构建对核酸序列进行分类的模型方法及其应用
文献发布时间:2023-06-19 10:27:30
技术领域
本发明涉及生物技术领域,具体涉及构建对核酸序列进行分类的模型方法及其应用。
背景技术
目前核酸序列分类检测主要有三类方法:1)基于序列相似性比对的方法;2)基于统计学的方法;3)基于系统发育树的方法。由于目前数据库均是基于已知分类序列进行构建,通过序列相似性比对方法对新出现的核酸序列则不能准确地进行分类。而基于系统发育树进行分类的方法相比其他两种方法虽然灵敏度和特异性更高,但是会需要更多的运算时间和计算资源。
发明内容
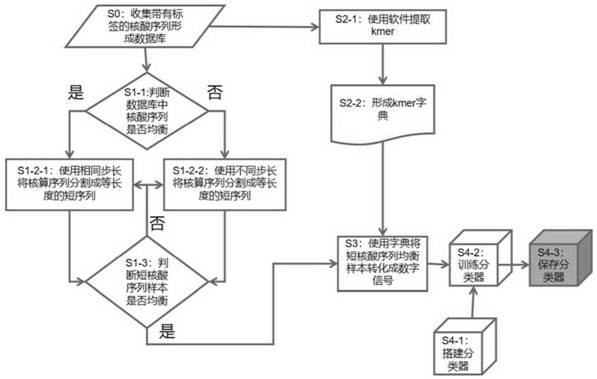
为了解决上述问题,本发明构建对核酸序列进行分类的模型方法,所述方法包括以下步骤:
S0:收集带有准确分类标签的核酸序列构建核酸序列数据库;
S1:将核酸序列数据库中所有核酸序列分割成等长度的短序列,从而形成短核酸序列均衡样本,该短序列的长度为4bp~核酸序列数据库中最短序列的长度,短核酸序列均衡样本适合于机器学习,短核酸序列均衡样本是指最多分类标签数目的短核酸序列数量N
S2:根据所述核酸序列数据库中的核酸序列,提取核酸Kmer,核酸Kmer长度为4≤Kmer长度≤所述短序列的长度,形成核酸序列的Kmer字典;
S3:使用S2中的Kmer字典将S1中的短核酸序列均衡样本转化成数字信号;
S4:使用S3中的数字信号训练一个能对带有分类标签的核酸序列进行多分类的神经网络分类器,该神经网络分类器用于识别和判断待检测核酸序列在核酸数据库中核酸分类。
在一种实施方式中,所述S1包括以下步骤:
S1-1:判断核酸序列数据库中所有核酸序列是否均衡;
S1-2-1:如果均衡,则使用相同分割步长将核酸序列数据库中所有核酸序列分割成等长度的短序列,所述相同分割步长是分割时后移长度,该后移长度不大于所述短序列的长度,分割核酸序列数据库中所有核酸序列获得短序列样本;
S1-2-2:如果不均衡,则对核酸序列数据库中每类标签核酸序列使用各自分割步长将每类标签核酸序列分割成等长度的短序列,每类标签核酸序列分割步长为分割各类标签核酸序列时后移步长,该后移长度不大于所述短序列的长度,分割核酸序列数据库中所有核酸序列获得短序列样本;
S1-3:判断S1-2-1或者S1-2-2获得短核酸序列的样本是否为均衡并适合于机器学习的样本,如果是,则停止S1-2-1或者S1-2-2,如果不是,则重复S1-2-1或者S1-2-2,直到获得均衡并适合于机器学习的短核酸序列的样本。
在一种实施方式中,所述S1-2-2中,包括以下步骤:
(1)获得核酸序列数据库中带有准确分类标签的核酸序列中每类核酸序列数目与特定值的比率,特定值的选择范围在核酸序列数据库中核酸序列中最多标签数目的核酸序列数量N
(2)根据每类核酸的比率值求得此类核酸的分割步长,分割步长=短序列长度÷比率值。
在一种实施方式中,S2包括:
S2-1:使用软件提取核酸序列数据库中所有的Kmer组成集合;
S2-2:构建Kmer字典,将S2-1中的所有Kmer按字母顺序从小到大排序,并且排序好后的Kmer中的第一位置插入
5.根据权利要求4所述的方法,其特征在于,所述字典的查询方式为Kmer:Kmer所在字典中的位置。
在一种实施方式中,S2-1中软件是jellyfish软件。
在一种实施方式中,所述S4包括以下步骤:
S4-1:搭建一个含有输入层、Embedding层、最大池化层、平均池化层、中间合并层、全连接层一、全连接层二、输出层的模型;
S4-2:把S3得到的数字信号输入到S4-1搭建的模型中进行训练,直到该神经网络成为对输入核酸序列进行多分类的神经网络分类器,分类标签为S0核酸数据库中的分类标签;
