放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物及其制备方法和应用
文献发布时间:2024-04-18 19:48:15
技术领域
本发明涉及放射性药物化学与核医学技术领域,具体地说,涉及放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物及其制备方法和应用。
背景技术
成纤维细胞激活蛋白(fibroblast activation protein FAP)是Ⅱ型跨膜丝氨酸蛋白酶,属于脯氨酰寡肽酶家族,广泛表达于肿瘤微环境中的癌症相关成纤维细胞和肿瘤相关巨噬细胞的表面上,如肝癌、结直肠癌、胰腺癌和卵巢癌等上皮性肿瘤中高表达,在以间质组织活化为特征的病变,如感染性、炎症性、纤维化疾病及愈合伤口中也呈高表达,而在正常组织、良性肿瘤间质中无表达或表达较低。因此,FAP是反映肿瘤微环境特征、肿瘤治疗靶点和评价疗效的一个可靠生物标志物。基于FAP开发的放射性药物,有望为肿瘤的诊断和治疗提供重要手段。
基于FAP开发的放射性探针,最早起源于
基于该结构,2018年,Anastasia Loktev等人开发了放射性碘标记FAPI-01。同时通过引入连接喹啉环的DOTA螯合剂开发出用
为了克服现有FAP显像剂无法应用于全身诊疗一体化的问题,本发明在(4-喹啉基)甘氨酰基-2-氰基吡咯烷的基础上引入
发明内容
本发明首先提供了为了实现上述目的,本发明提供了一种提供的放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物,
优选的,所述衍生物具有I的结构,
所述I的结构式如下:
其中,R为
本发明提供了一种提供的放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物,
优选的,所述衍生物具有Ⅲ的结构,
所述Ⅲ的结构式如下:
其中,R为
任选地,R为
任选地,R为
任选地,R为
任选地,R为
本发明还提供一种式I化合物的制备方法,所述方法包含以下步骤:使用含活化基的标记前体式II的化合物,经过放射性标记得到式I的化合物;
所述II的结构式如下:
其中,R为SnMe
本发明还提供式Ⅲ化合物的制备方法,所述方法包含以下步骤:使用含活化基的标记前体式Ⅳ的化合物,经过放射性标记得到式Ⅲ的化合物。
所述Ⅳ的结构式如下:
其中,R为SnMe3、
本发明还提供一种产品,所述产品包含衍生物I和Ⅲ。
优选的,所述产品为药剂或药物或试剂盒或混合物。
优选的,所述药剂为疾病的成像显像剂;所述药物为肿瘤或动脉粥样硬化的治疗药物;所述试剂盒包含一个瓶或多个瓶;所述混合物包含式I、式Ⅱ、式Ⅲ和式Ⅳ中的一种或者多种化合物。
本发明还提供一种衍生物I和Ⅲ的化合物、或其制备方法、或包含上述化合物的产品的应用;
优选的,所述应用为(1)制备药剂或药物或试剂盒或混合物;或(2)用于疾病的成像显像或治疗疾病,且该应用为非诊断和治疗目的的。
优选的,
当R为
当R为
当R为
当R为
本发明的上述技术方案的有益效果如下:
本发明提供一类高亲和FAP的(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物。该类化合物高亲和力,属于诊断和治疗FAPI相关疾病的全新化合物,所述衍生物制备所得到的靶向FAPI的显像或者治疗与已报道的靶向FAPI的放射性药物相比,本发明报道的化合物分子较小,可以应用于包括脑在内的全身显像和治疗。
附图说明
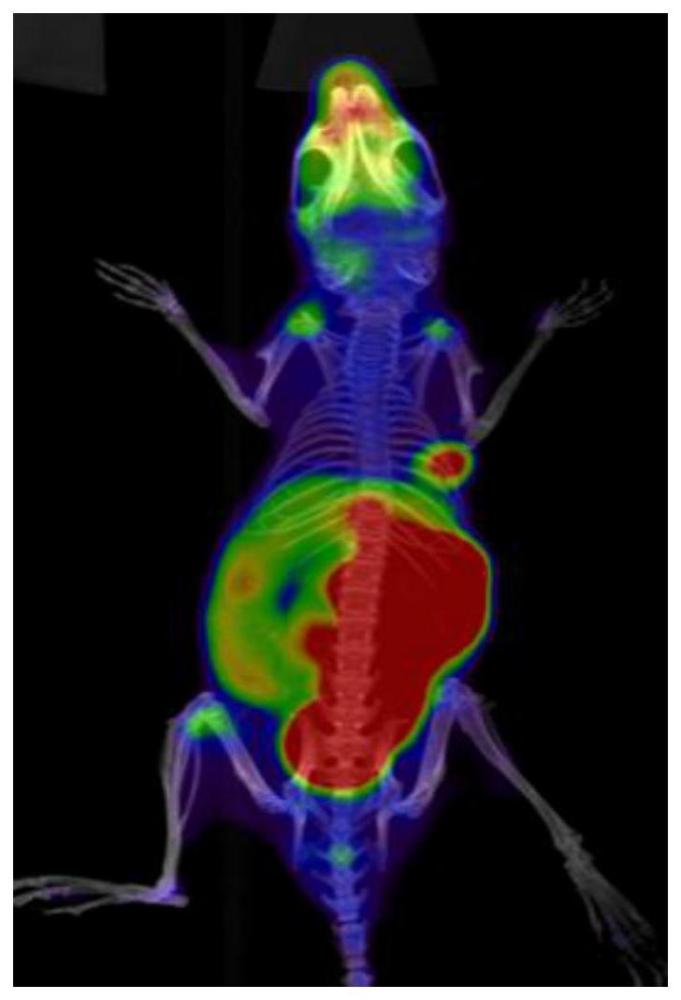
图1为在对U87MG的MicroPET显像;白色圆圈区域为肿瘤。
具体实施方式
为使本发明要解决的技术问题、技术方案和优点更加清楚,下面将结合附图及具体实施例进行详细描述。
实施例1(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-氟喹啉-4-甲酰胺
