表示CN401或IN0385,实验中实际所用“CN401/IN0385”药物均为PI3K抑制剂CN401(Tenalisib),若用其代谢物IN0385替代也可以取得类似的疗效。
实施例1-1CN401/IN0385与来那度胺和泽布替尼在人弥漫性大B细胞淋巴瘤细胞系中的联合用药研究
本发明研究了在人弥漫性大B细胞淋巴瘤细胞系(TMD8,Dohh2,OCI-ly3)中,PI3K抑制剂CN401(Tenalisib)与小分子免疫调节剂来那度胺(Lenalidomide)和小分子BTK抑制剂泽布替尼(Zanubrutinib)的体外联合治疗作用。
材料和方法
本实验所用TMD8细胞购自吉满生物科技(上海)有限公司,Dohh2和OCI-ly3细胞购自南京科佰生物科技有限公司,于37℃、含5% CO
实验分组设计如表1所示,每个细胞系均设置对照组、单药处理组、双药联用组及三药联用组,药物浓度见各细胞系对应实施例。
表1
实施例1.1CN401//IN0385与来那度胺和泽布替尼在TMD8细胞系中的联合用药研究
人DLBCL细胞系TMD8在RPMI1640培养基(含10% FBS和1×P/S)中进行培养,实验时收集细胞到离心管中,300×g离心5min,弃上清后用新鲜培养基重悬混,取20μL进行台盼蓝染色计数,调整活细胞密度至8E3/90μL,以每孔90μL的体积接种于96孔细胞培养板(白壁底不透)中。药物使用终浓度为:CN401/IN0385(0.5μM),来那度胺(50μM),泽布替尼(3nM),实验分组设计见表1。根据分组,配制药物混合液(10×),10μL/孔加入到已接种细胞的96孔培养板中,在培养箱中继续培养72h,使用CTG进行细胞活力的检测。以对照组细胞活力均值为100%,计算每个孔细胞的相对活力值。
结果:不同分组的TMD8细胞活力变化情况见图1A-图1C,联合用药组相对单药处理组的p值分析显示在表2中。在本研究中,CN401与来那度胺联用对TMD8细胞生长的抑制有协同作用(图1A),CN401与泽布替尼联用对TMD8细胞生长的抑制有协同作用(图1B),CN401,来那度胺和泽布替尼三药联用对TMD8细胞生长的抑制相比于单药、二组分联用有更强的协同作用(图1C)。
表2
注:401表示CN401/IN0385,L表示来那度胺,Z表示泽布替尼。
实施例1.2CN401//IN0385与来那度胺和泽布替尼在Dohh2细胞系中的联合用药研究
人DLBCL细胞系Dohh2在RPMI1640培养基(含10% FBS和1×P/S)中进行培养,实验时收集细胞到离心管中,300×g离心5min,弃上清后用新鲜培养基重悬混,取20μL进行台盼蓝染色计数,调整活细胞密度至7E3/90μL,以每孔90μL的体积接种于96孔细胞培养板(白壁底不透)中。药物使用终浓度为:CN401/IN0385(5μM),来那度胺(50μM),泽布替尼(1μM),实验分组设计见表1。根据分组,配制药物混合液(10×),10μL/孔加入到已接种细胞的96孔培养板中,在培养箱中继续培养72h,使用CTG进行细胞活力的检测。以对照组细胞活力均值为100%,计算每个孔细胞的相对活力值。
结果:不同分组的Dohh2细胞活力变化情况见图2A-图2C,联合用药组相对单药处理组的p值分析显示在表3中。在本研究中,CN401/IN0385与来那度胺联用对Dohh2细胞生长的抑制有协同作用(图2A),CN401/IN0385与泽布替尼联用对Dohh2细胞生长的抑制有协同作用(图2B),CN401/IN0385,来那度胺和泽布替尼三药联用对Dohh2细胞生长的抑制相比于单药或者二组分联用有更强的协同作用(图2C)。
表3
注:401表示CN401/IN0385,L表示来那度胺,Z表示泽布替尼。
实施例1.3CN401/IN0385与来那度胺和泽布替尼在OCI-ly3细胞系中的联合用药研究
