分析用试样的制备方法、分析方法和分析用试样的制备用试剂盒
文献发布时间:2023-06-19 10:54:12
技术领域
本发明涉及分析用试样的制备方法、分析方法和分析用试样的制备用试剂盒。
背景技术
唾液酸是在生物体内大量存在的糖。唾液酸在生物体内也包含在与蛋白质结合的糖链中,多存在于糖链的非还原末端。因此,唾液酸由于在这种糖蛋白分子中配置在分子的外侧、可被其他分子直接识别,因此具有重要的作用。
唾液酸与邻接的糖之间的连接方式(linkage type)有时会不同。例如,已知人的N-连接型糖链(N型糖链)主要是α2,3-和α2,6-的连接方式,O-连接型糖链(O型糖链)和鞘糖脂在前述连接方式的基础上还有α2,8-和α2,9-的连接方式。根据这种连接方式的区别,唾液酸可以被不同的分子识别、具有不同的作用。
质谱分析等中,作为针对含有唾液酸的糖链的前处理,进行唾液酸的修饰。其将具有负电荷的唾液酸的羧基通过酯化或酰胺化等而中性化,从而克服抑制电离和唾液酸脱离等缺点。关于唾液酸的内酯化,根据连接方式而生成的内酯的稳定性不同,因此能够利用该稳定性的差异,连接方式特异性地进行唾液酸的修饰和解析。
此处,内酯极不稳定,在水中也容易水解,在酸性或碱性条件下更迅速地水解。因此,报告了将通过前处理中的修饰而生成的内酯通过酰胺化而稳定化(参照专利文献1、非专利文献1和非专利文献2)。适宜地将通过内酯化而生成的分子中的环状结构称为内酯结构。内酯结构也存在于生物体中的糖链和抗体药物的糖链等,进行它们的解析时也可以进行稳定化。另外,非专利文献2中记载的内酯的直接酰胺化能够进行内酯的迅速修饰,未来的利用受到期待。
内酯化为酯化的一种。唾液酸的修饰中,也进行除内酯化以外的酯化。非专利文献3中,在溶解于甲醇的游离糖链中添加脱水缩合剂4-(4,6-二甲氧基-1,3,5-三嗪-2-基)-4-甲基吗啉鎓盐酸盐(DMT-MM)。由此,将α2,6-唾液酸甲酯化,将α2,3-唾液酸内酯化。
非专利文献4中,在游离糖链中添加溶解于乙醇等的N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺(EDC)和1-羟基苯并三唑(HOBt)等脱水缩合剂。由此,将α2,6-唾液酸酯化,将α2,3-唾液酸内酯化。
非专利文献5中,在结合于固相载体的糖蛋白中添加包含乙醇和脱水缩合剂的溶液,将α2,6-唾液酸酯化,将α2,3-唾液酸内酯化。然后,如非专利文献5的插图(Scheme)1(b)所示,为了将内酯水解,在试样中添加pH10的Tris缓冲液使其反应1小时,然后在试样中添加包含甲胺盐酸盐的溶液,进而在其中添加脱水缩合剂使其反应30分钟。
非专利文献6中,在结合于固相载体的糖蛋白中添加包含EDC盐酸盐和HOBt的乙醇,将α2,6-唾液酸酯化后,将对甲苯胺溶液与脱水缩合剂一起添加到试样中,将α2,3-唾液酸酰胺化。
非专利文献7中,在结合于固相载体的糖蛋白中添加包含EDC盐酸盐和HOBt的乙醇,将α2,6-唾液酸酯化后,将乙二胺溶液与脱水缩合剂一起添加到试样中,将α2,3-唾液酸酰胺化。
非专利文献8中,对糖链添加包含EDC和HOBt的乙醇,将α2,6-唾液酸酯化并将α2,3-唾液酸内酯化后,然后添加氨水并在脱水缩合剂存在下反应,从而将内酯水解后使其酰胺化。
现有技术文献
专利文献
专利文献1:日本特许第6135710号公報
非专利文献
非专利文献1:Nishikaze T,Tsumoto H,Sekiya S,Iwamoto S,Miura Y,TanakaK."Differentiation of Sialyl Linkage Isomers by One-Pot Sialic AcidDerivatization for Mass Spectrometry-Based Glycan Profiling"AnalyticalChemistry,(美国),ACS Publications,2017年2月21日、Volume 89,Issue 4,pp.2353-2360
非专利文献2:Hanamatsu H,Nishikaze T,Miura N,Piao J,Okada K,Sekiya S,Iwamoto S,Sakamoto N,Tanaka K,Furukawa JI."Sialic Acid Linkage SpecificDerivatization of Glycosphingolipid Glycans by Ring-Opening Aminolysis ofLactones"Analytical Chemistry,(美国),ACS Publications,2018年10月29日、Volume90,Issue 22,pp.13193-13199
非专利文献3:Wheeler SF,Domann P,Harvey DJ."Derivatization of sialicacids for stabilization in matrix-assisted laser desorption/ionization massspectrometry and concomitant differentiation of alpha(2-->3)-and alpha(2-->6)-isomers"Rapid communications in mass spectrometry,(英国),John Wiley AndSons Ltd.,2009年1月、Volume 23,Issue 2,pp.303-12