S4-3:把S4-2中训练好的模型保存,用于后续对待检测核酸序列进行分析。
在一种实施方式中,使用S3中50%-90%数字信号作为训练集,S3中10%-50%数字信号作为测试集,将训练集输入S4-2中搭建的神经网络,并对该网络进行反复训练,并利用测试集不断测试,直到该网络的分类表现达到成为对输入核酸序列进行多分类的神经网络分类器的要求,其分类标签为核酸数据库中核酸序列分类标签。
在一种实施方式中,本发明提供上述方法构建的模型在核酸分类中的应用。
在一种实施方式中,所述应用包括以下步骤:
F1:将一条或不同样本的多条待检测核酸序列保存到文件中,并且分别命名;
F2:使用S2中字典将F1中的待检测核酸序列转化成数字信号;
F3:使用S4中已经训练好的网络分类器对F2中的数字信号进行分析,得到每条核酸序列的分类结果;
F4:对所述分类结果进行分析,判定待检测核酸序列的标签是由数据库中哪几种标签重组而成。
在一种实施方式中,所述应用包括以下步骤:
P1:将来源于同一样本基因组的多个基因序列片段或者来源于不同样本基因组的多个基因序列片段作为待检测序列保存到文件中,并且分别命名;
P2:使用S2中字典将P1中的待检测核酸序列转化成数字信号;
P3:使用S4中已经训练好的网络分类器对P2中的数字信号进行分析,得到每条核酸的分类结果;
P4:对所述分类结果进行分析,使用分类结果中同一样本基因组的不同核酸序列片段的结果确定每条核酸序列在数据库中的标签。
在本发明方法中,在步骤S1中,根据数据库中各类别核酸的数目设定相应的步长对各类别的核酸进行分割,从而获得短序列均衡样本,使其适合于机器学习并得到可靠的神经网络分类器,短序列的长度可根据后续待检测样本的长度灵活选择。
在S2步骤中,使用Kmer提取软件直接提取数据库中所有的Kmer组成字典,可以减少后续步骤中与字典对应的Embedding数组的运算量,减少模型的存储空间,增加模型的可移植性。
在S4步骤中,训练一个核酸序列分类器并保存,可以快速将待检测核酸序列与数据库中的核酸序列标签进行对应分类。
通过本发明方法构建的模型,能够准确快速地将待检测核酸序列与数据库中的核酸序列标签进行对应分类,并展示每个标签的可靠性分值,为后续研究提供参考,增加了模型的使用价值。
当数据库中核酸序列不均衡时,本发明方法对核酸序列数据库中每类标签核酸序列使用各自分割步长将每类标签核酸序列分割成等长度的短序列组成短核酸序列均衡样本,避免因数据库核酸序列不平衡导致数据库不可用或需要付出额外努力增加核酸样本的现象,增加了机器学习对核酸序列进行分类的适用范围。
由于某一长度的Kmer的全部数目为4k/2+1(k值为Kmer的长度),因此当k值足够大时,Kmer的全部数目会变得非常大,本地计算机资源难以处理。例如当k=20,Kmer的数目约为5497亿,而与之对应的Embedding数组储存的数据则会更加庞大。本发明方法使用Kmer提取软件直接提取数据库中所有的Kmer组成字典,可以减少后续步骤中与字典对应的Embedding数组的运算量,减少模型的存储空间,增加模型的可移植性。
通过本发明方法构建的模型,能够准确快速地将待检测核酸序列与数据库中的核酸序列标签进行对应分类,并展示每个标签的可靠性分值,根据可靠性分值来判定待检测核酸序列是否是由数据库中的多个标签重组而成;或者根据同一样本基因组上的多个不同区域的核酸片段可靠性分值确定样本的标签,增加结果的可靠性。由于本方法是在本地计算机上运行,因此对数据的处理量没有限制,并且完全可以保证临床数据的保密性。
附图说明