结构式如下:
合成路线如下:
1.(S)-(2-(2-氨基-4-二氟吡咯烷-1-基)-2-氧乙基)氨基甲酸叔丁酯
将(S)-4,4-二氟吡咯烷-2-甲酰胺盐酸盐(0.38g,2mmol)和(叔丁氧羰基)甘氨酸(0.35g,2mmol)溶于8mL DMF中,随后加入DIPEA(3mL,17mmol)室温搅拌15分钟,最后加入HBTU(1.9g,5mmol),氮气保护下室温搅拌过夜。反应液加入NaHCO
2.(S)-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)氨基甲酸叔丁酯
将起始物(0.48g,1.56mmol)溶于THF(6mL)中,氮气保护下,0℃加入吡啶(0.9mL,10.9mmol)搅拌10分钟,随后加入DCM(2mL)和三氟乙酸酐(0.26mL,2mmol)的混合溶液缓慢滴加至上述反应液,室温搅拌过夜。旋干DCM,将反应液溶于乙酸乙酯中,用盐酸(2mol/L)洗涤3次,有机相用饱和碳酸氢钠溶液(30mL)洗涤1次,再用饱和食盐水洗涤1次。将上述有机相干燥浓缩,FC纯化得到白色固体(0.25g,51%)。HRMS calcd for C12H17F2N3O3
3.(S)-4,4-二氟-1-甘氨基吡咯烷-2-腈
将起始物(0.25g,0.86mmol)溶于三氟乙酸(3mL)中,室温搅拌3h。随后除去三氟乙酸,得到黄色油状物化合物(0.13g,81%)。该化合物直接进行下一步反应。
4.(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-氟喹啉-4-甲酰胺
将6-氟喹啉-4-羧酸(0.05g,0.26mmol),EDCI(0.15g,0.78mmol)和HOBt(0.02g,0.13mmol)溶于DCM(5mL)中,随后滴加TEA(0.2mL,1.3mmol)室温搅拌30min。将起始物(0.03g,0.16mmol)溶于DCM(1mL)中,滴加TEA(0.1mL)室温搅拌30min后滴加至上述反应液,氮气保护下室温过夜。停止反应后,反应液水洗1次,水相再用DCM萃取3次,合并有机相,FC纯化得到黄色油状物实例1目标物(0.012g,21%)
实施例2(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-氟喹啉-4-甲酰胺
结构式如下:
合成路线如下:
化合物合成:
7-氟喹啉-2,4-二羧酸
将NaOH(3.05g,55mmol)溶于水(21mL)中,随后依次将6-氟吲哚啉-2,3-二酮(2.1g,13mmol)和丙酮酸钠(1.75g,16mmol)加入到上述反应液中,加热回流100℃,反应8h。停止反应后冷却到室温,用盐酸(2mol/L)调pH值至1~2,抽滤得到棕色固体(1.67g,51%)。HRMS calcd for C11H7FNO4
7-氟喹啉-4-羧酸
向起始物(1.67g,6.6mmol)加入10mL水,置于高压反应釜中,205℃,4h后停止反应。反应液旋干后,FC纯化得到棕色固体化合物(0.76g,61%)。HRMS calcd forC10H6FNO2,191.0383[M+H]
(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-氟喹啉-4-甲酰胺
将起始物(0.06g,0.31mmol),EDCI(0.18g,0.94mmol),和HOBt(0.024g,0.16mmol)溶于DCM(5mL)中,随后滴加TEA(0.22mL,1.6mmol),室温下搅拌30min。将(S)-4,4-二氟-1-甘氨基吡咯烷-2-腈(0.07g,0.37mmol)溶于DCM(2mL)中,加入TEA(0.1mL)室温搅拌30min后,滴加至上述反应液中。氮气保护下室温过夜。停止反应后,反应液水洗1次,水相再用DCM萃取3次,合并有机相,FC纯化得到油状物实施例2(0.022g,20%)。