非专利文献4:Reiding KR,Blank D,Kuijper DM,Deelder AM,Wuhrer M."High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employinglinkage-specific sialic acid esterification"Analytical Chemistry,(美国),ACSPublications,2014年6月、Volume 86,Issue 12,pp.5784-93
非专利文献5:Li H,Gao W,Feng X,Liu BF,Liu X."MALDI-MS analysis ofsialylated N-glycan linkage isomers using solid-phase two step derivatizationmethod,"Analytica Chimica Acta,(荷兰),Elsevier B.V.,2016年6月14日、Volume 924,pp.77-85
非专利文献6:Yang S,Jankowska E,Kosikova M,Xie H,Cipollo J."Solid-Phase Chemical Modification for Sialic Acid Linkage Analysis:Application toGlycoproteins of Host Cells Used in Influenza Virus Propagation"AnalyticalChemistry,(美国),ACS Publications,2017年9月、Volume 89,Issue17,pp.9508-17
非专利文献7:Yang S,Wu WW,Shen RF,Bern M,Cipollo J"Identification ofSialic Acid Linkages on Intact Glycopeptides via Differential ChemicalModification Using IntactGIG-HILIC"Journal of the American Society for MassSpectrometry.,(美国),Springer,2018年4月12日、Volume 29,Issue 6,pp.1273-1283
非专利文献8:Lageveen-Kammeijer GSM,de Haan N,Mohaupt P,Wagt S,FiliusM,Nouta J,Falck D,Wuhrer M."Highly sensitive CE-ESI-MS analysis of N-glycansfrom complex biological samples"Nature communications,(英国),NaturePub.Group,2019年5月13日、Volume 10,Issue 1,p.2137
发明内容
期望提出为了分析糖链所包含的唾液酸而修饰糖链所包含的唾液酸的新方法。
本发明的第一方式涉及一种分析用试样的制备方法,其为由包含糖链的试样制备分析用试样的方法,所述制备方法具备如下操作:进行酯化反应,所述酯化反应将前述糖链所包含的唾液酸的至少一部分供于除内酯化以外的酯化;使前述试样与酰胺化反应溶液接触,进行酰胺化反应,所述酰胺化反应溶液包含与通过前述酯化而进行了修饰的唾液酸发生反应的选自由氨、胺、肼、肼衍生物和羟胺以及它们的盐组成的组中的至少一种化合物,所述酰胺化反应将通过前述酯化而进行了修饰的唾液酸的酯化修饰转化为酰胺化修饰。
本发明的第二方式涉及一种分析方法,其具备如下操作:通过第一方式的分析用试样的制备方法制备试样;以及,进行所制备的前述分析用试样的分析。
本发明的第三方式涉及一种分析用试样的制备用试剂盒,其用于第一方式的分析用试样的制备方法。
根据本发明,能够提供用于分析糖链所包含的唾液酸的、通过新机理来修饰唾液酸的方法。
附图说明
图1为示出一实施方式的分析方法的流程的流程图。
图2为示出分析用试样的制备用试剂盒的概念图。
图3为示出变形例的分析方法的流程的流程图。
图4A为示出糖链GD1a的结构的概念图。
图4B为示出糖链GD1b的结构的概念图。
图4C为示出糖链A2GN1的结构的概念图。
图5为示出变形例的分析方法的流程的流程图。
图6为示出实施例中使用的糖链的结构的概念图。
图7为示出进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a);以及在进行甲酯化反应后分别利用1%(b)、10%(c)和40%(d)甲胺水溶液进行酰胺化反应而制备的分析用试样的质谱图的图。
图8为示出在进行甲酯化反应后分别使用5.6%氨水(a)、10%甲胺溶液(b)、17.5%乙胺溶液(c)、16.7%二甲胺溶液(d)、20%三甲胺溶液(e)进行酰胺化反应而制备的分析用试样的质谱图的图。
图9为示出在进行甲酯化反应后使用5%甲胺水溶液进行酰胺化反应而制备的分析用试样的质谱图(a);以及分别将甲醇(b)、二甲基亚砜(c)和乙腈(d)作为溶剂,使用6%甲胺溶液进行酰胺化反应而制备的分析用试样的质谱图的图。