人DLBCL细胞系OCI-ly3在IMDM培养基(含20% FBS和1×P/S)中进行培养,实验时收集细胞到离心管中,300×g离心5min,弃上清后用新鲜培养基重悬混,取20μL进行台盼蓝染色计数,调整活细胞密度至7E3/90μL,以每孔90μL的体积接种于96孔细胞培养板(白壁底不透)中。药物使用终浓度为:CN401/IN0385(5μM),来那度胺(50μM),泽布替尼(20μM),实验分组设计见表1。根据分组,配制药物混合液(10×),10μL/孔加入到已接种细胞的96孔培养板中,在培养箱中继续培养72h,使用CTG进行细胞活力的检测。以对照组细胞活力均值为100%,计算每个孔细胞的相对活力值。
结果:不同分组的OCI-ly3细胞活力变化情况见图3A-图3C,联合用药组相对单药处理组的p值分析显示在表4中。在本研究中,CN401/IN0385与来那度胺联用对OCI-ly3细胞生长的抑制有协同作用(图3A),CN401/IN0385与泽布替尼联用对OCI-ly3细胞生长的抑制有协同作用(图3B),CN401/IN0385,来那度胺和泽布替尼三药联用对OCI-ly3细胞生长的抑制相比于单药、二组分联用有更强的协同作用(图3C)。
表4
注:401表示CN401/IN0385,L表示来那度胺,Z表示泽布替尼。
实施例1-2,PI3K抑制剂与阿扎胞苷以及PI3K抑制剂与阿扎胞苷和维奈托克在人急性髓系白血病细胞系中的联合用药研究
试验设计
本发明研究了在人急性髓系白血病细胞系(MV4-11)中,PI3K抑制剂CN401(Tenalisib)与小分子BCL-2抑制剂维奈托克(Venetoclax)和胞嘧啶核苷类似物阿扎胞苷(Azacytidine)的体外联合治疗作用,并与其它PI3K抑制剂(IPI549、Alpelisib,Umbralisib)进行了横向比较。
材料和方法
本实验所用MV4-11细胞购自ATCC,于37℃、含5% CO
实验分组设计如表5所示,每个实施例均设置对照组、单药处理组、双药联用组及三药联用组,药物浓度见具体实施例。
表5
实施例1-2-1&实施例1-2-2,CN401/IN0385与维奈托克和阿扎胞苷在MV4-11细胞系中的联合用药研究
人急性髓系白血病细胞系MV4-11在IMDM培养基(含10% FBS和1×P/S)中进行培养,实验时收集细胞到离心管中,300×g离心5min,弃上清后用新鲜培养基重悬混,取20μL进行台盼蓝染色计数,调整活细胞密度至7E3/90μL,以每孔90μL的体积接种于96孔细胞培养板(白壁底不透)中。药物使用终浓度为:CN401/IN0385(10μM),维奈托克(5nM),阿扎胞苷(1.2μM),实验分组设计见表5。根据分组,配制药物混合液(10×),10μL/孔加入到已接种细胞的96孔培养板中,在培养箱中继续培养72h,使用CTG进行细胞活力的检测。以Control组细胞活力均值为100%,计算每个孔细胞的相对活力值。
结果:不同分组的MV4-11细胞活力变化情况见图4A-图4C,联合用药组相对单药处理组的p值分析显示在表6中。在本研究中,CN401/IN0385与维奈托克联用对MV4-11细胞生长的抑制有协同作用(图4A),CN401/IN0385与阿扎胞苷联用对MV4-11细胞生长的抑制有协同作用(图4B),CN401/IN0385,维奈托克和阿扎胞苷三药联用对MV4-11细胞生长的抑制同样有协同作用(图4C)。
表6
注:401表示CN401/IN0385,V表示维奈托克,A表示阿扎胞苷。
实施例1-2-3不同PI3K抑制剂与维奈托克和阿扎胞苷在MV4-11细胞系中的联合用药比较
人急性髓系白血病细胞系MV4-11在IMDM培养基(含10% FBS和1×P/S)中进行培养,实验时收集细胞到离心管中,300×g离心5min,弃上清后用新鲜培养基重悬混,取20μL进行台盼蓝染色计数,调整活细胞密度至7E3/90μL,以每孔90μL的体积接种于96孔细胞培养板(白壁底不透)中。药物使用终浓度为:PI3K抑制剂(CN401/IN0385或IPI549或Alpelisib或Umbralisib,均为10μM),维奈托克(5nM),阿扎胞苷(1.2μM),实验分组设计见表5。根据分组,配制药物混合液(10×),10μL/孔加入到已接种细胞的96孔培养板中,在培养箱中继续培养72h,使用CTG进行细胞活力的检测。以对照组细胞活力均值为100%,计算每个孔细胞的相对活力值。
