一种用于多组学联合分析展示的方法和装置及其应用
文献发布时间:2024-04-18 19:58:53
技术领域
本发明涉及生物信息技术领域,具体地,涉及一种用于多组学联合分析展示的方法和装置及其应用。
背景技术
近年来,随着二代测序的迅猛发展,各种组学层出不穷,而不同的组学代表基因组不同侧面的信息,如何整合不同侧面的数据来真正阐释生物学问题将是生物学家不得不面对的问题。在此背景下,多组学数据分析可视化软件将是研究者必备的工具,为研究者从海量数据挖掘生物学核心问题提供了有效的帮助。
三维基因组能够反映基因组在空间的信息,比如在DNA层面上,远端的调控元件(如增强子)招募转录因子形成loop结构在空间上拉近与目标基因的距离,从而对目标基因的表达进行调控。而由多个loop缠绕在一起形成的拓扑结构域TAD(topologicallyassociating domains)被认为是哺乳动物染色体结构的基本单元。已有研究表明拓扑结构域边界的破坏与多种疾病的发生发展有关。
对于二维表观方向的组学(如ChIP和ATAC),我们可以用IGV和UCSC这些基因浏览器来查看相应的数据,但对于三维基因组(如Hi-C)这种特殊结构的数据,增加了一个额外的数据维度,传统的基因浏览器无法兼容,为整合以三维基因组为首的多组学分析带来了困难。随着Hi-C数据的飞速增长,逐渐衍生了3D Genome Browser,Juicebox,WashUEpiGenome Browser等一系列针对三维浏览器。这些三维基因组浏览器极大的辅助了非生信人员对于测序数据的分析。3D Genome Browser由岳峰课题组和王艇课题组开发,该款可视化浏览器支持众多实验类型的数据展示,包括Hi-C,DNase Hi-C,ChIA-PET,PLAC-seq,HiChIP和Capture Hi-C等,数据库包含人类和小鼠的公共数据。主要通过加载BLUST格式的数据展示相应区域的交互信息。
Juicebox由Erez LiebermanAiden课题组开发,该款工具如同IGV(IntergretiveGenome Viewer,整合基因组学查看器)一样有本地化软件,界面友好,具有放大和缩小的功能,实现从全基因组的互作热图到局部的loop结构的展示,也可用于Hi-C辅助组装。通过加载高度压缩的二进制.hic文件,可以看到核心区域互作热图的信息而且支持多种校正方法和矩阵形式,可实时地改变矩阵。此外分辨率的大小和colorbar也可实时地调整。WashU(The New WashU Epigenome Browser,WashU浏览器)EpiGenome Browser由王艇课题组开发,相当于“IGV升级版”,数据展现形式与IGV一致,同样支持众多实验类型的数据展示,可添加感兴趣的tracks联合展示。也是通过加载高度压缩的二进制.hic文件通过检索基因、SNP或基因组位置,可以看到相应区域的互作热图。除了方便非生信人员对数据进行查看的基因组浏览器之外,还有一些生信人员串写流程针对多个区域进行批量可视化的软件如HiCExploer以及pyGenomeTrack(pyGenomeTrack基因组Track个性化定制工具)。它们都以压缩的二进制文件.h5格式作为输入文件。
但是Juicebox只能在Mac端本地使用,局限性比较大。3D Genome Browser、WashUEpiGenome Browser都需要使用浏览器进行分型展示,而如果用户上传数据量较大的数据,会产生处理速度慢和上传失败等问题。
发明内容