图10为示出在进行甲酯化反应后使用17.5%乙胺溶液进行酰胺化反应而制备的分析用试样的质谱图(a)、以及在进行乙酯化反应后使用10%甲胺溶液进行酰胺化反应而制备的分析用试样的质谱图(b)的图。
图11为示出对于包含GD1a糖链的试样进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a)、对于包含GD1a糖链的试样进行甲酯化反应后使用10%甲胺溶液进行酰胺化反应而制备的分析用试样的质谱图(b)、对于包含GD1b糖链的试样进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(c)、以及对于包含GD1b糖链的试样进行甲酯化反应后使用10%甲胺溶液进行酰胺化反应而制备的分析用试样的质谱图(d)的图。
图12为示出实施例中使用的糖链的结构的概念图。
图13为示出对于源自血清糖蛋白的N型糖链进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a);以及在进行甲酯化反应后分别利用5%(b)、10%(c)、20%(d)和40%(e)乙胺溶液(通过真空抽吸来通液)进行酰胺化反应而制备的分析用试样的质谱图的图。
图14为示出对于源自血清糖蛋白的N型糖链进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a);以及在进行甲酯化反应后分别利用5%(b)、10%(c)、20%(d)和40%(e)乙胺溶液(通过自然落下来通液)进行酰胺化反应而制备的分析用试样的质谱图的图。
图15为示出对于源自血清糖蛋白的N型糖链进行甲酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a);以及在进行甲酯化反应后在液相下分别利用2.5%(b)、5%(c)、10%(d)、20%(e)和40%(f)乙胺溶液进行酰胺化反应而制备的分析用试样的质谱图的图。
图16为对于GD1a糖链和A2GN1糖链进行乙酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(a);以及在进行乙酯化反应后,将溶剂设为乙腈,分别使用甲胺溶液(b)、乙胺溶液(c)和丙胺溶液(d)进行酰胺化反应而制备的分析用试样的质谱图的图。
图17为对于GD1b糖链和A2GN1糖链进行乙酯化反应且未进行酰胺化反应而制备的分析用试样的质谱图(e);以及在进行乙酯化反应后,将溶剂设为乙腈,分别使用甲胺溶液(f)、乙胺溶液(g)和丙胺溶液(h)进行酰胺化反应而制备的分析用试样的质谱图的图。
图18为示出对于GD1a(a)和GD1b(b)糖链进行乙酯化反应后,将溶剂设为乙腈,使用丙胺溶液进行酰胺化反应而制备的分析用试样的MS/MS谱的图。
图19为示出对于GD1a糖链进行乙酯化反应后,将溶剂设为乙腈,分别使用0M、0.125M、0.25M、0.5M、1M和3M的各浓度的丙胺溶液进行酰胺化反应而制备的分析用试样的质谱图的图。
图20为示出对于GD1b糖链进行乙酯化反应后,将溶剂设为乙腈,分别使用0M、0.125M、0.25M、0.5M、1M和3M的各浓度的丙胺溶液进行酰胺化反应而制备的分析用试样的质谱图的图。
10…酯化剂、11…容器、100…分析用试样的制备用试剂盒。
具体实施方式
以下,参照图对用于实施本发明的方式进行说明。以下的实施方式中,记载为“酯化”时,在没有特别注释的情况下是指除内酯化以外的酯化。
-第一实施方式-
图1为示出本实施方式的分析用试样的制备方法的分析方法的流程的流程图。本实施方式的分析用试样的制备方法中,将试样所包含的糖链的唾液酸酯化后,将酯化的唾液酸连接方式特异性地酰胺化。在步骤S1001中,准备包含糖链的试样。
(关于试样)
包含糖链的试样没有特别限定,可以含有选自由糖链、糖肽和糖蛋白以及糖脂组成的组中的至少一种的分子。肽和糖肽可以具备由2以上且不足50的氨基酸形成的肽主链,蛋白质和糖蛋白可以具备由50以上的氨基酸形成的肽主链。但是,也有惯例上的例外,肽和蛋白质的范围的界限以及糖肽和糖蛋白的范围的界限不限定于此。本实施方式的分析用试样的制备方法中,进行糖链所包含的唾液酸的连接方式特异性的修饰。试样中的糖链优选包含N-连接型糖链(N型糖链)或O-连接型糖链(O型糖链)、或者糖脂型糖链等有可能在末端或末端以外的位置具有唾液酸的糖链。另外,试样中的糖链更优选包含或有可能包含α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸中的至少一者,进一步优选除此之外还包含或有可能包含α2,6-唾液酸。
试样包含游离糖链的情况下,可以使用自糖蛋白、糖肽或糖脂游离的糖链。作为该游离的方法,可以采用使用N-糖苷酶、O-糖苷酶、或鞘糖脂内切糖苷酶(Endoglycoceramidase)等的酶处理、肼分解、基于碱处理的β脱离等方法。使N-连接型糖链自糖肽和糖蛋白的肽链游离的情况下,可适宜使用利用肽-N-糖苷酶F(PNGase F)、肽-N-糖苷酶A(PNGase A)、或内切-β-N-乙酰葡糖胺糖苷酶(Endo M)等的酶处理。另外,可以适宜进行糖链的还原末端的吡啶氨基化(PA标记)等修饰。在酶处理前,也可以进行后述的糖肽或糖蛋白的肽链的切断。