结果:不同分组的MV4-11细胞活力变化情况见图5,不同PI3K抑制剂间(单药或联用)的p值分析显示在表7中。在本研究中,CN401单药对MV4-11细胞生长的抑制效果要优于相同浓度下的IPI549、Alpelisib和Umbralisib;与Venetoclax、Azacytidine或Venetoclax+Azacytidine联合使用时,在对MV4-11细胞生长的抑制上,CN401同样优于相同浓度下的IPI549、Alpelisib和Umbralisib。
表7
注:401表示CN401/IN0385。
实施例1-3
本发明研究了在Hut78和HH人T细胞淋巴瘤动物皮下模型中PI3K抑制剂CN401与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)联合治疗的作用。
材料和方法
本动物实验按照AAALAC要求进行,并经过必凯动物实验中心IACUC批准进行此动物实验。从江苏集萃药康生物科技股份有限公司采购6-8周龄的NCG小鼠(18-20克),每只老鼠皮下接种200万个Hut78或HH细胞,接种在老鼠的背部右前侧。当肿瘤的体积生长到约140-160mm
研究设计如表8所示。
空白对照组(1):生理盐水静脉注射,第一天给1次;0.5%MC口服,一天两次,一共给药3周;
对照组(2):CHOP(C:环磷酰胺40毫克/千克,H:多柔比星3.3毫克/千克,O:紫杉醇0.5毫克/千克,P:泼尼松龙0.2毫克/千克),CHO第一天给药一次,尾静脉注射,P口服,一天一次,从第一天开始连续给5天;
对照组(3):CN401(tenalisib)150毫克/千克,口服,一天两次,一共给药3周;
实验组(4):CN401 150毫克/千克,口服,一天两次,一共给药3周;且CHOP(C:环磷酰胺40毫克/千克,H:多柔比星3.3毫克/千克,O:紫杉醇0.5毫克/千克,P:泼尼松龙0.2毫克/千克),CHO第一天给药一次,尾静脉注射,P口服,一天一次,从第一天开始连续给5天;
对照组(5):1/2CHOP(C:环磷酰胺20毫克/千克,H:多柔比星1.65毫克/千克,O:紫杉醇0.25毫克/千克,P:泼尼松龙0.1毫克/千克),CHO第一天给药一次,尾静脉注射,P口服,一天一次,从第一天开始连续给5天;
实验组(6):CN401 150毫克/千克,口服,一天两次,一共给药3周;且1/2CHOP(C:环磷酰胺20毫克/千克,H:多柔比星1.65毫克/千克,O:紫杉醇0.25毫克/千克,P:泼尼松龙0.1毫克/千克),CHO第一天给药一次,尾静脉注射,P口服,一天一次,从第一天开始连续给5天。
每天检查小鼠并记录不良临床反应,肿瘤体积和动物体重一周测量三次。
表8
实施例1-3-1PI3K抑制剂CN401与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)的联合用药在Hut78模型中的研究
CN401(Tenalisib)与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)在Hut78人T细胞淋巴瘤动物皮下模型中联合用药研究(试验设计见表9)。
6-8周龄NCG小鼠(18-20g)购自江苏集萃药康生物科技股份有限公司,每只老鼠皮下接种200万个Hut78细胞,接种在老鼠的背部右前侧。当肿瘤的体积生长到约140mm
表9
结果:在本研究中,CN401与CHOP联合在Hut78人T细胞淋巴瘤动物皮下模型中对肿瘤抑制有协同作用(p=0.00004,TGI=80%),小鼠对所给药物表现出良好的耐受性。肿瘤生长曲线和体重生长曲线的结果见图6A-图6F;在第25天与空白组相比,治疗组的TGI、p值和完全反应小鼠的数量显示在表10中。
表10
实施例1-3-2PI3K抑制剂CN401与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)的联合用药在HH模型中的研究
CN401(Tenalisib)与CHOP(C:环磷酰胺,H:多柔比星,O:紫杉醇,P:泼尼松龙)在HH人T细胞淋巴瘤动物皮下模型中联合用药研究(试验设计见表11)。