针对上述现有技术存在的仅能在Mac端本地使用,或者仅能在浏览器上进行分析展示,当数据量大时存在处理速度慢或分析失败的问题,本发明提供一种新的用于多组学联合分析展示的方法和装置,本发明的方法和装置为基于linux服务器运行的工具,运用资源更灵活,支持多样本,大数据量处理和可视化,界面更友好,应用更广泛。
具体来说,本发明涉及如下用于多组学联合分析展示的方法和装置及其应用。
1、一种用于多组学联合分析展示的方法,该方法包括如下步骤:
步骤1、获取原始下机数据;
步骤2、将所述原始下机数据的Hi-C部分进行第一数据整理;将所述原始下机数据的Hi-C以外部分进行第二数据处理;其中,
所述第一数据处理的步骤包括:
步骤3.1、在服务器上,采用Hic-Pro进行基因比对,并对比到到酶切片段,生成vaildpairs数据;
步骤3.2、在服务器上,采用juicerpre将所述vaildpairs数据生成.hic文件;
步骤3.3、在服务器上,用juicer dump读取.hic文件;
步骤3.4、在服务器上,用HiCCUPs进行call loop处理。
2、根据项1所述的方法,Hi-C以外部分选自ATAC、CHIP和RNA中的一种或多种。
3、根据项1所述的方法,所述第二数据处理的步骤包括:
步骤4.1、在服务器上,采用Hic-Pro进行基因比对,并获得bam文件;
步骤4.2、在服务器上,采用deeptools解释bam文件,读取信号值并绘图。
4、根据项1-3中任一项所述的方法,所述服务器为Linux。
5、根据项1-3中任一项所述的方法,在步骤1之后且步骤2之前,所述方法还包括:进行数据拆分;
优选地,所述数据拆分的方法为bcl2fastq。
6、一种用于多组学联合分析展示的装置,包括:数据获取单元、第一数据处理单元和第二数据处理单元,其中,
所述数据获取单元,用于获取原始下机数据;
所述第一数据处理单元,用于将所述原始下机数据的Hi-C部分进行第一数据整理;
所述第二数据处理单元,用于将所述原始下机数据的Hi-C以外部分进行第二数据处理;其中,
所述第一数据处理单元包括第一基因比对单元、生成.hic文件单元、读取.hic文件单元和call loop处理单元,
所述第一基因比对单元,用于在服务器上,采用Hic-Pro进行基因比对,并对比到到酶切片段,生成vaildpairs数据;
所述生成.hic文件单元,用于在服务器上,采用juicerpre将所述vaildpairs数据生成.hic文件;
所述读取.hic文件单元,用于在服务器上,用juicer dump读取.hic文件;
所述call loop处理单元,用于在服务器上,用HiCCUPs进行call loop处理。
7、根据项6所述的装置,Hi-C以外部分选自ATAC、CHIP和RNA中的一种或多种。
8、根据项6所述的装置,所述第二数据处理单元包括第二基因比对单元和解释单元,其中,
所述第二基因比对单元,用于在服务器上,采用Hic-Pro进行基因比对,并获得bam文件;
所述解释单元,用于在服务器上,采用deeptools解释bam文件,读取信号值并绘图。
9、根据项6-8中任一项所述的装置,所述服务器为Linux。
10、项1-5中任一项所述的方法或项6-9中任一项所述的装置在生物信息中的应用。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
本发明第一方面提供了一种用于多组学联合分析展示的方法,该方法包括如下步骤:
步骤1、获取原始下机数据;