试样包含糖肽或糖蛋白的情况下,如后述的“关于糖肽和糖蛋白的副反应的抑制”的部分所述,可以适宜进行用于抑制肽部分的副反应的处理。另外,肽链的氨基酸的残基数多的糖肽或糖蛋白优选通过酶的切断等来将肽链切断而使用。例如制备质谱分析用的试样的情况下,肽链的氨基酸残基数优选为30以下,更优选为20以下,进一步优选为15以下。另一方面,在要求明确糖链所结合的肽的来源的情况下,肽链的氨基酸残基数优选为2以上,更优选为3以上。
作为将糖肽或糖蛋白的肽链切断时的消化酶,可使用胰蛋白酶、赖氨酸内肽酶、精氨酸内肽酶、胰凝乳蛋白酶、胃蛋白酶、嗜热菌蛋白酶、蛋白酶K、或链霉蛋白酶E等。可以将这些消化酶中的2种以上组合使用。切断肽链时的条件没有特别限定,可采用与所使用的消化酶相应的适当的规程。在该切断前,可以进行试样中的蛋白质和肽的改性处理或烷基化处理。改性处理或烷基化处理的条件没有特别限定。可以通过化学切断等而非酶切断来将肽链切断。
步骤S1001结束后,进入步骤S1003。
(酯化反应)
步骤S1003中,使试样与用于酯化的反应溶液(以下称为酯化反应溶液)接触,进行将糖链所包含的唾液酸的至少一部分酯化的酯化反应(以下记载为酯化反应时,在没有特别提及的情况下,是指步骤S1003的酯化反应)。酯化反应为将糖链所包含的唾液酸供于除内酯化以外的酯化的反应。但是,酯化反应中,只要对一部分的唾液酸发生除内酯化以外的酯化,则使另一部分的唾液酸内酯化也不会有妨碍。以下的实施方式中,“除内酯化以外的酯化”是指,酯化反应溶液的成分的一部分与唾液酸的羧酸键合,形成羧酸酯。即,相当于酯化反应溶液的成分的一部分被导入到表示羧酸酯的-COOR的R的部分。酯化反应中,优选连接方式非特异性地将糖链所包含的唾液酸酯化。酯化反应中,适宜将α2,3-唾液酸、α2,6-唾液酸、α2,8-唾液酸和α2,9-唾液酸的羧基酯化。
酯化反应溶液只要将糖链所包含的唾液酸、特别是至少除α2,6-唾液酸以外的、例如α2,3-唾液酸等唾液酸的至少一部分酯化,则其组成没有特别限定。从进行连接方式特异性的修饰的观点出发,优选对α2,6-唾液酸也进行酯化。酯化反应溶液优选包含醇和酯化剂中的至少一者。但是,酯化的方法不限定于使用醇或酯化剂的方法,可以使用任意的酯化的方法。
(酯化反应溶液包含醇的情况)
酯化反应溶液包含醇的情况下,酯化反应溶液优选还包含缩合剂,更优选除了缩合剂之外还包含添加剂。酯化反应溶液可以包含甲醇或乙醇等醇且包含盐酸等酸。
酯化反应溶液所包含的醇没有特别限定,可以采用任意的元数的醇。酯化反应溶液所包含的醇优选一元的醇,更优选甲醇、乙醇、丙醇、丁醇、戊醇或己醇,进一步优选甲醇或乙醇。
酯化反应溶液所包含的缩合剂没有特别限定。该缩合剂可以包含碳二亚胺、羰基二咪唑、光气衍生物、鏻系缩合剂、脲鎓系缩合剂、甲脒鎓系缩合剂和三氟甲磺酸盐试剂中的至少一者。
该缩合剂中的碳二亚胺的例子包括:1-环己基-3-(2-吗啉基乙基)碳二亚胺甲基-对甲苯磺酸盐(1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate)(CMC)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(EDAC)、N,N’-二环己基碳二亚胺(DCC)、N-(3-二甲基氨基丙基)-N’-乙基碳二亚胺(EDC)、N,N’-二异丙基碳二亚胺(DIC)、1-叔丁基-3-乙基碳二亚胺(BEC)、N,N’-二叔丁基碳二亚胺、1,3-二对甲苯基碳二亚胺、双(2,6-二异丙基苯基)碳二亚胺、双(三甲基甲硅烷基)碳二亚胺和1,3-双(2,2-二甲基-1,3-二氧戊环-4-基甲基)碳二亚胺(BDDC)。
该缩合剂中的羰基二咪唑的例子包括:1,1’-羰基二咪唑(CDI)、1,1’-羰基二(1,2,4-三唑)(CDT)和1,1’-草酰二咪唑。
该缩合剂中的光气衍生物的例子包括:碳酸双(五氟苯基)酯、碳酸二-2-吡啶酯、二(N-琥珀酰亚胺)碳酸酯、硫光气、三光气和O,O’-二-2-吡啶硫代碳酸酯。
该缩合剂中的鏻系脱水缩合剂的例子包括:(苯并三唑-1-基氧基)三-(二甲基氨基)鏻六氟磷酸盐(BOP)、苯并三唑-1-基氧基三(吡咯烷基)鏻六氟磷酸盐(PyBOP)、溴代三(二甲基氨基)鏻六氟磷酸盐(BroP)、溴代三(吡咯烷基)鏻六氟磷酸盐(PyBroP)、(7-氮杂苯并三唑-1-基氧基)三(吡咯烷基)鏻六氟磷酸盐(PyAOP)和氯代三吡咯烷基鏻六氟磷酸盐(PyCloP)。这些统称为“BOP试剂”。
该缩合剂中的脲鎓系脱水缩合剂的例子包括:(1-氰基-2-乙氧基-2-氧代乙叉基氨基氧基)二甲基氨基-吗啉代-碳鎓六氟磷酸盐(COMU)、2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐(HBTU)、2-(7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐(HATU)、2-(1H-苯并三唑-1-基)-1,1,3,3-四甲基脲四氟硼酸盐(TBTU)、2-(5-降冰片烯-2,3-二甲酰亚胺)-1,1,3,3-四甲基脲四氟硼酸盐(TNTU)和O-(N-琥珀酰亚胺基)-1,1,3,3-四甲基脲四氟硼酸盐(TSTU)。
该缩合剂中的三氟甲磺酸盐试剂的例子包括:2-[N,N-双(三氟甲磺酰基)氨基]-5-氯吡啶、N-(2-吡啶)双(三氟甲磺酰亚胺)、三氟甲磺酸4-硝基苯酯、N-苯基双(三氟甲磺酰亚胺)、三氟甲磺酰氯、三氟甲磺酸酐、三氟甲烷磺基苯胺和1-(三氟甲磺酰基)咪唑。