实施例3(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-(三甲基锡基)喹啉-4-甲酰胺
结构式如下:
合成路线如下:
(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-(三甲基锡基)喹啉-4-甲酰胺
(S)6-溴-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)喹啉-4-甲酰胺
将6-溴喹啉-4-羧酸(0.26g,1mmol),EDCI(0.4g,2.1mmol),HOBt(0.05g,0.34mmol)溶于DCM中,随后滴加TEA(0.5mL,3.4mmol)室温搅拌30分钟,将起始物(0.13g,0.69mmol)溶于DCM(3mL)中,滴加TEA(0.5mL)室温搅拌30min后滴加至上述反应液中,氮气保护,室温过夜。停止反应后,反应液水洗1次,水相再用DCM萃取3次,合并有机相,FC纯化得到黄色油状物目标化合物(0.09g,31%)
(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-(三甲基锡基)喹啉-4-甲酰胺
将起始物(0.2g,0.47mmol)、四(三苯基磷)钯(0.08g,0.07mmol)、氯化锂(0.03g,0.71mmol)和六甲基二锡(0.78g,2.4mmol)溶于甲苯(8mL)中,加热回流110℃,反应1.5h。旋干甲苯后,将剩余物溶于DCM中,硅藻土过滤,用DCM淋洗,合并有机相,用水洗涤1次,水层DCM萃取2次,浓缩有机相,FC纯化得到实例3(0.05g,21%)
实施例4(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-(三甲基锡基)喹啉-4-甲酰胺
结构式如下:
合成方法参考实施例3,产率为25%,HRMS calcd for Chemical Formula:C20H23F2N4O2Sn+509.0806,[M+H]
实施例5(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-碘喹啉-4-甲酰胺
将实施例4(0.2g,0.39mmol)、碘(0.09g,0.39mmol)、溶于二氯甲烷中室温搅拌0.5h。后加入NaHSO
实施例6(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-(氟-
反应方程式如下:
实验步骤:a)将[
b)于100℃、氮气吹扫下蒸干上述洗脱液;向蒸发残余物中加入1mL无水乙腈并再次蒸干,重复上述过程三次,制得干燥的[
c)冷却反应液,向上述干燥的络合物中先加入0.06mL吡啶(1M in DMA)和0.04mLCu(OTf)
d)将反应溶液经有机滤膜过滤后,经半制备HPLC纯化(Phenomenex Gemini-NxC18 110A(250×4.6mm×5μm,乙腈/0.1%甲酸水=4/6),出峰时间为10.4min,纯度大于95%。
实施例7(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-(氟-
结构式如下:
反应方程式如下:
实验步骤参考实施例6,经半制备HPLC纯化(Phenomenex Gemini-Nx C18 110A(250×4.6mm×5μm,乙腈/0.1%甲酸水=4/6),出峰时间为12.5min,纯度大于95%。
实施例8(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-6-(碘-
反应方程式如下:
实验步骤:a)依次加入50μL乙醇、40μL Na
b)将混合溶液缓慢加入至1.5mL饱和NaHCO
