无副产物固体杂化电解质的方法
文献发布时间:2023-06-19 19:28:50
引用并入
与本说明书同时提交PCT请求表作为本申请的一部分。本申请要求的在同时提交的PCT申请表中所确认的权益或优先权的每个申请通过引用将其整体并入本文并且用于所有目的。
技术领域
本公开涉及包含离子传导无机材料和原位交联的基体的杂化电解质组合物。本文还描述包括此类组合物的方法和设备。
背景技术
对于一次电池和二次电池,固态电解质相对于液体电解质表现出各种优点。例如,在锂离子二次电池中,无机固态电解质可能比常规的液体有机电解质更不易燃。固态电解质还因阻止枝晶形成可促进锂金属电极的使用。固态电解质还可在一些条件下表现出高能量密度、良好的循环稳定性和电化学稳定性的优点。然而,在固态电解质的大规模商业化中存在各种挑战。一个挑战是维持电解质和电极之间的接触。例如,虽然无机材料例如无机硫化物玻璃和陶瓷在室温下具有高离子传导率(大于10
对于固态电池的大规模生产和商业化,需要在室温下具有高离子传导率并且足够柔顺以被加工成薄的致密膜而不牺牲离子传导率的材料。
发明内容
本公开涉及杂化电解质组合物。在第一方面,组合物包含:约60重量%至约95重量%的离子传导无机材料;和约5重量%至约40重量%的原位交联的基体。
在一些实施方案中,离子传导无机材料包含锂。在其它实施方案中,离子传导无机材料为硫化物基材料。
在一些实施方案中,原位交联的基体包含粘合剂和多种交联剂。非限制性粘合剂包含聚合物主链、共聚物主链、或接枝共聚物主链。其它非限制性粘合剂可包含全氟醚、环氧化物、聚丁二烯、聚(苯乙烯-b-丁二烯)、聚烯烃、聚硅氧烷、聚四氢呋喃、聚苯乙烯、聚乙烯、聚丁烯、聚(苯乙烯-丁二烯-苯乙烯)(SBS)、聚(苯乙烯-乙烯-丁烯-苯乙烯)(SEBS)、聚(苯乙烯-异戊二烯-苯乙烯)(SIS)、丙烯腈丁二烯橡胶、乙烯丙烯二烯单体聚合物、以及其共聚物。
在其它实施方案中,粘合剂包含多个无机笼。非限制性无机笼可包含二氧化硅、倍半硅氧烷、氢基倍半硅氧烷、或部分缩合的倍半硅氧烷。在一些实施方案中,多个无机笼包含(SiO
原位交联的基体可包含多种交联剂。在一些实施方案中,交联剂在基体内形成热可逆键,其中热可逆键没有产生副产物。在特定实施方案中,热可逆键通过狄尔斯-阿尔德(Diels-Alder)环加成反应、Huisgen环加成反应、硫醇-烯(thiol-ene)反应、迈克尔(Michael)加成反应、开环反应或点击化学反应的方式形成。
在其它实施方案中,交联剂具有以下结构:-L
L
X
在一些实施方案中,L
在其它实施方案中,X
和/>
在第二方面,本公开涉及包括杂化电解质组合物(例如本文所述的任一者)的膜。在一些实施方案中,膜的弹性模量为约0.2Gpa至约3GPa。
在第三方面,本公开涉及形成杂化电解质组合物(例如本文所述的任一者)的方法,该方法包括:提供包含与具有第一反应性基团的第一连接体结合的粘合剂组分和离子传导无机材料的混合物;以及使粘合剂组分与连接剂反应以形成原位交联的基体。
在一些实施方案中,方法还包括:将杂化电解质组合物浇注为膜;以及任选地通过加热至约100℃至约190℃的温度使膜修复。
在一些实施方案中,连接剂包含构造为与第一反应性基团一起反应以在基体内形成热可逆键的第二反应性基团,其中热可逆键没有产生副产物。在特定实施方案中,第一反应性基团和第二反应性基团一起反应形成Diels-Alder环加成产物、Huisgen环加成产物、硫醇-烯反应产物、Michael加成产物、或开环反应产物。
在其它实施方案中,第一反应性基团和第二反应性基团选自以下对中之一:二烯和亲双烯体;1,3-偶极和亲偶极体;硫醇和任选取代的烯烃;硫醇和任选取代的炔烃;亲核体和具有张力的(strained)杂环亲电体;亲核体和任选取代的α,β-不饱和羰基化合物;或亲核体和任选取代的具有张力的环状化合物。在又一其它实施方案中,第一反应性基团和第二反应性基团选自任选取代的1,3-丁二烯、任选取代的烯烃、任选取代的炔烃、任选取代的α,β-不饱和醛、任选取代的不饱和α,β-硫醛、任选取代的α,β-不饱和酮、任选取代的叠氮化物、任选取代的硫醇、任选取代的不饱和环烷基、任选取代的不饱和杂环基、任选取代的α,β-不饱和亚胺、任选取代的醛、任选取代的亚胺、任选取代的亚硝基化合物、任选取代的重氮烯、任选取代的硫酮、任选取代的α,β-不饱和酮、任选取代的α,β-不饱和醛、任选取代的阴离子亲核体和任选取代的具有张力的环氧基。
粘合剂组分可提供任何可用的粘合剂并包含任何可用的单体。在一些实施方案中,粘合剂组分包含与具有第一反应性基团的第一连接体结合的单体。在其它实施方案中,粘合剂组分包含以下结构:-[R
在其它实施方案中,单体包括任选取代的苯乙烯单体、任选取代的乙烯单体、任选取代的丙烯单体、任选取代的丁烯单体、任选取代的丁二烯单体、任选取代的全氟烷烃单体、任选取代的全氟醚单体、任选取代的异戊二烯单体、任选取代的乙叉降冰片烯单体、或任选取代的二烯单体。
在一些实施方案中,粘合剂组分包括以下结构:-[R
在其它实施方案中,粘合剂组分包含与具有第一反应性基团的第一连接体结合的无机笼。在特定实施方案中,粘合剂组分具有以下结构:R
在本文的任何实施方案中(例如在粘合剂组分中),至少一个L
在本文的任何实施方案中,R
任何可用的连接剂可用于形成原位交联的基体。在一些实施方案中,连接剂还包含第三反应性基团,其中第一反应性基团和第二反应性基团中至少一个一起反应以在基体内形成热可逆键,并且其中另一第一反应性基团和第三反应性基团一起反应以形成另一热可逆键。在特定实施方案中,第二反应性基团和第三反应性基团相同。
在一些实施方案中,连接剂具有以下结构:R
在本文的任何实施方案中(例如在连接剂中),L
在本文的任何实施方案中,热可逆键通过Diels-Alder环加成反应、Huisgen环加成反应、硫醇-烯反应、Michael加成反应、开环反应、或点击化学反应的方式形成。在特定实施方案中,热可逆键包含Diels-Alder环加成产物、Huisgen环加成产物、硫醇-烯反应产物、Michael加成产物、或开环反应产物。在其它实施方案中,热可逆键包含硫基、任选取代的杂环基、或任选取代的环烷基。在又一其它实施方案中,热可逆键包含选自以下的结构部分:
和/>
在第四方面,本公开包括电池,所述电池包括本文描述的任何组合物或任何膜。
在第五方面,本公开包括电极,所述电极包括本文描述的任何组合物或任何膜。
在第六方面,本公开包括电极,所述电极包含:原位交联的基体;电化学活性材料;和离子传导颗粒。在一些实施方案中,电极包含任选碳添加剂。在特定实施方案中,碳添加剂是导电碳基添加剂(例如活性炭、碳纳米管、石墨烯、石墨、碳纤维、炭黑、或本文所述的任一者)。在其它实施方案中,电极是阳极或阴极。在又一其它实施方案中,将碳添加剂提供给阳极、阴极、或两者。
在一些实施方案中,原位交联的基体包含粘合剂和多种交联剂,其中交联剂在基体内形成热可逆键,并且其中热可逆键没有产生副产物。
在第七方面,本公开包括组合物,所述组合物包括:分隔体,所述分隔体包含离子传导无机材料和原位交联的第一基体;和电极。在一些实施方案中,电极包含原位交联的第二基体,其中第一基体和第二基体包含粘合剂和多种交联剂,其中交联剂在基体之间形成热可逆键,并且其中热可逆键没有产生副产物。
在第八方面,本公开包括方法,所述方法包括:提供电极和分隔体组合物;和使电极的粘合剂组分和分隔体组合物与连接剂反应以在电极和分隔体组合物之间形成原位交联的基体。在一些实施方案中,电极和分隔体组合物各包含与具有第一反应性基团的第一连接体结合的粘合剂组分。在其它实施方案中,连接剂包含构造为与第一反应性基团一起反应以在基体内形成热可逆键的第二反应性基团,其中热可逆键没有产生副产物。附加详细信息如下。