6-8周龄NCG小鼠(18-20g)购自江苏集萃药康生物科技股份有限公司,每只老鼠皮下接种200万个HH细胞,接种在老鼠的背部右前侧。当肿瘤的体积生长到约158mm
表11
结果:在本研究中,CN401与CHOP联合在HH人T细胞淋巴瘤动物皮下模型中对肿瘤抑制有协同作用(p=0.000005,TGI=70%),CN401与1/2CHOP联合在HH人T细胞淋巴瘤动物皮下模型中对肿瘤抑制有协同作用(p=0.000003,TGI=64%),小鼠对给所给药物表现出良好的耐受性。肿瘤生长曲线和体重生长曲线的结果见图7A-图7H;在第21天与空白组相比,治疗组的TGI、p值和完全反应小鼠的数量显示在表12中。
表12
实施例2不同盐型晶型的(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮的合成及表征
一、分析方法
1.1X射线粉末衍射仪(XRPD)
利用X-粉末衍射仪对样品进行晶型分析。样品的2θ扫描角度为3°至40°,扫描步长为0.02°,每步的扫描时间为0.2s。光管电压和电流分别为40kV和40mA。制样时将适量样品放在载样盘上,确保样品表面光滑平整。
1.2差示扫描量热分析(DSC)
采用TA instruments Q200 DSC对样品进行分析。将称量过的样品(0.5mg-5mg)放入载样盘中,在氮气(50mL/min)的保护下将样品以10℃/min的速率升高到最终温度。
1.3热重分析(TGA)
采用TA instruments Q500对样品进行分析。将样品放入去掉皮重的铂金坩埚中,系统自动称重,然后在氮气(40mL/min)的保护下将样品以10℃/min的速率升高到最终温度。
1.4偏振光显微镜(PLM)
利用偏光显微镜对样品进行分析,通过调节不同的放大倍数,得到晶体的形貌和微观结构。
1.5动态水分吸附(DVS)
动态水分吸附采用TA Instruments Q5000 SA进行。将大约1-10mg样品置于样品盘中并悬挂于样品室内。室内温度由水浴保持在恒定的25±1℃。在step模式下,样品在0%RH-80%RH的相对环境湿度进行循环测试。以10%RH/step进行分析。设置保持各湿度的时间为90min,使样品与室内环境达到平衡。
1.6液态核磁氢谱(
利用Bruker Ascend 500MH对样品进行分析,溶剂为氘代二甲基亚砜。
二、制备方法
2.1(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮游离碱化合物(CN-401)的制备方法
参照专利文献CN105358560A实施例的方法制备(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮游离碱化合物(CN-401)。
2.2(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮苯磺酸盐晶型的制备
步骤(1):将约200mg由实施例2.1制备得到的游离碱化合物,加入2mL异丙醇60℃水浴溶清,得到溶液1;
步骤(2):将约94mg苯磺酸超声加热溶于0.4mL异丙醇,得到溶液2;
步骤(3):将将溶液2滴加至搅拌中的溶液1中,得到溶液3;
步骤(4):溶液3搅拌约1天未析出固体,加3mL正庚烷析出固体,搅拌后成油,继续搅拌约2天,得到混悬液。
步骤(5):将混悬液减压抽滤,用异丙醇冲洗滤饼表面,所得固体在室温下真空干燥过夜,得到苯磺酸盐结晶(约200mg)。
对产物进行表征,其XRPD谱图如附图8所示。
2.2(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮己二酸盐结晶形式的制备
步骤(1):将约100mg由实施例2.1制备得到的游离碱化合物加入2.0mL乙酸乙酯,得到混悬液1;
步骤(2):将约40mg己二酸超声加热溶于0.4mL乙醇,得到溶液1;
步骤(3):将溶液1滴加至搅拌中的混悬液1中,溶清,4℃下搅拌过夜,未析出固体,得到溶液2;