为了促进利用缩合剂的缩合、且抑制副反应,优选除了缩合剂之外还使用亲核性高的添加剂。亲核性高的添加剂的例子包括:1-羟基苯并三唑(HOBt)、1-羟基-7-氮杂-苯并三唑(HOAt)、4-(二甲基氨基)吡啶(DMAP)、2-氰基-2-(羟基亚氨基)乙酸乙酯(Oxyma)、N-羟基-琥珀酰亚胺(HOSu)、6-氯-1-羟基-苯并三唑(Cl-HoBt)和N-羟基-3,4-二氢-4-氧代-1,2,3-苯并三嗪(HOOBt)。
(酯化反应溶液包含酯化剂的情况)
酯化反应溶液所包含的酯化剂没有特别限定,酯化剂之中优选烷基化剂,更优选甲基化剂、乙基化剂、异丙基化剂等丙基化剂、叔丁基化剂等丁基化剂、或苄基化剂,进一步优选甲基化剂或乙基化剂。
该酯化剂中的甲基化剂的例子包括:溴甲烷、碳酸二甲酯、硫酸二甲酯、N,N-二甲基甲酰胺二甲基缩醛、N,N’-二异丙基-O-甲基异脲、氟磺酸甲酯、碘甲烷、甲磺酸甲酯、1-甲基-3-对甲苯基三氮烯、原甲酸三甲酯、四甲基氯化铵、对甲苯磺酸甲酯、四甲基氢氧化铵、3-(三氟甲基)苯基三甲基氢氧化铵、三甲基氧鎓四氟硼酸盐、三甲基氢氧化锍和三氟甲磺酸甲酯。
该酯化剂中的乙基化剂的例子包括:溴乙烷、碳酸二乙酯、硫酸二乙酯、N,N-二甲基甲酰胺二乙基缩醛、1-乙基-3-对甲苯基三氮烯、O-乙基-N,N’-二异丙基异脲、碘乙烷、甲磺酸乙酯、原甲酸三乙酯、三氟甲磺酸乙酯和三乙基氧鎓四氟硼酸盐。
该酯化剂中的丙基化剂的例子包括:1-溴丙烷、2-溴丙烷、N,N-二甲基甲酰胺二丙基缩醛、硫酸二丙酯、硫酸二异丙酯、1-碘丙烷、2-碘丙烷、1-异丙基-3-对甲苯基三氮烯、甲磺酸异丙酯、原甲酸三异丙酯、甲磺酸丙酯和O,N,N’-三异丙基异脲。
该酯化剂中的丁基化剂的例子包括:1-溴丁烷、2-溴-2-甲基丙烷、2-碘-2-甲基丙烷、叔丁基2,2,2-三氯乙酰亚胺酯、O-叔丁基-N,N’-二异丙基异脲、N,N-二甲基甲酰胺二丁基缩醛、N,N-二甲基甲酰胺二叔丁基缩醛、硫酸二丁酯、1-碘丁烷和原甲酸三丁酯。
该酯化剂中的苄基化剂的例子包括:苄基溴、苄基氯、1-苄基-3-对甲苯基三氮烯、苄基2,2,2-三氯乙酰亚胺酯、2,2,2-三氟-N-苯基乙酰亚胺酸苄酯和O-苄基-N,N’-二异丙基异脲。
该酯化剂的例子除了上述之外还可以包括任意的烷基化剂。
酯化反应溶液优选包含三氮烯衍生物。酯化反应溶液所包含的三氮烯衍生物优选1-甲基-3-对甲苯基三氮烯(MTT)、1-乙基-3-对甲苯基三氮烯(ETT)、1-异丙基-3-对甲苯基三氮烯、1-苄基-3-对甲苯基三氮烯或1-(4-硝基苄基)-3-对甲苯基三氮烯,更优选MTT或ETT。
酯化反应溶液不限定于上述酯化剂,可以包含甲磺酸2-氯乙酯、N,N-二甲基甲酰胺二新戊基缩醛、硫酸二戊酯等任意的酯化剂。另外,酯化反应溶液也可以包含上述酯化剂的盐。
(酯化反应溶液中的醇或酯化剂等的浓度)
酯化反应中,优选调整所用的醇或酯化剂等的浓度,使得至少α2,3-唾液酸,α2,8-唾液酸和α2,9-唾液酸中的至少一者进行酯化。从进行连接方式特异性地修饰的观点出发,优选调整所用的醇或酯化剂等的浓度,使得α2,6-唾液酸进行酯化。另外,即使试样中的糖链的全部唾液酸进行酯化,也能够进行连接方式特异性的修饰,因此只要在其它方面没有障碍,则可以较高地设定酯化反应溶液中的醇、缩合剂或者添加剂、或酯化剂的浓度。
作为一例,利用酯化剂进行酯化反应的情况下,MTT或ETT等酯化剂的浓度优选10mM~10M、更优选50mM~5M、进一步优选100mM~1M。使用醇进行酯化反应的情况下,缩合剂的浓度例如可以设为1mM~5M等。组合使用缩合剂与添加剂的情况下,可以将各自的浓度设为上述范围。醇的浓度优选0.01~20M、更优选0.1M~10M。反应温度优选-20℃~100℃左右、更优选-10℃~50℃。
(进行酯化反应的相)
酯化反应在液相中或在固相中都能进行。只要能使试样与酯化反应溶液接触,则发生酯化反应时的试样的状态没有特别限定。
在固相中进行反应的情况下,作为固相载体,只要能够固定糖链、糖肽或糖蛋白等,就可以没有特别限制地使用。例如,为了固定糖肽或糖蛋白,可以使用具有环氧基、甲苯磺酰基、羧基、氨基等作为配体的固相载体。另外,为了固定糖链,可以使用具有酰肼基、氨基氧基等作为配体的固相载体。另外,也优选使糖链吸附于亲水性相互作用色谱法(Hydrophilic Interaction Chromatography;HILIC)用的载体、即固定相,进一步优选该HILIC用的载体包含酰胺基。
通过在将试样固定于固相载体的状态下进行反应,反应溶液的去除和脱盐纯化变得更容易,能够将试样的制备简化。另外,在糖蛋白或糖肽的状态下将试样固定于固相载体的情况下,若在酯化反应后进行利用PNGase F等糖苷酶等的切断,则也可以以游离糖链的形式回收酯化反应后的试样。将糖链固定于具有酰肼基、氨基氧基等作为配体的固相载体的情况下,可以将酯化反应后的试样通过酸处理等进行游离来回收。
酯化反应后的试样也可以根据需要通过公知的方法等进行纯化、脱盐、增溶等处理。后述酰胺化反应的前后也同样。
在酯化反应后使试样从固相载体游离的情况下,可以采用针对后述的酰胺化反应说明的条件。通过在将试样固定于固相载体的状态下进行反应,酯化反应后的酯化反应溶液的去除等变得容易,可以效率良好地进行唾液酸的修饰。
步骤S1003结束后,前进至步骤S1005。