为了更清楚地说明本申请实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本申请中记载的一些实施例,对于本领域普通技术人员来说,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1是本发明构建对核酸序列进行分类的模型方法示意图;
图2是本发明实施例的核酸序列数据库中各亚型所对应的数目示意图,共184个亚型,根据亚型的数目从大到小排序,截取前26种亚型名称及其数目;
图3是通过本发明实施例核酸数据库中各亚型的数目求得每种亚型相较于某一特定值的比率结果图,共184种亚型的比率值,根据比率值从大到小排序,截取前20种亚型的名称及比率值;
图4是将本发明实施例核酸数据库中184种亚型的全长DNA序列分割后,根据短序列的亚型数目排序结果图示意图,截取前21种亚型名称及所对应的短序列的数目;
图5是本发明实施例使用jellyfish软件提取本发明核酸数据库中的所有20mer形成的txt文件的示意图,截取前13种20mer和数目,‘>49’表示下面20mer(‘AAAAAAAAAAAAAAAAAAAA’)序列在数据库中的数目;
图6是本发明将20mer集合按字母顺序从小到大排列形成20mer字典示意图,不存在的20mer序列标记为
图7显示本发明实施例前20个1000bp DNA序列的数字信号示意图,转化后每个序列的信号长度为981,9284表示为20mer字典中第9824位20mer表示的序列;
图8是本发明实施例1的神经网络分类器构建示意图;
图9是本发明实施例的模型训练过程示意图;
图10是实施例2中使用的NCBI数据库中13条HIV序列ID;
图11是实施例2中待检测的DNA序列保存到fasta文件部分展示;
图12是实施例2中分析结果示意图;
图13是实施例3中使用的NCBI数据库中12条HIV序列ID;
图14是实施例3中待检测的DNA序列保存到fasta文件部分展示;和
图15是实施例3中分析结果示意图。
具体实施方式
为了使本领域技术人员更好地理解本申请中的技术方案,下面将结合实施例对本发明作进一步说明,显然,所描述的实施例仅仅是本申请一部分实施例,而不是全部的实施例。基于本申请中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都应当属于本申请保护的范围。下面结合附图及实施例对本发明作进一步描述。
实施例1构建对HIV-1型核酸序列分类的模型
如图1所示,本发明构建对HIV-1型核酸序列进行分类的模型方法,具体方法如下:
S0:收集带有准确分类标签的核酸序列构建核酸序列数据库:收集带有准确分类标签的HIV基因组DNA数据构建基础数据库,在本实施例中分类标签是HIV各种亚型标签。
本数据库来源于HIV sequence database(https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html)中的12825条完整的HIV-1型DNA序列(截止到2020年9月6日),然后将其中每种亚型含有的DNA序列数目多于2的基因组DNA序列构建核酸序列数据库,共计184种亚型。
S1:将核酸序列数据库中所有核酸序列分割成等长度的短序列,从而形成短核酸序列均衡样本,该短序列的长度为4bp~核酸序列数据库中最短序列的长度,短核酸序列均衡样本适合于机器学习,短核酸序列均衡样本是指最多分类标签数目的短核酸序列数量N
S1-1:判断核酸序列数据库中所有核酸序列是否均衡。在本发明实施例中,由于带有HIV-1_B亚型标签的序列有5703条,带有HIV-1_AHJU亚型标签的序列有2条(未展示),所以数据是不均衡的,如图2所示。