步骤2、将所述原始下机数据的Hi-C部分进行第一数据整理;将所述原始下机数据的Hi-C以外部分进行第二数据处理;其中,
所述第一数据处理的步骤包括:
步骤3.1、在服务器上,采用Hic-Pro进行基因比对,并对比到到酶切片段,生成vaildpairs数据;
步骤3.2、在服务器上,采用juicerpre将所述vaildpairs数据生成.hic文件;
步骤3.3、在服务器上,用juicer dump读取.hic文件;
步骤3.4、在服务器上,用HiCCUPs进行call loop处理。
根据本发明所述的方法,优选地,Hi-C以外部分选自ATAC、CHIP和RNA中的一种或多种。
根据本发明所述的方法,优选地,所述第二数据处理的步骤包括:
步骤4.1、在服务器上,采用Hic-Pro进行基因比对,并获得bam文件;
步骤4.2、在服务器上,采用deeptools解释bam文件,读取信号值并绘图。
根据本发明所述的方法,优选地,所述服务器为Linux。
根据本发明所述的方法,优选地,在步骤1之后且步骤2之前,所述方法还包括:进行数据拆分;
根据本发明所述的方法,优选地,所述数据拆分的方法为bcl2fastq。例如,针对原始的下机数据,采用bcl2fastq进行数据拆分。
根据本发明所述的方法,优选地,在数据拆分之后进行数据过滤,过滤掉低质量reads。例如采用fqtools_plus进行过滤。
本发明第二方面提供了一种用于多组学联合分析展示的装置,包括:数据获取单元、第一数据处理单元和第二数据处理单元,其中,
所述数据获取单元,用于获取原始下机数据;
所述第一数据处理单元,用于将所述原始下机数据的Hi-C部分进行第一数据整理;
所述第二数据处理单元,用于将所述原始下机数据的Hi-C以外部分进行第二数据处理;其中,
所述第一数据处理单元包括第一基因比对单元、生成.hic文件单元、读取.hic文件单元和call loop处理单元,
所述第一基因比对单元,用于在服务器上,采用Hic-Pro进行基因比对,并对比到到酶切片段,生成vaildpairs数据;
所述生成.hic文件单元,用于在服务器上,采用juicerpre将所述vaildpairs数据生成.hic文件;
所述读取.hic文件单元,用于在服务器上,用juicer dump读取.hic文件;
所述call loop处理单元,用于在服务器上,用HiCCUPs进行call loop处理。
根据本发明所述的装置,优选地,Hi-C以外部分选自ATAC、CHIP和RNA中的一种或多种。
根据本发明所述的装置,优选地,所述第二数据处理单元包括第二基因比对单元和解释单元,其中,
所述第二基因比对单元,用于在服务器上,采用Hic-Pro进行基因比对,并获得bam文件;
所述解释单元,用于在服务器上,采用deeptools解释bam文件,读取信号值并绘图。
根据本发明所述的装置,优选地,所述服务器为Linux。
根据本发明所述的装置,优选地,所述装置还包括数据拆分单元,用于将所述原始下机数据进行拆分,然后再进行第一数据处理和第二数据处理。
根据本发明所述的装置,优选地,所述数据拆分的方法为bcl2fastq。例如,针对原始的下机数据,采用bcl2fastq进行数据拆分。
根据本发明所述的装置,优选地,所述装置还包括数据过滤单元,用于在数据拆分之后进行数据过滤,过滤掉低质量reads。例如采用fqtools_plus进行过滤。
本发明第三方面提供了上述的方法或上述的装置在生物信息中的应用。
本发明具有以下优点:
(1)对于传统的在浏览器上分析并显示的方法,本发明的方法和装置基于基于Linux服务器,运用资源更灵活,支持多样本,大数据量处理和可视化,界面更友好,应用更广泛。
(2)相对于传统的matrix和二进制的h5格式,.hic可以同时容纳多个分辨率的交互矩阵,而且存储小,读取快的优点,本发明基于.hic文件构建了一款针对于生信人员用于多组学联合分析展示的软件。相对于HiCExploer以及pyGenomeTrack,本发明的方法和装置采用了格式更为友好的.hic文件格式。并增加了局部差减hic以及针对特定区域virtual4C的可视化,并对已有的展示方式进行了优化。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
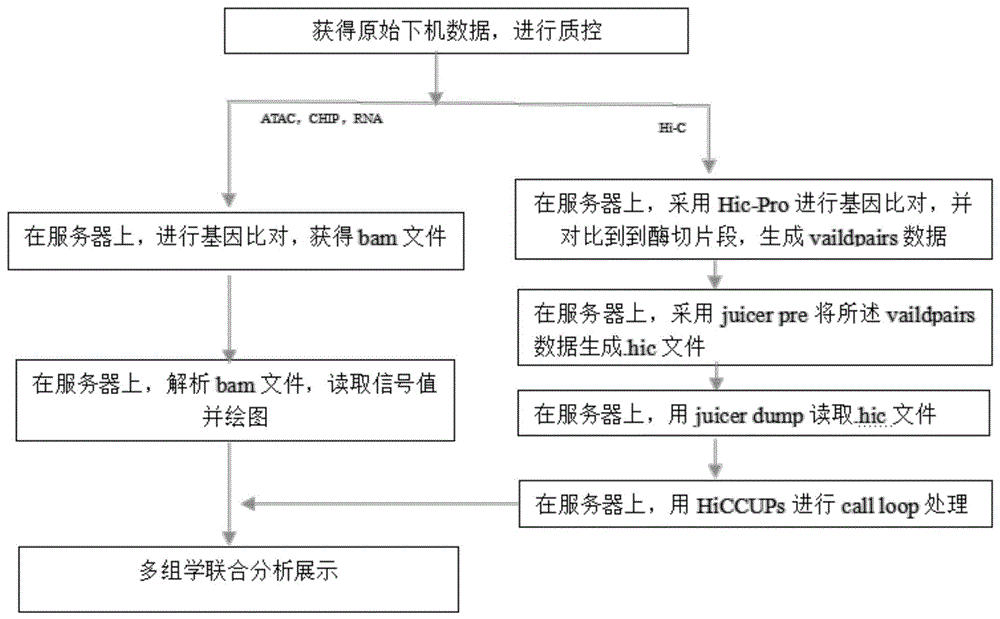
图1为本发明实施例1提供的用于多组学联合分析展示的方法的流程图。
图2为本发明实施例1提供的多组学联合分析展示的结果图。
图3为基于图2增加了解释说明的多组学联合分析展示的结果图。
下面参考具体实施例,对本发明进行说明,需要说明的是,这些实施例仅仅是说明性的,而不能理解为对本发明的限制。
【实施例1】
一种用于多组学联合分析展示的方法,流程如图1所示。
步骤1、获取小鼠细胞系C2C12的CHIP,RNA,Hi-C原始下机数据,结果如表1所示。
表1
采用bcl2fastq进行数据拆分,采用fqtools_plus进行过滤,过滤掉低质量reads。结果如表2所示:
表2
步骤2、将原始下机数据的Hi-C部分进行第一数据整理;将所述原始下机数据的CHIP,RNA部分进行第二数据处理;其中,
第一数据处理的步骤为:
步骤3.1、在Linux服务器上,采用Hic-Pro进行基因比对,并对比到到酶切片段,生成vaildpairs数据,结果如表3所示:
表3
步骤3.2、在Linux服务器上,采用juicer pre将所述vaildpairs数据生成.hic文件;
步骤3.3、在Linux服务器上,用juicer dump读取.hic文件;
步骤3.4、在Linux服务器上,用HiCCUPs进行call loop处理;
第二数据处理的步骤为:
步骤4.1、在Linux服务器上,采用Hic-Pro进行基因比对,并获得bam文件;
步骤4.2、在Linux服务器上,采用deeptools解释bam文件,读取信号值并绘图。多组学联合分析展示结果如图2所示。
图2为显示染色体互作热图的视图,可以帮助研究人员了解基因组中不同区域之间的相互作用,用于查看和分析各种测序数据,包括基因表达、染色体互作和基因组结构等。可以显示两个基因组区域之间的交互频率和联系强度。为了便于解释说明,在图2的基础上,发明人在图2的右侧增加了“上部”、“中部”和“下部”,得到图3。图3中上部对应的每个矩形代表一个小的基因组区域,矩形的颜色和亮度表示该区域与其他区域之间的交互频率和联系强度。如果两个区域之间存在较强的交互作用,那么它们之间的矩形会显示为深色;反之,如果两个区域之间的交互作用较弱,那么它们之间的矩形会显示为浅色。图3中上部对应的是基因组该位置上的基因,中部对应的是该位置的不同组蛋白或其他信号,下部对应的表示loop结构。通过图2和图3能够看出,本发明的方法可以同时容纳多个分辨率的交互矩阵,大数据量处理和可视化,不依赖于浏览器。而且本发明方法存储小,读取快。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。