附图说明
图1示出提供交联剂的非限制性示例的示意,包含化合物(I-1)至(I-8)。此类化合物可以为在硫醇-烯聚合中使用的硫醇和烯烃/炔烃。
图2A-2B示出提供(A)包括化合物(II-1)至(II-10)的全碳二烯和(B)包括化合物(II-11)至(II-14)的杂原子二烯的非限制性示例的示意,其可进行Diels-Alder反应。
图3A-3B示出提供(A)包括化合物(III-1)至(III-11)的全碳亲双烯体和(B)包括化合物(III-12)至(III-19)的杂原子亲双烯体的非限制性示例的示意,其可进行Diels-Alder反应。
图4A-4D示出提供(A)具有二烯/亲双烯体作为聚合物主链的端基的聚合物,(B)在聚合物主链的主链上具有二烯/亲双烯体的聚合物,(C)在从聚合物主链延伸出的链接枝上具有二烯/亲双烯体的聚合物,其中在聚合期间所述二烯/亲双烯体可直接并入,和(D)可进行后官能化以包含改性反应性基团的二烯/亲双烯体的聚合物的非限制性示例的示意。
图5示出提供单体和交联剂的非限制性示例的示意,包括化合物(V-1)至(V-7)。
图6为示出聚苯乙烯-b-聚(乙烯-ran-丁烯)-b-聚苯乙烯-g-马来酸酐(SEBS-gMA)的热重分析(TGA)的图。
图7为示出SEBS-gMA和糠基改性的SEBS(SEBS-gFA)的傅里叶变换红外光谱仪(FTIR)光谱的图。
图8为示出在700MHz仪器上在CDCl
图9为示出以0.05英寸/分钟速率测试的SEBS膜(粗黑线)、SEBS-gMA膜(细黑线)、SEBS-gFA膜(虚线)和SEBS-gFA+0.5BMI膜(灰线)的应力-应变曲线的图。
图10为示出以0.05英寸/分钟速率测试的非限制性杂化电解质组合物的应力-应变分析的图,其中用75:25=Li
图11A至11C示出根据本发明一些实施方案的非限制性电芯的示意图。提供了包括(A)设置在集流体102和电解质/分隔体106之间的阳极104;(B)与电解质/分隔体106相邻的集流体102;和(C)设置在集流体102和电解质/阴极双层112之间的阳极104的电芯。
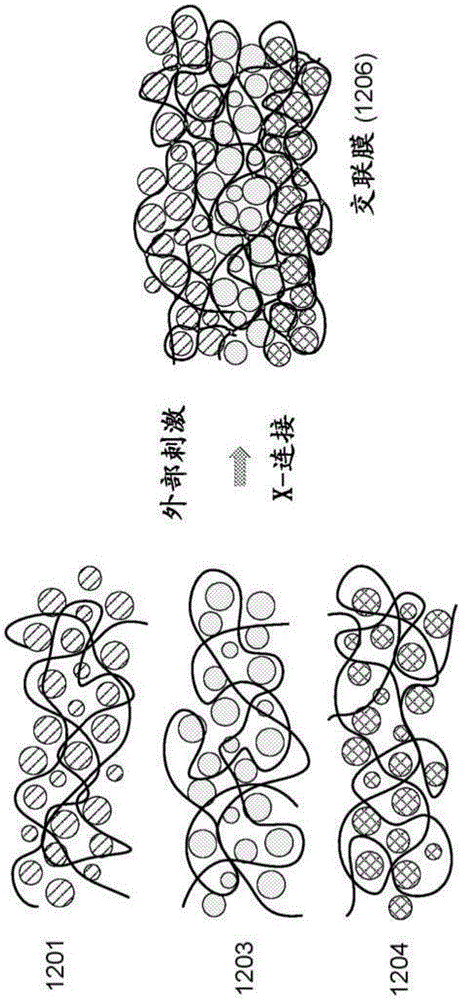
图12示出提供交联膜1206的交联组分的示意。
具体实施方式
本发明的一方面涉及离子传导固态组合物,其包含在有机材料的基体中的离子传导无机颗粒。所得复合材料具有高的离子传导性和促进加工的机械性质。在特定实施方案中,离子传导固态组合物柔顺并可浇注为膜。
本发明的另一方面涉及电池,其包含本文所述的离子传导固态组合物。在本发明的一些实施方案中,提供包含离子传导固态组合物的固态电解质。在本发明的一些实施方案中,提供包含离子传导固态组合物的电极。
本文所述的主题的特定实施方案可具有以下优点。在一些实施方案中,可将离子传导固态组合物用容易按比例放大的制造技术加工为各种形状。制造的复合物是柔顺的,允许与电池或其它装置的其它部件的良好粘附。固态组合物具有高离子传导率,允许组合物用作电解质或电极材料。在一些实施方案中,离子传导固态组合物能够由于抵抗枝晶而能够使用锂金属阳极。在一些实施方案中,离子传导固态组合物不溶解聚硫化物并能够使用硫阴极。
以下描述根据本发明实施方案的离子传导固态组合物、固态电解质、电极、和电池的进一步细节。
离子传导固态组合物在此可被称作杂化组合物。术语“杂化”在此用于描述包含无机相和有机相的复合材料。术语“复合物”在此用于描述无机材料和有机材料的复合物。
在一些实施方案中,复合材料由在与无机颗粒混合之后原位聚合的前体形成。可在施加的压力下发生引起颗粒与颗粒接触的聚合。一旦聚合,施加的压力可通过被聚合物基体固定的颗粒除去。在一些实施方式中,有机材料包括交联聚合物网络。该网络可约束无机颗粒并防止它们在包含该复合材料的电池或其它装置的运行期间移动。
在一些实施方案中,聚合可在没有施加外部压力的情况下引起颗粒与颗粒接触。例如,包括交联的一些聚合反应可导致在聚合期间在没有施加压力的情况下实现颗粒与颗粒的接触和高传导率的。
聚合物前体和聚合物基体与固态离子传导颗粒相容、非挥发且不与电池部件例如电极反应。聚合物前体和聚合物基体的特征还可以为非极性或具有低极性。聚合物前体和聚合物基体可与无机相相互作用使得组分均匀且微观良好地混合,而不影响至少无机相本体的组成。相互作用可包括物理相互作用或化学相互作用中一种或两种。物理相互作用的示例包括氢键、范德华键、静电相互作用、和离子键。化学相互作用是指共价键。通常对无机相不反应的聚合物基体仍可与颗粒的表面形成键,但没有分解或改变无机相的本体组成。在一些实施方案中,聚合物基体可与无机相机械地相互作用。
关于固态组合物的特定组分(例如,高分子量聚合物粘合剂)的术语“数均分子量”或“M
其中M
关于固态组合物的特定组分(例如高分子量聚合物粘合剂)的术语“重均分子量”或“M
其中M
“烷氧基”意指-OR,其中R为任选取代的烷基基团,如本文所述。示例性烷氧基基团包括甲氧基、乙氧基、丁氧基、三卤代烷氧基例如三氟甲氧基等。烷氧基基团可为取代的或未取代的。例如,烷氧基基团可用一个或多个取代基取代,如本文关于烷基所述。示例性未取代的烷氧基基团包括C
如本文单独使用或作为另一基团的一部分所使用的术语“烷基”是指含有任何数量的碳原子数并且在主链中不包括双键或三键的直链或支链的烃。如本文所用,“低级烷基”是烷基的子集并且是指含有1至6个碳原子的直链或支链的烃基。除非另有说明,术语“烷基”和“低级烷基”包括取代和未取代的烷基或低级烷基。低级烷基的示例包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基等。
烷基基团也可为取代的或未取代的。例如,烷基基团可被一个、两个、三个取代基或在两个碳或更多个的烷基基团的情况下被四个取代基取代,所述取代基独立地选自:(1)C
“亚烷基”意指烷基基团的多价(例如二价)形式,如本文所述。示例性亚烷基基团包括亚甲基、亚乙基、亚丙基、亚丁基等。在一些实施方案中,亚烷基基团为C
如本文所用,术语“芳基”是指包括单环和双环芳族基团的基团。示例包括苯基、苄基、蒽基(anthracenyl)、蒽基(anthryl)、苯并环丁烯基、苯并环辛烯基、联苯基、
“亚芳基”是指芳基基团的多价(例如二价)形式,如本文所述。示例性亚芳基基团包括亚苯基、亚萘基、亚联苯基、三亚苯基、二苯基醚、亚苊基、亚蒽基或亚菲基。在一些实施方案中,亚芳基基团为C
“碳环”是指其中所有环成员都是碳原子的环状化合物。碳环可为取代的或未取代的。示例性取代基包括氰基、羟基、卤基、硝基、羧基醛、羧基、烷氧基、氧基或烷基。非限制性碳环包括环己烯、降冰片烯、萘、四氢萘(例如1,2,3,4-四氢萘)、氢蒽醌(例如1,4,4a,5,8,8a,9a,10a-八氢蒽-9,10-二酮)和桥连的多环结构(例如四环[6.6.1.02,7.09,14]五癸-4,11-二烯)。
“羧基醛”是指-C(O)H基团。
“羧基”是指-CO
“氰基”是指-CN基团。
“环烷基”是指具有3至10个碳的单价饱和或不饱和的非芳族环状烃基(例如C
“卤基”是指F、Cl、Br或I。
“杂亚烷基”是指亚烷基基团的二价形式,如本文所定义,含有一个、两个、三个或四个非碳杂原子(例如独立地选自氮、氧、磷、硫、硒或卤素)。杂亚烷基基团可为取代的或未取代的。例如,杂亚烷基基团可被一个或多个取代基取代,如本文关于烷基所述。