S1-2-2:对核酸序列数据库中每类标签核酸序列使用各自分割步长将每类标签核酸序列分割成等长度的短序列,每类标签核酸序列分割步长为分割各类标签核酸序列时后移步长,该后移长度不大于所述短序列的长度,分割核酸序列数据库中所有核酸序列获得短序列样本。
(1)获得核酸序列数据库中带有准确分类标签的核酸序列中每类核酸序列数目与特定值的比率,特定值的选择范围在核酸序列数据库中核酸序列中最多标签数目的核酸序列数量N
(2)根据每类核酸的比率值求得此类核酸的分割步长,分割步长=短序列长度÷比率值。
在本实施例中短序列长度为1000bp,不同步长的计算方法如下:
(1)通过核酸序列数据库中每种亚型的数目求得每种亚型与特定值的比率。特定值的选择范围在数据库中含有核酸序列最多的标签数目(N
(2)根据每类核酸的比率值求得此类核酸的分割步长,分割步长=短序列长度÷比率值。例如,HIV-1_B亚型的步长为step=1000bp÷1.0=1000bp,HIV-1_01BC亚型的步长为step=1000bp÷9.94=100.603≈100bp。
根据每种亚型的步长将核酸序列数据库中对应亚型的全长基因组DNA序列分割成1000bp短DNA序列。分割核酸序列数据库后可得到HIV-1_C亚型含1000bp短DNA序列最多,数目为5066条;HIV-1_AHJU亚型含1000bp短DNA序列最少,数目为2954条(在图中未显示),如图4所示。
S1-3:判断S1-2-1或者S1-2-2获得短核酸序列的样本是否为均衡并适合于机器学习的样本,如果是,则停止S1-2-1或者S1-2-2,如果不是,则重复S1-2-1或者S1-2-2,直到获得均衡并适合于机器学习的短核酸序列的样本。在本实施例中,由于N
S2-1:使用软件提取核酸序列数据库中所有的Kmer组成集合。在本实施例中,使用jellyfish软件提取数据库种的所有20mer。Kmer的取值范围为:4≤Kmer长度≤短序列长度,在本实施例中短序列长度为1000bp,Kmer选择20mer,如图5所示。
S2-2:构建Kmer字典,将S2-1中的所有Kmer按字母顺序从小到大排序,并且排序好后的Kmer中的第一位置插入
在本实施例中,构建20mer字典。将S2-1中的所有20mer形成集合并按字母顺序从小到大排序,考虑到在待检测序列中的20mer可能不存在S2-1中的20mer集合中,因此在排序好后的20mer中的第一位置插入
S3:使用S2中的Kmer字典将S1中的短核酸序列均衡样本转化成数字信号。在本实施例中,使用20mer字典将S1-3中的均衡样本转化成数字信号,根据样本序列的Kmer在Kmer字典中的位置将样本转化成数字信号,如图7所示。
S4:使用S3中的数字信号训练一个能对带有分类标签的核酸序列进行多分类的神经网络分类器,该神经网络分类器用于识别和判断待检测核酸序列在核酸数据库中核酸分类。
S4-1:搭建一个含有输入层、Embedding层、最大池化层、平均化层、中间合并层、全连接层一、全连接层二、输出层的模型。在S4-1中,输入层维度为1×981,维度计算公式为:短DNA序列长度-Kmer+1,在此实施例中得到的信号长度为1000-20+1=981;Embedding层维度为6942463×100,其中6942463为构建字典中Kmer的种类;最大池化层为获取embs(981×100)中每一列的最大值,得到一维数组max_pool(1×100);平均池化层为获取embs中每一列的平均值,得到一维数组avg_pool(1×100);中间合并层为将max_pool与avg_pool两者通过numpy模块中的concatenate函数进行拼接,得到一维数组input(1×200);全连接层一含有150个神经元;全连接层二含有150个神经元;输出层含有184个神经元,最后采用softmax激活函数把输出层每个神经元的值转化成概率值。如图8所示。