步骤(4):加入约20mL正庚烷至溶液2中,析出固体,4℃下搅拌约8h,得到混悬液;
步骤(5):将混悬液离心,所得固体在室温下真空干燥过夜,得到己二酸盐结晶。
对产物进行表征,其XRPD谱图如附图9所示。
2.3(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮富马酸盐结晶的制备方法
步骤(1):称取约200mg由实施例2.1制备得到的游离碱化合物,加入至1mL的异丙醇中,得到混悬液1;
步骤(2):称取约63mg富马酸,超声加热溶于0.4mL异丙醇和0.4mL水混合溶液中,得到溶液1;
步骤(3):将步骤(2)所得到的溶液1滴加至由步骤(1)所得的搅拌中的混悬液1中,得到混悬液2;
步骤(4):将步骤(3)所得到的混悬液2搅拌后立即溶清,向溶液中加入16mL正庚烷,在4℃下搅拌过夜析出固体,得到混悬液3;
步骤(5):将步骤4所得到的混悬液3离心,所得固体在室温下真空干燥过夜,得到约198mg产物。
对产物进行表征,其XRPD谱图如附图10所示。
2.4(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮半富马酸盐结晶形式1的制备
方法一:
步骤(1):称取约200mg由实施例2.1制备得到的游离碱化合物加入4mL乙腈搅拌,得到混悬液1;
步骤(2):约63mg富马酸超声加热溶于0.8mL乙腈和0.4mL水的混合溶液中,得到溶液1;
步骤(3):将溶液1滴加至搅拌中的混悬液1中,得到混悬液2;
步骤(4):混悬液2搅拌后先溶清,然后析出固体,室温搅拌过夜,得到混悬液3;
步骤(5):将混悬液3离心,所得固体在室温下真空干燥过夜,得到约119mg半富马酸盐结晶形式1;
方法二:
步骤(1):称取约200mg由实施例2.1制备得到的游离碱化合物,加入2mL乙腈搅拌,得到混悬液1;
步骤(2):约64mg富马酸超声加热溶于0.8mL乙腈和0.4mL水的混合溶液中,得到溶液1;
步骤(3):将溶液1滴加至搅拌中的混悬液1中,得到混悬液2;
步骤(4):混悬液2搅拌后先溶清,然后析出固体,室温搅拌3天,得到混悬液3;
步骤(5):将混悬液3减压抽滤,用水冲洗滤饼表面,所得固体在室温下真空干燥过夜,得到约210mg半富马酸盐结晶形式1。
对产物进行表征,XRPD谱图如附图11所示。
2.5(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮半富马酸盐结晶形式2的制备
方法一:
步骤(1):称取约202mg由实施例2.1制备得到的游离碱化合物,加入1mL异丙醇,得到混悬液1;
步骤(2):约65mg富马酸超声加热溶于0.6mL异丙醇和0.4mL水混合溶液中,得到溶液1;
步骤(3):将溶液1滴加至搅拌中的混悬液1中,得到混悬液2;
步骤(4):混悬液2搅拌后立即溶清,室温搅拌过夜未析出固体,经室温小孔挥发一天后瓶壁上析出少量透明晶体,加无孔盖,室温搅拌3小时析出白色固体,继续搅拌1天,得到混悬液3;
步骤(5):将混悬液3减压抽滤,用水轻轻冲洗滤饼表面,所得固体在室温下真空干燥过夜,得到约77mg半富马酸盐结晶形式2;
方法二:
步骤(1):称取约501mg由实施例2.1制备得到的游离碱化合物,加入2.5mL异丙醇,得到混悬液1;
步骤(2):约156mg富马酸超声加热溶于1.5mL异丙醇和1mL水混合溶液中,得到溶液1;
步骤(3):将溶液1滴加至搅拌中的混悬液1中,得到混悬液2;
步骤(4):混悬液2搅拌后立即溶清,继续搅拌10分钟,经室温小孔挥发约1天,瓶口析出白色固体,加无孔盖,室温搅拌1小时析出白色固体,继续搅拌2天,得到混悬液3;
步骤(5):将混悬液3减压抽滤,用水轻轻冲洗滤饼表面,所得固体在室温下真空干燥约5天,得到约240mg半富马酸盐结晶形式2。
对产物进行表征,XRPD谱图如附图12所示。
实施例3不同离子盐型的物理性质比较
不同盐型的物理性质比较如表13、表14和表15所示。
表13
表14
表15