“杂环”是指具有一个或多个杂环基结构部分的化合物。杂环可为取代的或未取代的。示例性取代基包括氰基、羟基、卤基、硝基、羧基醛、羧基、烷氧基、氧基或烷基。非限制性杂环包括四氢吡啶(例如,1,2,3,4-四氢吡啶、1,2,3,6-四氢吡啶、或2,3,4,5-四氢吡啶),四氢吡嗪(例如1,2,3,4-四氢吡嗪);四氢嘧啶(例如1,4,5,6-四氢嘧啶),二氢吡喃(例如3,4-二氢-2H-吡喃或3,6-二氢-2H-吡喃),二氢噻喃(例如3,4-二氢-2H-噻喃或3,6-二氢-2H-噻喃),二氢噁嗪(例如5,6-二氢-4H-1,3-噁嗪或3,4-二氢-2H-1,4-噁嗪),二氢噻嗪(例如5,6-二氢-4H-1,3-噻嗪或5,6-二氢-4H-1,4-噻嗪),杂双环庚烯(例如7-氧杂双环[2.2.1]庚-2-烯),桥连异吲哚酐(例如3a,4,7,7a-四氢-4,7-环氧异吲哚-1,3-二酮),桥连苯并呋喃酐(例如3a,4,7,7a-四氢-4,7-环氧异苯并呋喃-1,3-二酮),四氢邻苯二甲酸酐(例如1,2,3,6-四氢邻苯二甲酸酐)、杂降冰片烯(例如7-硫杂降冰片烯或7-氮杂降冰片烯)、环酐(例如3元环、4元环、5元环、6元环或7元环(例如5元环、6元环或7元环),除非另有说明,在环内具有-C(O)-O-C(O)-基团)、或环状酰亚胺(例如,3元环、4元环、5元环、6元环或7元环(例如5元环、6元环或7元环),除非另有说明,在环内具有-C(O)-NR
“杂环基”是指3元环、4元环、5元环、6元环或7元环(例如5元环、6元环或7元环),除非另有说明,含有一个、两个、三个或四个非碳杂原子(例如独立地选自氮、氧、磷、硫、硒或卤素)。3元环具有0至1个双键,4元环和5元环具有0至2个双键,6元环和7元环具有0至3个双键。术语“杂环基”还包括双环、三环和四环基团,其中任何上述杂环环与一个、两个或三个环稠合。
“羟基”是指-OH。
“硝基”是指-NO
“氧基”是指=O基团。
“硫基”是指-S-基团。
有机相
有机相含有一种或多种类型的聚合物,还可被称作聚合物基体或聚合物粘合剂。在一些实施方案中,有机基体可含有单个聚合物链而在聚合物链之间没有明显的或任何交联。在一些实施方案中,有机基体可为或者包括由连接聚合物链的节点为特征的聚合物网络。这些节点可在聚合过程中通过交联而形成。有机基体通过前体在混合物中与无机离子传导颗粒的原位聚合而形成。有机基体的聚合物的特征可以在于主链和一个或多个官能团。
有机基体聚合物具有非挥发性的聚合物主链。聚合物粘合剂是高分子量聚合物或不同高分子量聚合物的混合物。高分子量是指至少30kg/mol的分子量,可为至少50kg/mol或至少100kg/mol。分子量分布可为单峰、双峰和/或多峰。
聚合物或聚合物粘合剂具有可被官能化的主链。在一些实施方案中,聚合物主链是相对非极性的。示例包括共聚物(嵌段、梯度、无规等)例如苯乙烯-丁二烯-苯乙烯(SBS)、苯乙烯-异戊二烯-苯乙烯(SIS)、苯乙烯-乙烯/丙烯-苯乙烯(SEPS)、苯乙烯-乙烯-丁烯-苯乙烯(SEBS)、苯乙烯丁二烯橡胶(SBR)、乙烯丙烯二烯单体(EPDM)橡胶,以及均聚物例如聚丁二烯(PBD)、聚乙烯(PE)、聚丙烯(PP)、和聚苯乙烯(PS)。在一些实施方案中,聚合物是相对极性的,其示例包括丙烯腈-丁二烯-苯乙烯(ABS)、丁腈橡胶(NBR)、乙烯乙酸乙烯酯(EVA)共聚物、氧化聚乙烯。另外的示例包括氟化聚合物例如PVDF、聚四氟乙烯和全氟聚醚(PFPE)和硅氧烷例如聚二甲基硅氧烷(PDMS)。
聚合物可由任何可用的单体或单体的组合形成。在一些实施方案中,单体可为任选取代的苯乙烯单体、任选取代的乙烯单体、任选取代的丙烯单体、任选取代的丁烯单体、任选取代的丁二烯单体、任选取代的全氟烷烃单体、任选取代的全氟醚单体、任选取代的异戊二烯单体、任选取代的乙叉降冰片烯单体、或任选取代的二烯单体。
在粘合剂是共聚物的实施方案中,构成聚合物可以以任何适当方式分布,使得粘合剂可为嵌段共聚物、无规共聚物、统计共聚物、接枝共聚物等。聚合物主链可为线性或非线性的,其中示例包括支链、星型、梳型、和瓶刷聚合物。此外,在共聚物的构成聚合物之间的过渡可为尖锐的、锥形的或无规的。
有机基体的存在量相对高(例如固体复合物的2.5-60重量%),可提供具有期望机械性质的复合材料。根据各种实施方案,复合物是软的并可被加工为各种形状。另外,有机基体还可填充复合物中的空隙,导致致密的材料。
有机基体还可含有能够在以下描述的原位聚合反应中形成聚合的官能团。端基的示例包括氰基、硫醇基、酰胺、氨基、磺酸、环氧基、羧基或羟基。端基还可与无机相的颗粒具有表面相互作用。以下讨论另外的官能团。
聚合物前体和无副产物原位聚合
根据各种实施方案,原位聚合通过混合离子传导颗粒、聚合物前体和任何引发剂、催化剂、交联剂和其它添加剂(如果存在)并然后引发聚合来进行。这可在溶液中或热压的。聚合可在施加的压力下引发和进行以建立紧密的颗粒与颗粒接触。然而,一些原位聚合方法可形成副产物,可导致极化可能提高,并因此电芯的寿命和性能降低。
聚合物前体可为小分子单体、低聚物、聚合物、或粘合剂。聚合反应可由前体形成单个聚合物链(或由聚合物前体形成较长的聚合物链)和/或在聚合物链之间引入交联以形成聚合物网络。聚合物前体可包括官能团,其属性取决于使用的聚合方法。
聚合物前体可为上文所述的上述聚合物主链中任一者(例如聚硅氧烷、聚乙烯、聚烯烃、聚四氢呋喃、PFPE、环烯烃聚合物(COP)或环烯烃共聚物(COC)或其它非极性或低极性聚合物)或其构成单体或低聚物。取决于聚合方法,聚合物前体可为末端和/或主链官能化的聚合物。
离子传导无机颗粒(特别是硫化物玻璃)的反应性对于原位聚合存在挑战。聚合反应应为不分解硫化物玻璃或其它类型颗粒并且不导致有机组分的不受控或过早聚合的聚合反应。特别地,玻璃硫化物对极性溶剂和有机分子敏感,这可引起分解或结晶,其后者可导致离子传导率的明显降低。使用金属催化剂的方法还与硫化物基离子导体不相容。高含量的硫可导致催化剂中毒,从而防止聚合。因此,不可使用例如用于硅橡胶形成的铂介导的硅氢化方法。
无副产物的反应是一类形成主产物而不形成二次副产物的方法。由于它们的经济和性能益处,这些是期望的方法。不需要处理副产物的方法更具成本效益,因为不需要与副产物去除有关的纯化或额外的处理步骤。此外,即使在广泛纯化之后,二次产物也可能残留,充当杂质并导致材料的性能降低或甚至失效。
无副产物的反应是可通过以下反应方案描述的任何方法:
A+B→C
存在大量无副产物的化学反应,包括各种Michael加成或开环方法。环氧树脂、自由基和聚氨酯合成仅仅是许多无副产物的聚合途径中的几种。示例性Michael加成反应包括在亲核体(例如碳负离子或其它亲核体)和α,β-不饱和羰基化合物之间的反应;以及示例性开环反应使用亲核体和具有张力的杂环亲电体(例如环醚、环状碳酸酯、环状环烯烃、环状三硅氧烷、内酯、丙交酯等)。
一些聚合技术没有产生副产物,包括Diels-Alder化学反应和“点击”化学途径。这些类型的反应可导致有机或杂化基体的期望的机械性质,其仍允许使用低压力的加工工具,提供单体和组合物的广泛选择。另外,通过这些途径产生的一些聚合材料呈现出自修复性质以在热处理下自动修复物理损伤,并且因此可提高它们并入其中的电池的安全指数和使用寿命。
在一些实施方案中,聚合物前体被官能团官能化以允许无副产物反应。官能团可在聚合步骤和/或在聚合后官能化步骤期间并入。聚合物还可由一个或多个类型的官能团制备,取决于粘合剂的目标特征。性质包括但不限于:在有机溶剂中的溶解性、与无机颗粒的粘附性、与集流体的粘附性、无机物的分散性、机械性能、离子传导率、电化学和化学稳定性以及电子传导率。