(酰胺化反应)
步骤S1005中,使试样与反应溶液(以下称为酰胺化反应溶液)接触,进行酰胺化反应,所述酰胺化反应将通过步骤S1003的酯化而进行了修饰的唾液酸酰胺化(以下记载为酰胺化反应时,在没有特别提及的情况下,是指步骤S1005的酰胺化反应)。酰胺化反应中,在步骤S1003中酯化的唾液酸的酯化修饰转化为酰胺化修饰(以下适宜称为酯酰胺转化)。酰胺化反应中,将α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸中的至少一者、特别是α2,3-唾液酸和α2,8-唾液酸中的至少一者酰胺化。
发明人等发现了,与通过在试样中添加包含脱水缩合剂和胺等的溶液来将唾液酸的羧基酰胺化的技术常识完全不同,将唾液酸中形成的酯迅速直接酰胺化的方法。进而,出人意料的是,该直接酰胺化在对α2,3-唾液酸和α2,8-唾液酸等一部分连接方式的唾液酸进行的情况下,与在对α2,6-唾液酸等其它连接方式的唾液酸进行的情况下反应的效率不同。因此,通过对于包含酯化的这些连接方式的唾液酸的糖链进行酰胺化反应,能够进行连接方式特异性的修饰。
另外,如非专利文献2所示,内酯化的唾液酸也能够利用本实施方式的酰胺化反应溶液迅速地酰胺化。因此,即使通过步骤S1003的酯化反应而将α2,3-、α2,8-或α2,9-唾液酸进行了内酯化,也与利用酰胺化反应溶液进行酯化的情况同样地发生酰胺化。因此,在酰胺化反应前,优选不进行用于使通过酯化反应而生成的内酯结构裂解的操作。
例如,非专利文献4那样的方法中,在第一阶段的α2,6-唾液酸的酯化中使用甲醇来代替乙醇时,酯化的反应速度提高,因此对于α2,3-唾液酸,不仅发生内酯化,还发生甲酯化,变得无法与α2,6-唾液酸相区别。本实施方式的方法中,在这种情况下也能够将甲酯化或内酯化的唾液酸两者都酰胺化。因此,本实施方式的方法中,能够更可靠地进行唾液酸的连接方式特异性修饰。
以下的实施方式中,在提及唾液酸中形成的内酯的情况下,除了在唾液酸和邻接于该唾液酸的单糖间形成的内酯之外,还指在唾液酸的内部等形成的内酯。
酰胺化反应溶液中包含:含有与通过酯化而进行了修饰的唾液酸发生反应的氨、胺或它们的盐的反应剂(以下称为酰胺化反应剂)。酰胺化反应剂用于通过其至少一部分键合于唾液酸来进行基于酰胺化的修饰。酰胺化反应剂例如为亲核试剂。优选的是,酰胺化反应仅通过使试样与酰胺化反应溶液接触来进行,以简便的操作将内酯稳定化。
需要说明的是,酰胺化反应中,脱水缩合剂不是必需的,可以不包含,但酰胺化反应溶液中也可以包含脱水缩合剂。例如,也可以在不去除步骤S1003中添加到试样中的酯化反应溶液的情况下,将氨、胺或它们的盐添加到试样中,从而制备酰胺化反应溶液。如此,酰胺化反应能够以简便的操作进行。或者,在用于去除步骤S1003中添加到试样中的酯化反应溶液的操作后,仅通过使试样与酰胺化反应溶液接触来进行酰胺化反应。另外,酰胺化反应中,使试样与酰胺化反应溶液接触后,可以不进行使试样与脱水缩合剂反应的操作,但也可以为了例如其它目的而进行。
对于通过本实施方式的分析用试样的制备方法所得到的分析用试样,通过质谱分析来进行分析的情况下,选择酯化反应溶液所包含的醇或酯化剂、和酰胺化反应剂,使得通过酯化反应形成的酯的修饰体与通过酰胺化反应形成的酰胺的修饰体的质量不同。选择酯化反应溶液所包含的醇或酯化剂、和酰胺化反应剂,使得与质谱分析的质量分辨率相应地对所得到的2种修饰体精度良好地进行质量分离。为了通过单次质谱分析而明确区别2种修饰体,优选采用甲酯/乙酰胺、乙酯/甲酰胺的组合等来充分设置质量差,或通过稳定同位素标记来扩大质量差。
通过酯化反应形成的酯的修饰体与通过酰胺化反应形成的酰胺的修饰体的组合可以根据所用的试样而适宜变更。例如,唾液酸中,除了代表性的Neu5Ac以外,还存在比它大16Da的Neu5Gc。这些唾液酸可以混合存在,因此例如二唾液酸双链糖链的情况下,作为唾液酸的组合存在Neu5Ac x2、Neu5Ac+Neu5Gc、Neu5Gc x2这三种(以下,x2表示包含2个相同唾液酸)。对它们混合存在的试样进行甲酯化和乙酰胺化时,可检测到以下的表1所示的9种峰。(以下的表1和表2中,示出以Neu5Ac x2、α2,6-x2的情况下的质量为基准的质量差)
表1
此处,+13与+16、+26与+29、+29与+32、+42与+45的质量相近,也考虑同位素分布时,峰部分重复地被检测到,因此存在无法正确地进行数据解析的可能性。这种情况下,通过将丙胺作为酰胺化反应剂来进行甲酯化后的酰胺化,能够防止峰的重复,正确地进行数据解析。对于Neu5Ac x2、Neu5Ac+Neu5Gc、Neu5Gc x2这三种混合存在的试样进行甲酯化和丙酰胺化时,可检测到以下的表2所示的9种峰。
表2
此时,峰适宜地分离。对包含3个以上的唾液酸的糖链也同样。如此,根据试样所包含的糖链的复杂性,可以适宜选择酯化与酰胺化的组合。
对于通过本实施方式的分析用试样的制备方法所得到的分析用试样,通过色谱法进行分析的情况下,选择酯化反应溶液所包含的醇或酯化剂、和酰胺化反应剂,使得与色谱法的分辨率相应地对所得到的2种修饰体精度良好地进行分离。
(酰胺化反应中的胺)
以下的实施方式中,“胺”的术语包括肼、肼衍生物和羟胺,不包括氨和氨的盐。酰胺化反应中使用胺的情况下,酰胺化反应剂所包含的胺优选为选自伯胺、肼、肼衍生物和羟胺以及它们的盐中的至少一种化合物。α2,3-唾液酸等的羧基与α2,6-唾液酸的羧基相比处于位阻较大的位置,因此认为伯胺与其它胺相比容易选择性地与α2,3-唾液酸等反应。