实施例9(S)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)-7-(碘-
实施例9与实施例8的反应条件类似,经半制备HPLC纯化(Phenomenex Gemini-NxC18 110A(250×4.6mm×5μm,乙腈/0.1%甲酸水=4/6),出峰时间为13.2min,纯度大于95%。
实施例10(S)-7-(asta-211At)-N-(2-(2-氰基-4,4-二氟吡咯烷-1-基)-2-氧乙基)喹啉-4-甲酰胺
实施例10与实施例9的反应条件类似,经半制备HPLC纯化(Phenomenex Gemini-NxC18 110A(250×4.6mm×5μm,乙腈/0.1%甲酸水=4/6),出峰时间为13.4min,纯度大于95%。
123I、124I、125I、131I、或者211At等(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物的制备与实施例9-10类似,出峰时间大概为13min左右,纯度大于95%。在这里不在一一例举。
实施例11亲和性实验
为了进一步探究化合物对FAPI的亲和性,我们进行了体外稳定性实验。使用测定缓冲液(25mM Tris,250mM NaCl,pH=7.4)进行稀释和反应。重组人FAP蛋白购自Bio-Techne Co.LTD.(Cat.3715-SE-010),初始浓度为0.2mg/mL,稀释至0.4μg/mL。底物Gly-Pro-AMC(Macklin)稀释至40μM。候选探针的浓度从40μM稀释至4pM,以8个梯度。将探针(25μL)和底物(25μL)等量加入96孔板中,然后加入FAP蛋白(50μL)并在37℃孵育1h。每个浓度一式三份。测量荧光强度(Ex/Em=380/460nm)。IC50值定义为在测定条件下导致活性降低50%的抑制剂浓度。数据通过GraphPad Prism处理,并使用剂量响应计算IC50。从表中可以看出,本发明所涉及化合物对FAP亲和性均较高。
表1:化合物对FAP的亲和性
实施例12生物分布
为了进一步探究放射性化合物进脑、肿瘤摄取及药代动力学,我们进行了生物分布实验。通过U87MG肿瘤裸鼠体内分布实验评价了放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物。将放射性(4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物用生理盐水稀释到~20μCi/0.1mL。取U87MG肿瘤裸鼠,体重为16±2g,随机5只为一组。从尾静脉注入4-喹啉基)甘氨酰基-2-氰基吡咯烷衍生物,分别于注射后5、30、60、120min断头处死,取血、肿瘤、心、肝、脾、肺、肾、脑、肌肉、骨、皮毛等组织和脏器,擦净后称重,测定放射性计数,计算各脏器和组织的摄取量(%ID/g)及肿瘤/肌肉的比。结果见下表2。从表2可以看出,2min时,脑初始摄取较高,说明这些化合物均可以透过血脑屏障;且60min时,肿瘤摄取比肌肉摄取较高,说明这些化合物具有较高的靶向性。
表2:化合物的生物分布结果
实施例13显像实验
对U87MG肿瘤进行麻醉,将它们固定在鼠板上,麻醉后经尾静脉注射实例6,7(~1.5mCi/0.1mL),并于给药后进行2小时的micro PET/CT动态扫描,将采集重建后的PET/CT图像传至工作站进行图像融合、显示,判断实施例6,7的放射性分布以及与FAPI的结合情况等。从PET实验结果可知,实施例6,7在60min时的瘤肉比为9左右,这与生物分布结果一致。其中实施例6的PET显像如图1。实施例8,9的SPECT与实施例6,7的操作步骤类似,实施例8,9在60min时的瘤肉比为7左右。
实施例14药物疗效评估
将荷瘤U87MG裸鼠一定活度的实施例10或者
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。