又一其它点击化学反应可通过一对两个反应性基团(例如两个点击化学基团)之间的反应来描述。示例性的对包括炔基和叠氮基之间的Huisgen 1,3-偶极环加成反应,以形成含三唑的连接体;具有4π电子体系的二烯(例如任选取代的1,3-不饱和化合物,例如任选取代的1,3-丁二烯、1-甲氧基-3-三甲基甲硅烷氧基-1,3-丁二烯、环戊二烯、环己二烯或呋喃)和具有2π电子体系的亲双烯体或杂亲双烯体(例如任选取代的烯基或任选取代的炔基)之间的Diels-Alder反应;亲核体和具有张力的杂环亲电体的开环反应;硫醇和任选取代的炔;和硫代磷酸酯基团和碘基团的夹板连接(splint ligation)反应;亲核体和任选取代的α,β-不饱和羰基化合物;亲核体和任选取代的具有张力的环状化合物;以及醛基和氨基的还原胺化反应。
示例性和非限制性反应性基团包括任选取代的1,3-丁二烯、任选取代的烯烃、任选取代的炔烃、任选取代的α,β-不饱和醛、任选取代的不饱和α,β-硫醛、任选取代的α,β-不饱和酮、任选取代的叠氮化物、任选取代的硫醇、任选取代的不饱和环烷基、任选取代的不饱和杂环、任选取代的α,β-不饱和亚胺、任选取代的醛、任选取代的亚胺、任选取代的亚硝基化合物、任选取代的重氮烯、任选取代的硫酮、任选取代的α,β-不饱和酮、任选取代的α,β-不饱和醛、任选取代的阴离子亲核体和任选取代的具有张力的环氧基。任选的取代基可为本文所述(例如关于烷基或芳基)的任何取代基。
无副产物的途径
Diels-Alder反应
在一些实施方案中,聚合物基体通过Diels-Alder反应形成。Diels-Alder反应是用于制备六元环的方法。它还已知为[4+2]环加成反应。该过程发生在共轭二烯和烯烃或炔烃(已知为亲双烯体)之间。Diels-Alder环加成可分为两个子组。一个子组是正常电子需求的Diels-Alder(DA)(方案1A),其中二烯是富电子的,亲双烯体是贫电子的。在第二子组中,逆电子需求Diels-Alder(rDA)(方案1B),角色颠倒,二烯比亲双烯体更贫电子。在一些实施方案中,聚合物前体包括至少一个是二烯的官能团和至少一个是亲双烯体的官能团。
方案1:(A)正常电子需求Diels-Alder和(B)逆电子需求Diels-Alder的Diels-Alder[4+2]环加成反应的示意图
二烯和亲双烯体的化学结构决定反应发生容易程度。例如,在未取代的试剂(G
方案2:在丁二烯和丙烯腈(A)或四氰乙烯(B)之间的DA环加成反应
在一些实施方案中,二烯官能团可包括至少一个EWD取代基,例如:-SO
通过位于二烯反应物的给电子(EDG)取代基可实现正常的电子需求DA反应的类似活化效果。在涉及正常电子需求DA反应的一些实施方案中,可将给电子(EDG)取代基引入二烯(G
方案3:在丙烯醛和丁二烯(A)或1-甲氧基-1,3丁二烯(B)之间的DA环加成反应
在一些实施方案中,在聚合期间发生逆电子需求rDA反应。对于逆电子需求rDA反应,一种过程涉及在富电子亲双烯体(含有EDG官能度)和贫电子二烯(含有EWD基团)之间的环加成。通常,上文描述的EWD和EDG取代基可用于rDA反应(G
方案4:在丙烯醛和甲基乙烯基醚之间的rDA环加成反应
在一些实施方案中,正常电子需求Diels-Alder可通过路易斯酸催化,例如金属氯化物,例如氯化锡、氯化锌或三氟化硼。将催化剂与亲双烯体的结合增加了其亲电性,并因此提高反应性,从而降低热反应要求。
DA反应的一个益处在于它可为热可逆的。逆DA(retro DA)反应是六元环反应形成二烯和亲双烯体的过程,并且通常通过热处理完成。一些逆DA反应还可通过化学活化来促进,例如使用路易斯酸或碱介导。一些DA反应的热可逆性能够实现自修复性质,因为加热聚合物使DA交联离解,然后在随后的冷却时可重新形成DA交联。在一些实施方案中,聚合物前体被可进行逆DA以及正常DA或反向rDA的基团官能化。
1+3偶极环加成“点击”反应
在一些实施方案中,聚合物基体可通过[1+3]偶极环加成反应形成。[1+3]偶极环加成是通过1,3-偶极和亲偶极体的反应制备五元环的方法。一个示例是在叠氮化物和炔烃之间的[3+2]环加成,也已知为Huisgen环加成,其产生1,2,3-三唑(方案5)。
方案5:在叠氮化物和炔烃之间的Huisgen环加成
在一些实施方案中,1,3-偶极是烯丙基或炔丙基/丙二烯基型两性离子,例如甲亚胺叶立德和亚胺、硝酮、硝基化合物、羰基氧化物和酰亚胺、羰基叶立德和亚胺、叠氮化物、重氮烷烃、硫代锍化物等。在一些实施方案中,亲偶极体可为各种烯烃和炔烃以及羰基化合物和亚胺。在一些实施方案中,可使用金属催化剂例如铜基催化剂以提高反应动力学。在一些实施方案中,反应动力学也可在存在张力的亲偶极体例如环辛炔和其类似物和取代衍生物的情况下改善。在一些实施方案中,张力促进的环加成反应可在没有催化剂的情况下自发发生。
硫醇-烯“点击”反应
在一些实施方案中,聚合物基体可通过硫醇和烯烃或炔烃之间的硫醇-烯“点击”反应(方案6)形成硫化物而形成。该过程可经由自由基机制发生,通过自由基引发剂、UV光或温度催化,或经由Michael加成发生,并通过碱和亲核体加速。硫醇-烯“点击”途径可为非常有效的反应,其以高收率进行,使其成为用于各种应用的有吸引力的合成工具。
方案6:在硫醇和烯烃(A)和炔烃(B)之间的硫醇-烯加成
方案7示出可发生在各种实施方案中的各种硫醇-烯反应的示例。硫醇与许多烯烃和炔烃反应。例如,使用温度、UV光或自由基引发剂作为反应促进剂,将聚丁二烯可与不同的二硫醇“原位”交联,以形成交联的网络(方案7A)。该方法类似于橡胶的硫化,但是比使用硫的传统方法更有效并且需要更温和的条件。另外,反应性基团的广泛可得性使在硫醇-烯点击反应的制备中聚合物前体的后改性容易。例如,氢化聚丁二烯中的羟基端基可转化为硫醇反应性丙烯酸酯基团,其可进一步与硫醇交联剂反应以形成交联的网络(方案7B)。此外,硫醇-烯反应可用于控制不饱和聚合物的官能化。各种硫醇试剂的广泛可得性和反应的高效率使“硫醇-烯”方法成为聚合物的受控官能化的优异选择例如聚丁二烯(方案7C)或聚(苯乙烯-b-丁二烯)橡胶。
方案7:使用硫醇-烯反应的示例,包括(A)聚丁二烯交联,(B)具有硫醇反应性基团的氢化聚丁二烯的端基的丙烯酸酯改性,和(C)聚丁二烯的受控化学改性
图1示出可用于基于硫醇-烯的聚合/交联中的可商购得到的硫醇和烯烃/炔烃交联剂的代表性示例。在一些实施方案中,至少一些聚合物前体可包括或被至少一个硫醇基团和/或至少一个烯烃/炔烃官能化。非限制性化合物可包括图1中的化合物(I-1)至(I-8),其中化合物(I-4)中的环氧乙烷基团可为任何可用的数字n(例如1、2、3、4、5、6、7、8、9、10或更多)并且其中化合物(I-8)中的亚甲基基团可为任何可用的数字n(例如1、2、3、4、5、6、7、8、9、10或更多)。
杂化电解质中的Diels-Alde途径
Diels-Alder官能度可位于聚合物前体的粘合剂或小分子添加剂上。官能度(f)为2导致线性聚合物,然而f≥3允许交联聚合物。在一些实施方案中,至少一种聚合物前体携带二烯基团,和至少一种聚合物前体携带亲双烯体基团。通常,聚合物前体可每个分子携带至少一种类型的亲双烯体基团或二烯基团,或两种官能团。
在一些实施方案中,二烯基团可包括顺式构型的任何共轭二烯。二烯可分离成两个主要组别:全碳的(图2A)和基于杂原子的(图2B)。全碳的二烯含有仅由碳原子制成的不饱和共轭链,其包括线性和环状二烯例如丁二烯、环戊二烯、蒽、α-萜品烯、呋喃、硫代呋喃等。又一其它示例包括图2A中的化合物(II-1)至(II-10),其中R可为H、任选取代的烷基或任选取代的芳基,如本文所述。