作为酰胺化反应剂使用伯胺的情况下,更优选在键合于氨基的碳原子上直接键合有1个以下的碳原子的伯胺。这是因为,此时,即使在碳链上具有支链,若在距氨基较远的位置存在支链,则也能抑制酰胺化反应的效率降低。
酰胺化反应剂更优选具有直链烃基的伯胺,进一步优选具有直链烷基的伯胺。关于酰胺化反应剂,作为具有直链烷基的伯胺,优选碳数为10以下的伯胺,进一步优选碳数为6以下的伯胺、即甲胺、乙胺、丙胺、丁胺、戊胺和己胺,最优选甲胺。酰胺化反应溶液所包含的胺具备不具有支链(以下“支链”表示烃链的支链)的直链状的结构、或碳数少时,更高效地将酯化的唾液酸酰胺化,故而优选。
酰胺化反应剂所包含的肼衍生物没有特别限定。以下的实施方式中,乙酰肼、乙酸酰肼、苯并酰肼和苯甲酸酰肼等酰肼也包括在肼衍生物中,可以用作酰胺化反应剂。酰胺化反应剂所包含的肼衍生物可以采用选自由甲基肼、乙基肼、丙基肼、丁基肼、苯基肼和苄基肼、以及乙酰肼、乙酸酰肼、苯并酰肼和苯甲酸酰肼组成的组中的至少一种化合物。从提高或维持酰胺化反应的效率的观点出发,作为酰胺化反应剂的肼或其衍生物优选肼或甲基肼。
酰胺化反应剂为具有不饱和链式烃基的伯胺的情况下,该不饱和链式烃基优选包含双键,该不饱和链式烃基更优选包含烯丙基(Allyl),该胺进一步优选烯丙基胺(Allylamine)。酰胺化反应剂可以为包含羟基的伯胺,此时优选乙醇胺。酰胺化反应溶液所包含的胺也可以包含除烷基以外的各种官能团。作为酰胺化反应的结果,糖链以包含这种官能团的方式进行修饰,从而受到该修饰的糖链不仅变得更容易通过质谱分析进行分离,而且变得更容易通过色谱法等分离。
酰胺化反应剂可以包含氨、或作为酰胺化反应剂在上文中说明的胺的盐。作为酰胺化反应剂所包含的氨或胺的盐,可列举出氨或胺的无机酸盐或有机酸盐,优选碳酸盐、盐酸盐、硝酸盐、硫酸盐、磷酸盐或甲磺酸盐那样的无机盐,更优选碳酸盐、盐酸盐和硝酸盐,进一步优选盐酸盐。作为具有直链烃基的伯胺的盐酸盐,更优选甲胺盐酸盐、乙胺盐酸盐、丙胺盐酸盐、丁胺盐酸盐、戊胺盐酸盐,进一步优选甲胺盐酸盐或乙胺盐酸盐。
(酰胺化反应溶液的浓度)
酰胺化反应溶液中的、甲胺或乙胺等的酰胺化反应剂的浓度以重量/体积%计优选0.01%以上、更优选0.1%以上、进一步优选1%以上。酰胺化反应溶液中的酰胺化反应剂的浓度越高,越能够可靠地进行酯化的唾液酸的酰胺化。酰胺化反应溶液中的、甲胺或乙胺等的酰胺化反应剂的浓度以重量/体积%计优选不足40%、更优选不足20%。由此,能够抑制由高浓度导致的不希望的反应,或抑制将α2.6-唾液酸酰胺化,从而使连接方式特异性的修饰变得容易。
(酰胺化反应溶液的溶剂)
从可靠地发生酰胺化的观点出发,酰胺化反应溶液的溶剂优选水系溶剂、有机溶剂或水系溶剂与有机溶剂的混合溶剂。酰胺化反应溶液的溶剂例如可以采用水、甲醇或乙醇等醇、二甲基亚砜(DMSO)或乙腈或它们的混合液。连接方式特异性地修饰α2,6-唾液酸和其它唾液酸的情况下,从抑制α2,6-唾液酸的酰胺化的观点出发,酰胺化反应溶液的溶剂优选包含有机溶剂。
(酰胺化反应溶液的pH)
酰胺化反应溶液的pH为7.7以上。酰胺化反应溶液的pH优选8.0以上、更优选8.8以上、进一步优选10.3以上。酰胺化反应溶液的pH升高时,抑制水解等副反应,或通过使用各种酰胺化反应剂来更可靠地将酯化的唾液酸酰胺化,故而优选。
(用于发生酰胺化反应的时间)
酰胺化反应在数秒~数分钟以内完成。因此,为了通过酰胺化反应而将进行了酯化的唾液酸酰胺化,使试样与酰胺化反应溶液接触的时间(以下称为反应时间)优选不足1小时、更优选不足30分钟、进一步优选不足15分钟、进一步优选不足5分钟、最优选不足1分钟。可以适宜地仅将试样用酰胺化反应溶液清洗,或对保持于载体等的试样暂时通液。试样与酰胺化反应溶液接触的时间没有特别限定,从使反应充分完成等的观点出发,可以适宜设为0.1秒以上或1秒以上等。另外,也可以将试样与酰胺化反应溶液混合并直接干固,而不设置反应时间。通过较短地设置酰胺化反应的反应时间,能够更高效地进行试样的解析。
(进行酰胺化反应的相)
只要能够使试样与酰胺化反应溶液接触,则发生酰胺化反应时的试样的状态没有特别限定,可以为固相也可以为液相。在液相中进行酰胺化反应的情况下,可以如上所述在残留酯化反应后的溶液的状态下在试样中添加酰胺化反应溶液,也可以在酯化反应后进行纯化、脱盐、增溶等公知的前处理。在固定于固相的状态下进行酰胺化反应的情况下,可以将在固相中供于酯化反应的试样维持固定于固相的状态并进行酰胺化反应。另外,也可以在将试样供于酯化反应后,固定于固相并进行酰胺化反应。
在固相中进行酰胺化反应的情况下,作为固相载体,可以使用与关于酯化反应在上文中说明的固相载体同样的固相载体。关于试样向固相载体上的固定,可以使用关于酯化反应在上文中说明的条件。在固相中进行酰胺化反应的情况下,使固定于固相载体的试样与酰胺化反应溶液发生作用而进行酰胺化后,通过化学方法或酶反应等使试样从载体游离并回收即可。例如,可以利用PNGase F等糖苷酶或胰蛋白酶等消化酶将固定于载体的糖蛋白、糖肽进行酶切断并回收,可以利用弱酸性溶液使键合于具有酰肼基的固相载体的糖链游离并回收。HILIC中,可以利用以乙腈等作为溶剂的酰胺化反应溶液进行酰胺化反应,用水等水系溶液将试样洗脱。