S4-2:取S3数据集中80%的数据作为训练集,剩余20%的数据作为测试集。在选取时,需要保证训练集和测试集中各亚型的比例近似相同。将训练集送入S4-1中搭建的神经网络中,对网络进行反复训练,并利用测试集不断测试。直到该网络的分类表现达到要求,如图9所示。
S4-3:将上一步训练好的模型分类器保存到磁盘中。对后续待测核酸序列进行分析。
实施例2使用实施例1中的模型判定HIV病毒核酸序列的重组亚型
F1:选择NCBI数据库中13条HIV的基因序列作为待检测序列,其ID如图10所示。将待检测的DNA序列保存到fasta文件中,命名方式为“>ID名称”如图11所示。
F2:使用实施例1中的S2-2中的20mer字典将F1中的核酸序列转化成数字信号。
F3:使用实施例1中的S4-3中的已经训练好的网络分类器对F2中的数字信号进行分析。
F4:对分类结果进行分析。结果如图12所示,ID为fasta文件中每条序列的ID,Top-1至Top-5为这条ID序列前5种可能的亚型。括号中的数值为此种亚型的可靠性分值,分值越高则表明这条ID序列为此亚型的可靠性越高。若某一亚型的可靠性分值大于0.1则表明此ID包含此亚型,因此可以判断此ID是由哪几种亚型重组而成。例如ID值为“MN654105.1”的前2种可能的亚型分别是HIV-1_01B(0.84)和HIV-1_01AE(0.152)并且可靠性分值都大于0.1,因此“MN654105.1”是由01B和01AE重组而成。
实施例3使用实施例1中的模型分析同一HIV样本的三个不同基因序列片段判定样本的亚型
P1:选择NCBI数据库中12条HIV序列的gag,pol,env三种基因序列片段作为待检测序列,其ID如图13所示。将待检测的DNA序列保存到fasta文件中,命名方式为“>ID名称;片段名称”,如图14所示。例如‘>MK397789;gag’,其中‘MK397789’为样本的ID名称,‘gag’为此样本的片段名称,中间以‘;’隔开。
P2:使用实施例1中的S2-2中的20mer字典将P1中的核酸序列转化成数字信号。
P3:使用实施例1中的S4-3中的已经训练好的网络分类器对P2中的数字信号进行分析。
P4:对分类结果进行分析。结果如图15所示,subtype列为此ID样本的分类亚型,后面三列(gag,pol,env)为此ID相应片段下的亚型及可靠性值,ID样本亚型的判定规则为:
(1)若同一ID样本的三个片段所得出的亚型一致,则subtype列为此三个相同的亚型。
(2)若同一ID样本的三个片段中,只有两个片段得出的亚型一致且可靠性分值都超过0.5,则subtype列为此两个相同的亚型。
(3)若同一ID样本的三个片段所得出的亚型互不一致,则subtype列为可靠性分值得分最高的亚型。
(4)若某一ID样本的三个片段中某一片段不存在,则此片段下的显示结果为NaN。则subtype列为剩余两个片段中可靠性分值得分最高的亚型。
(5)除以上情况外则subtype列为Unclassified。
应该理解到披露的本发明不仅仅限于描述的特定的方法、方案和物质,因为这些均可变化。还应理解这里所用的术语仅仅是为了描述特定的实施方式方案的目的,而不是意欲限制本发明的范围,本发明的范围仅受限于所附的权利要求。
本领域的技术人员还将认识到,或者能够确认使用不超过常规实验,在本文中所述的本发明的具体的实施方案的许多等价物。这些等价物也包含在所附的权利要求中。
- 构建对核酸序列进行分类的模型方法及其应用
- 构建对核酸序列进行分类的模型方法及其应用