基于杂原子的二烯可在共轭二烯结构中包括至少一个杂原子例如O、N、S。杂原子二烯的示例包括α,β-不饱和醛和酮,和亚胺例如丙烯醛和硫代丙烯醛。又一其它示例包括图2B中的化合物(II-11)至(II-14),其中R可为H、任选取代的烷基或任选取代的芳基,如本文描述。
类似于二烯,亲双烯体基团可分为全碳的(图3A)和基于杂原子的(图3B)亲双烯体。全碳的亲双烯体包括各种基于烯烃和炔烃的化合物,例如丙烯醛、丙烯腈、富马酸酯、马来酸酯、马来酸酐和酰亚胺。又一其它示例包括图3A中的化合物(III-1)至(III-11),其中R可为H、任选取代的烷基或任选取代的芳基,如本文所述。
在反应性基团中具有杂原子的亲双烯体包括醛、亚胺、亚硝基化合物、重氮烯和硫酮。又一其它示例包括图3B中的化合物(III-12)至(III-19),其中R可为H、任选取代的烷基或任选取代的芳基,如本文所述。
在一些实施方案中,DA反应性聚合物通过使用直接或间接方法用不同浓度的官能团例如二烯或亲双烯体来改性。图4提供各种官能化聚合物的示例。DA不活泼单体与DA反应性单体或大分子单体的共聚可分别导致官能化的共聚物(图4B)和接枝共聚物(图4C)。在使用间接方法的实施方案中,在后官能化处理中聚合物可被DA基团官能化,其可包括特定基团例如端基(图4A)或官能化单体(图4D)的改性。
方案8示出在一些实施方案中可在不同聚合物与糠基基团的后官能化中使用的反应的一些示例。例如,聚丁二烯的羟基端基可通过异氰酸酯的反应来改性以形成氨基甲酸酯键(方案8A),与乙烯共聚的马来酸酐可与胺反应以形成环状酰胺(方案8B),和聚丁二烯中的不饱和键可与硫醇在硫醇-烯反应中反应(方案8C)。
方案8:(A)氢化的聚丁二烯端基、(B)马来酸酐-乙烯共聚物和(C)
聚丁二烯与糠基基团的后官能化
在一些实施方案中,除了官能化聚合物,有机基体可含有小分子单体和交联剂。图5示出小分子二烯和亲双烯体单体和交联剂的一些示例。在一些实施方案中,聚合物基体还可含有如图5示出的聚合物交联剂和单体,例如化合物(V-1)至(V-7),其中化合物(V-6)中的环氧乙烷基团或环氧丙烷基团可为任何可用的数字n(例如1、2、3、4、5、6、7、8、9、10或更多)。
实施例
实施例1:Diels-Alder交联的SEBS膜
热塑性弹性体例如SEBS、SBS或SIS可用作用于产生全固态薄膜电解质的粘合剂。此类粘合剂的低极性和疏水特性允许纯无机导体(例如锂磷硫化物(LPS)玻璃)的初始传导率的高保持,同时其基于嵌段的结构为在该方法中产生的杂化电解质提供好的机械性质。然而,此类粘合剂是基于热塑性的,这意味着它们形成通过非共价相互作用结合的物理交联的网络。
用糠基改性固体粘合剂,以使得能够在小分子双马来酰亚胺存在下进行DA交联。SEBS的DA交联使得能够将共价交联引入由粘合剂形成的物理交联的网络中,从而改善其机械强度并使其在好溶剂中耐溶解。
SEBS在软嵌段中掺杂有2重量%的马来酸酐(SEBS-gMA)并与糠基胺反应。通过使SEBS-gMA与过量的糠基胺反应来合成SEBS-gFA,如方案9.1中所示。
方案9.1:用糠基改性SEBS-gMA以形成SEBS-gFA
在氮气下操作的手套箱中,将30.0g(6.1mmol的马来酸酐)的聚苯乙烯-b-聚(乙烯-ran-丁烯)-b-聚苯乙烯-g-马来酸酐(SEBS-gMA,Sigma-Aldrich)和250g的干燥甲苯置于在145℃下预先干燥的500ml压力容器中。将容器密封,并在60℃加热板上搅拌混合物直至聚合物完全溶解。随后,将容器放回手套箱并冷却至室温,然后向混合物添加2.4g(24.7mmol)的糠基胺。然后将反应物在60℃下进一步搅拌18小时(hr)。此后,将反应混合物沉淀到甲醇中,将固体重新溶解在二氯甲烷中,并然后再次沉淀到甲醇中。将该过程再重复两次以获得糠基改性的SEBS(SEBS-gFA),为白色固体。然后,将SEBS-gFA在100℃下真空中干燥16小时。
使用热重分析测试SEBS-gMA的热稳定性和纯度。将SEBS-gMA在氮气下加热至500℃,实际上显示直至~370℃都没有重量损失,证明聚合物的高热稳定性以及没有明显挥发性杂质或水分含量(见图6)。
图7中示出SEBS-gMA和SEBS-gFA的FTIR光谱。与SEBS主链有关的重叠信号的高浓度使得光谱看起来相似。光谱之间的主要差别是羰基(-C=O)伸缩消失,~1790cm
使用700MHz仪器进行起始材料SEBS-gMA和产物SEBS-gFA的质子核磁共振(
其次,在Diels-Alder交联方法中使用1,1′-(亚甲基二-4,1-亚苯基)双马来酰亚胺(BMI)测试SEBS-gFA。将SEBS-gFA在甲苯中的溶液与BMI以糠基与马来酰亚胺基团2:1比率混合。配备有搅拌棒的20mL小瓶装有1.50g(0.037mmol的糠基基团)的SEBS-gFA、27.0mg(0.075mmol)的BMI和3.0g的1,2,4-三甲基苯。将混合物在40℃下搅拌直至所有组分溶解,然后冷却至室温。随后,使用刮刀将溶液浇注在Mylar上,并将薄膜空气干燥,然后转移至真空烘箱并在100℃下加热12小时。将膜切成三片并且其中之一在120℃下另外加热5小时。在从基材剥离之前将膜冷却至室温(方案9.2)。
方案9.2:在Diel-Alder过程中SEBS-gFA与BMI的交联
进行交联膜的拉伸测试以确定弹性模量、拉伸强度和断裂伸长率。SEBS-gFA+0.5BMI膜的性质是相对于在相同条件下加工的纯的SEBS(非官能化)、SEBS-gMA和SEBS-gFA膜进行测量的。所有膜切割成8mm×50mm的条,并使用微型拉伸测试仪对每个膜进行至少三次测量。由于仪器的短夹具分离,在材料失效之前达到仪器的极限时,无法测量拉伸强度和断裂伸长率。每个聚合物膜是非常有弹性的,达到>800%伸长率。图9示出以0.05英寸/分钟速率测试的SEBS(粗黑线)、SEBS-gMA(细黑线)、SEBS-gFA(虚线)和SEBS-gFA+0.5BMI(灰线)膜的应力-应变测试中获得的代表性曲线。表1总结SEBS、SEBS-gMA、SEBS-gFA和Diels-Alder交联的SEBS-gFA+0.5BMI膜的应力-应变曲线的弹性模量。
表1:不同聚合物膜的弹性模量
SEBS、SEBS-gMA、SEBS-gFA与交联的SEBS-gFA+0.5BMI测量的弹性模量彼此明显不同,提供总体组成和官能团类型的重要性的证据。向SEBS组合物添加2重量%的极性马来酸酐接枝大幅改善粘合剂的模量,示出比SEBS杂化物高出70%的值(20.82MPa)。将SEBS-gMA用糠基进一步改性,导致更具极性的SEBS-gFA粘合剂,以及甚至更高的模量26.82MPa。最后,当SEBS-gFA与BMI交联时,膜示出28.54MPa的模量,证明Diels-Alder反应发生,该方法中形成的另外的共价交联提高聚合物膜的整体韧性。
实施例2:Diels-Alder交联的杂化电解质
在测试纯SEBS、SEBS-gMA、SEBS-gFA和BMI交联的SEBS-gFA膜的机械性质之后,将聚合物并入复合电解质。测试在用80重量%的75:25=Li
测量复合物的传导率以评价粘合剂对纯75:25=Li