实施例4(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮苯磺酸盐、己二酸盐、富马酸盐、半富马酸盐结晶形式1、半富马酸盐结晶形式2与游离碱的药代动力学特征比较
(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮游离碱以及苯磺酸盐结晶形式口服给予Wistar大鼠的代动力学参数的比较,游离碱的口服给药的每组动物为4只,苯磺酸盐的口服给药的每组动物为3只,6-8周龄,雄性。口服给药10%Cremophor EL+90%(10%HP-β-CD in 1%HPMC(pH 2.2)in water,口服给药组动物隔夜禁食,给药4小时后恢复进食。口服给药组动物采血点为给药前及后0.25,0.5,1,2,4,8和24小时。颈静脉采血,每个采血点的采血量约150μL,EDTA-K2抗凝,采样后15分钟内4℃ 2000g离心5min,LCMSMS-28(Triple Quad 6500+)分析。
结果发现:与游离碱比较,苯磺酸盐结晶形式的Tmax和T
表16
本发明还研究了(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮游离碱以及己二酸盐结晶形式口服给予Wistar大鼠的代动力学参数的比较,游离碱的口服给药的每组动物为4只,己二酸盐的口服给药的每组动物为3只,6-8周龄,雄性。口服给药10%Cremophor EL+90%(10%HP-β-CD in 1%HPMC(pH 2.2)in water,口服给药组动物隔夜禁食,给药4小时后恢复进食。口服给药组动物采血点为给药前及后0.25,0.5,1,2,4,8和24小时。颈静脉采血,每个采血点的采血量约150μL,EDTA-K2抗凝,采样后15分钟内4℃2000g离心5min,LCMSMS-28(Triple Quad 6500+)分析。
结果发现,与游离碱比较,己二酸盐结晶形式的T
表17
本发明还研究了(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮游离碱以及富马酸盐结晶形式、半富马酸盐结晶形式1、半富马酸盐结晶形式2口服给予Wistar大鼠的代动力学参数的比较,游离碱的口服给药的每组动物为4只,各个盐型的口服给药的每组动物为3只,6-8周龄,雄性。口服给药10%Cremophor EL+90%(10%HP-β-CDin1%HPMC(pH 2.2)in water,口服给药组动物隔夜禁食,给药4小时后恢复进食。口服给药组动物采血点为给药前及后0.25,0.5,1,2,4,8和24小时。颈静脉采血,每个采血点的采血量约150μL,EDTA-K2抗凝,采样后15分钟内4℃ 2000g离心5min,LCMSMS-28(Triple Quad6500+)分析。
结果发现,半富马酸盐结晶形式1的Tmax、AUC和F%明显高于其它2个盐型;与游离碱比较,半富马酸盐结晶形式1的Tmax、T
表18
实施例5(S)-2-(1-(9H-嘌呤-6-基氨基)丙基)-3-(3-氟苯基)-4H-色烯-4-酮苯磺酸盐结晶、己二酸盐结晶、富马酸盐结晶、半富马酸盐结晶形式1、半富马酸盐结晶形式2与游离碱的针对AML(MV4-11)体外研究比较
CN401及其5种不同改造盐型的生物活性
试验设计
本发明利用人急性髓系白血病细胞系MV4-11,在体外研究比较了CN401及其本发明上述的苯磺酸盐结晶、己二酸盐结晶、富马酸盐结晶、半富马酸盐结晶形式1、半富马酸盐结晶形式2的生物活性。
材料和方法
本实验所用MV4-11细胞购自ATCC,使用IMDM培养基(含10%FBS和1×P/S),于37℃、含5%CO
结果:CN401及其5种改造盐型的分子形式见图13A,不同药物浓度下MV4-11细胞活力变化曲线见图13B,对MV4-11生长的半数抑制浓度显示在表19中。在本研究中,苯磺酸盐Form1与CN401的体外活性基本完全一致,富马酸盐Form1和半富马酸盐Form1和CN401的体外活性接近,半富马酸盐Form2的体外活性略优于CN401,己二酸盐的活性则弱于CN401。
表19IDC236021