当纯SEBS-gFA用作有机基体时,传导率仅低2.3倍。这表明添加BMI至体系中对有机基体有大的影响,并因此对所得杂化物的传导率有大的影响。在含有SEBS-gFA的杂化物与含有SEBS-gFA和BMI交联剂的杂化物之间的差异可与两种复合物中有机基体的粘度的差异有关。有机基体的较高粘度可导致热压期间颗粒的流动减小,并因此防止可导致电解质复合物的良好传导率性能的良好的颗粒与颗粒接触。在浇注含有SEBS-gFA和BMI的杂化物期间注意到浆料的粘度异常高,并需要高得多的稀释剂来浇注杂化膜。粘度的升高归因于浆料中糠基和马来酰胺基基团之间发生的Diels-Alder过程。这导致形成比起始SEBS-gFA具有高得多分子量的聚合物,因此,该聚合物具有更高粘度,在热压加工期间颗粒运动受阻。
表2:对于具有80重量%75:25=Li
其次,进行所有杂化物的机械测试以获得弹性模量、拉伸强度和断裂伸长率。在与纯聚合物膜的相同条件下进行机械测试。将每种杂化物的代表性应力-应变曲线示于图10中;并将提取模量、拉伸强度和断裂伸长率总结于表2中。
具有不同粘合剂的杂化物获得的应力-应变曲线的视觉比较示出在它们全部之间机械性质的明显不同。存在用较高极性粘合剂制备的杂化物的拉伸强度和断裂伸长率增加的趋势。在SEBS杂化物的情况下,样品在仅2.2%伸长率断裂(表2)。当仅仅2重量%的马来接枝物并入SEBS(SEBS-gMA)时,该值翻倍至4.7%。糠基基团的进一步改性(SEBS-gFA)将极性基团的重量%增加至3.5重量%。该改性将断裂伸长率急剧增加至17.0%,其分别高于SEBS和SEBS-gMA 8.5倍和4倍。对于膜的拉伸强度观察到相同趋势,对于SEBS、SEBS-gMA和SEBS-gFA粘合剂分别示出4.2、5.6和8.3MPa值,证明当较极性粘合剂并入有机基体时改善膜的耐断裂性(表2)。
BMI交联的SEBS-gFA杂化物的性质在SEBS-gMA和SEBS-gFA杂化物的那些之间(表2),示出与纯SEBS-gFA相比交联引起杂化物的性能降低。据推测,高负载量的无机颗粒可降低在糠基和马来酰亚胺基团之间的交联效率,影响机械性质。此外,Diels-Alder反应的低效率可导致更多部分地反应的BMI基团。因此,代替形成交联,此类基团将充当大体积的刚性官能团,其在与无机颗粒的表面配位方面可能效率较低。这可能不仅影响有机基体的机械性质,而且改变粘合剂与无机颗粒的粘附,因此影响杂化膜的机械性能。
实施例3:基于POSS纳米复合物的杂化电解质的合成
无机-有机杂化基体可基于多面体低聚物倍半硅氧烷(POSS)化合物,其为具有经验式R
FG-POSS通过使缩水甘油基(G)POSS与糠基胺(F)反应合成。将12.1g的G-POSS(9.0mmol,72.0mmol环氧基基团)在氩气下溶解在60ml二甲基甲酰胺中。将8.7g糠基胺(89.7mmol酰胺基基团)逐滴添加至溶液中。在60℃下反应1天之后,使用离心机(在-4℃下4500rpm)去除未反应的糠基胺和多余的溶剂,获得粘性透明液体。通过将5g FG-POSS溶解在40ml无水四氢呋喃(THF)中,之后添加化学计量量的1,1′-(亚甲基二-4,1-亚苯基)双马来酰亚胺(BMI),以获得杂化POSS基体。在室温下搅拌3小时后,通过离心缓慢去除THF。得到的粘性液体(重量%:15、25、30和35)与无机导体(例如锂离子传导硫银锗矿)在二氯苯中混合。将8个Φ=10mm的氧化锆球置于杯中作为混合介质。将杯封闭并用绝缘胶带紧密密封。将浆料在管式滚筒中以80rpm速度混合16小时。使用刮刀技术在镍箔上浇注薄膜。该浇注在配备有真空吸盘的涂布机上进行。膜在环境压力下在室温和45℃下干燥5小时,然后转移至预燃室并在真空下进一步干燥过夜。将干燥的薄膜切成50mm×70mm矩形试样。将单个膜片夹在FEP片之间并在立式压机中以15Mpa压制18小时,同时在100℃下加热样品。将样品冷却至40℃,然后释放压力并提取样品。
无机相
本文所述的复合材料的无机相传导碱金属离子。在一些实施方案中,它负责复合材料的所有离子传导率,提供通过复合材料的离子传导通路。
无机相是传导碱金属离子的颗粒状固态材料。在下文给出的示例中,主要描述锂离子传导材料,但可采用钠离子传导或其它碱金属离子传导材料。根据各种实施方案,材料可为玻璃颗粒、陶瓷颗粒或玻璃陶瓷颗粒。该方法特别适用于具有玻璃或玻璃陶瓷颗粒的复合物。特别地,如上所述,该方法可用于提供具有玻璃或玻璃陶瓷颗粒和极性聚合物的复合物而不诱导颗粒的结晶(或进一步结晶)。
本文所述的固态组合物不限于特定类型的化合物,可采用任何固态无机离子传导颗粒状材料,其示例在下面给出。
在一些实施方案中,无机材料是单离子导体,其具有接近1的迁移数。电解质中离子的迁移数是电解质中离子携带的总电流的分数。单离子导体具有接近1的迁移数。根据各种实施方案,固体电解质的无机相的迁移数为至少0.9(例如0.99)。
无机相可为氧化物基组合物、硫化物基组合物或磷酸盐基组合物,并可为结晶的、部分结晶的或无定形的。如上所述,方法的一些实施方案特别适用于硫化物基组合物,其可在极性聚合物存在下分解。
在一些实施方案中,可掺杂无机相以增加传导率。固体锂离子传导材料的示例包括钙钛矿(例如,Li
固体锂离子传导材料的示例包括钠超级离子导体(NASICON)化合物(例如Na
离子传导玻璃的另外示例公开于以下文献:Ribes et al.,J.Non-Cryst.Solids,1980,Vol.38-39(Pt.1),pp.271-276和Minami,J.Non-Cryst.Solids,1987,Vol.95-96,pp.107-118(其通过引用并入本文)。
根据各种实施方案,无机相可包括一种或多种类型的无机离子传导颗粒。无机相的颗粒尺寸可根据特定应用而变化,其中,对于大部分应用而言,组合物的颗粒的平均直径在0.1μm和500μm之间。在一些实施方案中,平均直径在0.1μm和100μm之间。在一些实施方案中,多峰尺寸分布可用于优化颗粒堆积。例如,可使用双峰分布。在一些实施方案中,使用尺寸为1μm或更小的颗粒,以使得复合物中平均最近颗粒距离不大于1μm。这可有助于防止枝晶生长。在一些实施方案中,平均颗粒尺寸小于10μm或小于7μm。在一些实施方案中,可使用具有平均尺寸小于7μm第一尺寸分布和大于10μm第二尺寸的多峰尺寸分布。较大的颗粒导致膜具有较稳健的机械性质和较好的传导率,而较小的颗粒产生具有较低孔隙率和较好密度的较致密均匀的膜。
无机相可通过任何适当的方法制造。例如,可使用不同的合成方法例如溶液、溶胶-凝胶和固态反应得到结晶材料。玻璃电解质可通过如以下文献:Tatsumisago et al.,J.Power Sources,2014,Vol.270,pp.603-607(其通过引用并入本文)中描述的淬火-熔融、溶液合成或机械研磨得到。
如本文所用,术语无定形玻璃材料是指至少半无定形的材料,尽管它们可具有小的结晶区。例如,无定形玻璃颗粒可以是完全无定形(100%无定形)的、至少95%(体积)无定形的、至少80%(体积)无定形的或至少75%(体积)无定形的。虽然这些无定形颗粒可具有一个或多个小的结晶区,但是穿过颗粒的离子传导是穿过大部分或完全各向同性的传导路径。
离子传导玻璃-陶瓷颗粒具有无定形区,但至少是半结晶的,例如具有至少75%(体积)结晶度。玻璃-陶瓷颗粒可使用在本文所述的复合物中,在此,玻璃-陶瓷颗粒具有相对大量的无定形特性(例如至少40%(体积)无定形),这在一些实施方案中可用于它们的各向同性传导路径。在一些实施方案中,可使用离子传导陶瓷颗粒。离子传导陶瓷颗粒是指大部分为晶体的材料,尽管它们可具有小的无定形区。例如,陶瓷颗粒可为完全结晶(100体积%结晶)的或至少95%(体积)结晶的。
在一些实施方案中,无机相包括硫银锗矿。硫银锗矿可具有通式:
A
其中A是碱金属,Hal选自氯(Cl)、溴(Br)和碘(I)。在特定实施方案中,x大于0。在其它实施方案中,x为3或更小。在又一其它实施方案中,0 在一些实施方案中,硫银锗矿可具有如上给出的通式,并进一步被掺杂。一个示例是掺杂有亲硫金属的硫银锗矿: A 其中A是碱金属;M是选自锰(Mn)、铁(Fe)、钴(Co)、镍(Ni)、铜(Cu)、锌(Zn)和汞(Hg)的金属;Hal选自氯(Cl)、溴(Br)和碘(I);z是金属的氧化态;0≤x≤2;并且0≤m<(7-x)/z。在一些实施方案中,A是锂(Li)、钠(Na)或钾(K)。在一些实施方案中,A是Li。金属掺杂的硫银锗矿进一步描述于美国专利申请第16/829962号,其公开为美国专利公开第20210047195号,通过引用并入本文。在一些实施方案中,复合物可包括氧化物硫银锗矿,例如,如美国专利申请第16/576570号中所述,其公开为美国专利公开第20200087155号,通过引用并入本文。碱金属硫银锗矿包括上文给出的式的硫银锗矿,以及在美国专利公开第20170352916号中所述的硫银锗矿,其包括Li 矿物硫银锗矿Ag Ag 在一个示例中,该矿物类型的含锂示例,Li 存在可进行保留整体硫银锗矿结构的取代的各种形式。例如,原始矿物具有两个当量的S 在其它示例中,将与上述的Li 本文所述的组合物中使用的硫银锗矿包括硫化物基离子导体,其中大量(至少20%,通常至少50%)的阴离子是含硫的(例如S 复合物 本文提供了包括有机相和非离子传导颗粒的复合物。在一些实施方案中,有机相基本上不具有离子传导率,并被称为“非离子传导的”。本文所述的非离子传导的聚合物具有小于0.0001S/cm的离子传导率。在一些实施方案中,有机相可包含在盐例如LiI的存在下是离子传导的聚合物。离子传导聚合物例如聚环氧乙烷(PEO)、聚环氧丙烷(PPO)、聚丙烯腈(PAN)、聚(甲基丙烯酸甲脂)(PMMA),其在盐的存在下是离子传导的、溶解或解离盐例如LiI。非离子传导聚合物不溶解或解离盐并且即使在存在盐的情况下也不是离子传导的。这是因为在没有溶解盐的情况下,不存在移动离子来传导。 在一些实施方案中,固相复合物中的聚合物负载量可相对高,例如以重量计至少为2.5%-30%。根据各种实施方案,它可在0.5重量%-60重量%的聚合物、1重量%-40重量%聚合物、或5重量%-30重量%之间。固相复合物形成连续膜。 如上所述,复合物包含官能化的聚合物主链粘合剂。粘合剂可为官能化聚合物粘合剂和非官能化聚合物粘合剂的混合物。例如,在一些实施方案中,粘合剂可为非极性聚合物(例如SEBS)和该聚合物的官能化形式的混合物,聚合物的官能化形式可如本文所述交联(例如SEBS-gFA、SEBS-gFA-0.5BMI)。根据各种实施方案,混合物可为1:9-9:1重量%聚合物:官能化聚合物,例如1:5-5:1或1:4-4:1。 根据各种实施方案,聚合物粘合剂可基本上是复合物的所有有机相,或者是复合物的至少95重量%、90重量%、至少80重量%、至少70重量%、至少60重量%、或至少50重量%的有机相。 在一些实施方案中,复合材料基本上由离子传导无机颗粒和有机相组成。然而,在替代的实施方案中,可将一种或多种额外的组分添加至固体复合物。 根据各种实施方案,固体组合物可或可不包含添加的盐。可添加锂盐(例如LiPF 在一些实施方案中,固态组合物可包含一种或多种传导增强剂。在一些实施方案中,电解质可包含一种或多种填充材料,包含陶瓷填料例如Al 在一些实施方案中,如下文进一步讨论的,复合物被并入或准备并入电极,并包含电化学活性材料,并且任选地包含电子传导添加剂。下面提供了电极的成分和组成的示例。 在一些实施方案中,电解质可包含可用于在电极的表面上形成钝化层的电极稳定剂。电极稳定剂的示例描述于美国专利第9093722号。在一些实施方案中,电解质可包含如上所述的传导增强剂、填料、或有机组分。 复合物可提供为自支撑膜、设置在离型膜上的自支撑膜、已层叠在电池或其它装置的组件例如电极或分隔体上的膜,或已浇注在电极、分隔体或其它组件上的膜。 复合材料膜可具有任何合适的厚度,取决于特定的电池或其它装置设计。对于许多应用,厚度可在1微米和250微米之间,例如30微米。在一些实施方案中,电解质可显著更厚,例如在毫米量级上。 在一些实施方案中,将复合物提供为浆料或糊剂。在这样的情况下,组合物包含稍后待蒸发的溶剂。此外,组合物可包含用于储存稳定性的一种或多种组分。这样的化合物可包括丙烯酸树脂。一旦准备进行加工,可适当地将浆料或糊剂浇注或铺在基材上并干燥。 在一些实施方案中,复合物作为可挤出的固体混合物提供。 装置 本文所述的复合材料可并入使用离子导体的任何装置,包括但不限于电池和燃料电池。例如在电池中,复合物可用作电解质分隔体。 电极组合物还包含电极活性材料和任选的传导添加剂。下文给出示例阴极组合物和阳极组合物。 对于阴极组合物,下表3给出组合物的示例。 根据各种实施方案,阴极活性材料是过渡金属氧化物,以锂镍锰钴氧化物(LiNiMnCoO 可使用任何适当的无机导体,如上文在无机导体的描述中所述。Li 电子传导率添加剂可用于具有低电子传导率的活性材料如NMC。炭黑是一种这样的添加剂的示例,但是可使用其它碳基添加剂,其包括其它炭黑、活性炭、碳纤维、石墨、石墨烯和碳纳米管(CNT)。小于1重量%可能不足以改善电子传导率,而大于5重量%导致能量密度的降低并干扰活性材料-硫银锗矿接触。 如上所述,可使用任何适当的有机相。小于1重量%可能不足以实现所期望的机械性质,而大于5重量%可导致能量密度的降低并干扰活性材料-无机导体-碳接触。在一些实施方案中,聚偏二氟乙烯(PVDF)与或不与非极性聚合物(例如聚苯乙烯或PS)一起使用。 对于阳极组合物,下表4给出组成的示例。 石墨可用作次级活性材料以改善Si阳极的起始库伦效率(ICE)。Si受制于低的ICE(例如在一些情况下小于80%),其低于NMC和其它阴极的ICE,在首次循环引起不可逆容量损失。石墨具有高的ICE(例如大于90%),能够实现全容量利用。使用Si和石墨两者作为活性材料的杂化阳极随着石墨含量提高提供更高的ICE,意味着阳极的ICE可通过调节Si/石墨比例来匹配阴极的ICE,从而防止在首次循环时的不可逆容量损失。ICE可随着其加工而变化,允许取决于特定阳极及其加工的相对宽范围的石墨含量。另外,石墨改善电子传导率并可有助于阳极的致密化。 可使用如上关于阴极所述的任何适当的无机导体。 一些实施方案可使用高表面积电子传导率添加剂(例如炭黑)。Si具有低的电子传导率,并且除了石墨(其为极好的电子导体但具有低表面积)之外,这样的添加剂是有帮助的。然而,硅-碳复合材料和含硅合金的电子传导率可以相当高,使得在一些实施方案中不需要使用添加剂。也可以使用其它高表面积的碳(炭黑、活性炭、石墨烯、碳纳米管)代替Super C。 可使用任何适当的有机相。在一些实施方案中,使用PVDF。 本文提供了碱金属电池和碱金属离子电池,其包括阳极、阴极、和如上所述的与阳极和阴极操作相关的柔顺固体电解质组合物。电池可包括用于物理地分离阳极和阴极的分隔体;这可为固体电解质组合物。 合适的阳极的示例包括但不限于由锂金属、锂合金、钠金属、钠合金、碳质材料例如石墨和它们的组合形成的阳极。合适的阴极的示例包括但不限于由过渡金属氧化物、掺杂的过渡金属氧化物、金属磷酸盐、金属硫化物、磷酸铁锂、硫和它们的组合形成的阴极。在一些实施方案中,阴极可为硫阴极。 在碱金属空气电池例如锂空气电池、钠空气电池、或钾空气电池中,阴极可透过氧(例如介孔碳、多孔铝等),并且阴极可任选含有并入其中的金属催化剂(例如锰、钴、钌、铂、或银催化剂、或它们的组合)以增强在阴极处锂离子和氧发生的还原反应。 在一些实施方案中,提供锂硫电芯,其包括锂金属阳极和含硫阴极。在一些实施方案中,本文所述的固态复合电解质通过防止枝晶形成而独特地实现锂金属阳极和通过不溶解在放电期间阴极处形成的多硫化物中间体而独特地实现硫阴极。 还可包括由可透过离子流的任何合适材料形成的分隔体以避免阳极和阴极彼此直接电接触。然而,由于本文所述的电解质组合物是固体组合物,所以它们可用作分隔体,特别是当它们为膜的形式时。 在一些实施方案中,固体电解质组合物用作碱金属离子电池中阳极和阴极之间的电解质,其依赖于循环期间碱金属离子的嵌入。 如上所述,在一些实施方案中,固体复合组合物可并入电池的阳极和阴极中一者或两者。电解质可为如上所述的柔顺固体电解质或任何其它适当的电解质,包括液体电解质。 在一些实施方案中,电池包括电极/电解质双层,其中每个层包含本文所述的离子传导的固态复合材料。 图11A示出根据本发明一些实施方案的电芯的示意图的示例。电芯包括负集流体102、阳极104、电解质/分隔体106、阴极108和正集流体110。负集流体102和正集流体110可为任何适当的电子传导材料,例如铜、铁、金、铂、铝和镍。在一些实施方案中,负集流体102是铜,正极流体110是铝。集流体可采用任何适当形式,例如片、箔、网状物、或泡沫。根据各种实施方案,阳极104、阴极108和电解质/分隔体106中一者或多者是包括如上所述的有机相和无机相的固态复合物。在一些实施方案中,阳极104、阴极108和电解质106中两者或更多者是包括如上所述的有机相和无机相的固态复合物。 在一些实施方案中,集流体是可嵌入相应电极中的多孔体。例如,它可为网状物。包括疏水性聚合物的电极可能不能很好地粘附到箔形式的集流体;然而网状物提供良好的机械接触。在一些实施方案中,可将如本文所述的两个复合膜压在网状集流体上以形成在电极中嵌入的集流体。在一些实施方案中,使用提供良好粘附的亲水性聚合物。 图11B示出根据本发明一些实施方案的组装的锂金属电芯示意图的示例。组装的电芯包括负集流体102、电解质/分隔体106、阴极108和正集流体110。锂金属产生于首次充电时并覆盖在负集流体102上以形成阳极。电解质106和阴极108中一者或两者可为如上所述的复合材料。在一些实施方案中,阴极108和电解质306一起形成电极/电解质双层。图11C示出根据本发明一些实施方案的电芯的示意图的示例。电芯包括负集流体102、阳极104、阴极/电解质双层112和正集流体110。双层中的每个层可包含硫化物导体。这样的双层可以例如通过制备电解质浆料并将其沉积在电极层上来制备。 根据已知的技术,电池的所有部件可包括在或封装在合适的刚性或柔性容器中,该容器具有用于与阳极和阴极建立电连接的外部引线或触点。 在一些实施方案中,复合分隔体包含经历原位无副产物聚合的有机相,如本文所述。在一些实施方案中,电池的一个或两个电极可具有可经历原位无副产物聚合的有机相。在一些实施方案中,复合分隔体和两个电极中每个独立形成并组装。 在一些实施方式中,复合分隔体和一个或两个电极通过如本文所述的无副产物反应交联。在这样的实施方案中,复合分隔体和一个或两个电极包含有机相,该有机相具有用无副产物反应性基团例如Diels-Alder反应性基团官能化的小分子和聚合物。在一些实施方案中,用Diels-Alder反应性基团官能化的分子可为分隔体和/或一个或两个电极的一部分。在这样的实施方案中,在聚合步骤期间,反应性基团可在复合分隔体和一个或两个电极之间交联。因此,复合分隔体和一个或两个电极具有基本上没有副产物的交联聚合物基体。该技术可导致具有原位分隔体的全电芯,所述分隔体具有较高机械性质,而没有形成副产物。 加工 固态组合物可通过任何适当的方法制备。根据各种实施方案,原位聚合通过混合离子传导颗粒、聚合物前体和任何粘合剂、引发剂、催化剂、交联剂和其它添加剂(如果存在)并然后引发聚合来进行。这可在溶液中或热压的,如后所述。可在施加的压力下引发和进行聚合从而建立紧密的颗粒与颗粒接触。 均匀的膜可通过溶液加工方法制备。在一个示例方法中,将所有组分通过使用实验室和/或工业装备例如超声波仪、均化器、高速混合器、旋转磨机、立式磨机和行星式球磨机来混合在一起。可添加混合介质以通过改善混合、破碎附聚物和聚集体来帮助均匀化,从而消除膜缺陷,例如针孔和高表面粗糙度。所得混合物是均匀混合的浆料形式,其粘度基于杂化组合物和溶剂含量而变化。用于浇注的基材可具有不同的厚度和组成。示例包括铝、铜和聚酯薄膜。无机颗粒可在添加交联剂之前或同时添加,但通常不在交联之后添加。 浆料在所选基材上的浇注可通过不同的工业方法实现。在一些实施方案中,可通过例如在辊之间压延、垂直平压或等静压的方法使膜的机械致密化(导致例如高达约50%厚度变化),从而降低孔隙率。致密化过程中涉及的压力迫使颗粒维持紧密的颗粒间接触。施加外部压力例如在大约1MPa至600MPa或1MPa至100MPa。在一些实施方案中,使用通过压延辊施加的压力。压力足以产生颗粒与颗粒的接触,但是保持足够低以避免未固化聚合物从压机中挤出。可包括交联的聚合可在压力下发生以形成基体。在一些实施方式中,使用热引发或光引发聚合技术,其中使用热能或紫外光的施加来引发聚合。离子传导无机颗粒被捕获在基体中并在释放外部压力时保持紧密接触。通过以上方法制备的复合物可为例如粒料或薄膜,且通过成熟的方法并入实际的固态锂电池。 在一些实施方案中,固态复合分隔体通过原位可热固化聚合物产生,在全电芯的制造过程期间不形成副产物。例如,用Diels-Alder反应性基团官能化的小分子和聚合物将在全电芯的压延步骤期间在给定的温度和压力(例如在60℃和140℃之间的温度,和在0.2吨/cm至3吨/cm之间的压力)下反应。聚合物可为分隔体和/或电极的一部分;并且用Diels-Alder反应性基团官能化的分子可为分隔体和/或电极的一部分。在全电芯的压延期间(在受控温度和压力下)聚合将导致具有较高机械性质的原位分隔体的全电芯而没有形成副产物。 在一些实施方案中,膜被干法加工而不是在溶液中加工。例如,膜可被挤出。挤出或其它干法加工可为溶液加工的替代,尤其是在有机相的较高负载量下(例如在有机相为至少30重量%的实施方案中)。 图12提供多个浇注膜的示意描述的示例,所述浇注膜包括离子传导无机颗粒在聚合物基体中经历原位聚合以交联聚合物链,例如在全电芯的压延期间。在图12的示例中,三个膜,第一电极1201、分隔体1203和第二电极1204各自包含聚合物基体中的各种颗粒。每个聚合物基体可被不形成副产物的反应性基团例如Diels-Alder反应性基团官能化。本文其它地方讨论第一电极、分隔体和第二电极的颗粒和其它组分。在一些实施方案中,膜可经受使膜致密化并迫使离子传导颗粒紧密接触的施加的压力。施加外部刺激从而引发聚合,在图12的示例中,聚合使每个膜1206的聚合物基体的聚合物链交联。具体地,聚合之后第一电极1201、分隔体1203和/或第二电极1204的聚合物基体可与单独膜的聚合物基体交联。在向膜施加压力的实施方案中,释放压力,交联的膜保持致密,离子传导颗粒紧密接触。在一些实施方案中,仅存在一个电极膜和分隔体,其中可使用相同的方法,导致在电极和分隔体之间交联的聚合物基体。 结论 尽管为了清楚理解的目的已经相当详细地描述了前述实施方案,但显而易见的是,可在所附权利要求书的范围内实施一些改变和修改。可在没有这些具体细节中的一些或全部的情况下实践本文公开的实施方案。在其它情况下,没有详细描述公知的过程操作,以免不必要地混淆所公开的实施方案。此外,虽然将结合具体实施方案描述所公开的实施方案,但应理解,具体实施方案不意在限制所公开的实施方案。应当注意,存在许多实现本实施方案的方法、系统和装置的替代方式。因此,本实施方案认为是说明性的而非限制性的,并且实施方案不限于本文给出的细节。