步骤S1005结束后,开始步骤S1007。步骤S1007中,制备分析用试样。分析用试样的制备的方法只要得到包含供于步骤S1005的酰胺化反应的糖链、且用于进行在步骤S1009中进行的分析的分析用试样,就没有特别限定。
例如,在步骤S1009中进行使用基质辅助激光解吸电离(Matrix Assisted LaserDesorption/Ionization;MALDI)的质谱分析的情况下,例如如以下那样制备分析用试样。将包含酰胺化反应后得到的试样的溶液滴加于MALDI用样品板,进而在板上的该溶液中滴入添加包含基质的溶液,使其干燥。由此,能够得到试样与基质的混合晶体作为质谱分析用试样。该质谱分析用试样的制备中,可以在将包含试样的溶液与包含基质的溶液混合后添加到MALDI用样品板中,也可以改变滴加这些溶液的顺序,还可以进一步使用基质的添加剂。进行色谱法等其它分析的情况下,也通过公知的方法等制备分析用试样。
通过上述制备方法所得到的分析用试样中,α2,6-唾液酸通过酯化反应而形成有酯化修饰。α2,3-、α2,8-和α2,9-唾液酸、特别是α2,3-和α2,8-唾液酸通过酯化反应而形成酯化修饰后,通过酰胺化反应将酯化修饰转化为酰胺化修饰。
步骤S1007结束后,开始步骤S1009。
步骤S1009中,对分析用试样进行分析。分析用试样优选通过质谱分析和色谱法中的至少一者进行分析。
通过上述酯化反应和酰胺化反应,通过各反应受到修饰的糖链彼此质量不同。因此,能够通过质谱分析将这些糖链根据唾液酸的连接方式进行分离。
质谱分析中的电离的方法没有特别限定,可以使用MALDI、电喷雾(Electrosprayionization;ESI)法、纳米电喷雾电离(nano-ESI)法等。电离的方法特别优选MALDI。质谱分析中的电离中,可以使用正离子模式和负离子模式中的任一者。质谱分析可以是单次质谱分析,也可以分多个阶段来进行,多阶段的质谱分析中,可以适宜解析除唾液酸的连接方式以外的糖链的结构或肽链的结构。作为质谱仪,可以组合使用至少一个以上的四极杆型、离子阱型和飞行时间型等任意的质谱仪。也可以适宜进行离子的离解或者原子或原子团向离子的加合等。
通过色谱法进行分析的情况下,优选液相色谱法。用于液相色谱法的柱没有特别限定,可以适宜使用C30、C18、C8、C4等疏水性反相柱或碳柱、HILIC用的正相柱等。通过液相色谱-质谱联用(Liquid Chromatography/Mass spectrometry;LC/MS)进行测定时,在通过多次的分离来精密地进行试样中的成分的分析的方面是优选的。
解析通过质谱分析或色谱法得到的数据,进行试样所包含的糖链中的唾液酸的解析等。该数据解析中,可以进行包括唾液酸的连接方式的糖链的结构的推定等。通过质谱分析或色谱法所得到的数据的解析方法没有特别限定。
步骤S1009结束后,处理结束。
(关于糖肽和糖蛋白的副反应的抑制)
在糖肽或糖蛋白中添加酯化反应溶液和酰胺化反应溶液,如上所述修饰唾液酸的情况下,有时在位于糖肽或糖蛋白所包含的氨基酸的侧链、主链末端的氨基和羧基之间发生分子内脱水缩合等副反应。此时,在唾液酸修饰前预先通过化学修饰等封闭氨基,从而能够在唾液酸修饰时抑制肽部分的副反应。详情参照以下的文献:Takashi Nishikaze,Sadanori Sekiya,Shinichi Iwamoto,Koichi Tanaka.“A Universal Approach tolinkage-Specific Derivatization for Sialic Acids on Glycopeptides,”Journal ofThe American Society for Mass Spectrometry,2017年6月,Volume 28,Issue1Supplement,海报编号MP091。例如,可以对于糖肽或糖蛋白进行二甲基标记化或胍基化等封闭氨基的反应,然后进行酯化反应和酰胺化反应。
(关于分析用试样的制备用试剂盒)
提供本实施方式的分析用试样的制备方法中适宜使用的分析用试样的制备用试剂盒(以下称为制备用试剂盒)。
图2为示出本实施方式的制备用试剂盒100的概念图。图2的例子中,制备用试剂盒100包含酯化反应中的酯化剂10。酯化剂10容纳于容器11。容器11的容量和形状等没有特别限定。制备用试剂盒100可以包含试剂、或者除试剂以外的上述分析所使用的任意的消耗品、或记载了用于制备本实施方式中的分析用试样的规程或者记载有该规程的Web站点的URL等的文件。例如,制备用试剂盒100可以包含:醇、缩合剂、添加剂或酯化剂等酯化反应溶液中所含的试剂;以及与通过酯化而进行了修饰的唾液酸发生反应的选自由氨、胺、肼、肼衍生物和羟胺以及它们的盐组成的组中的至少一种化合物。通过使用制备用试剂盒100制备分析用试样,能够更高效地制备分析用试样。
如下那样的变形也在本发明的范围内,可以与上述实施方式组合。以下的变形例中,关于表现与上述实施方式同样的结构、功能的部位,用同一符号参照,适宜地省略说明。
(变形例1)
上述实施方式中,除了通过酰胺化反应得到的试样的分析之外,还可以进行通过酯化反应得到的试样的分析。将通过酰胺化反应得到的试样的分析称为第一分析,将通过酯化反应得到的试样的分析称为第二分析。
图3为示出本变形例的分析用试样的制备方法的分析方法的流程的流程图。步骤S2001和S2003分别与上述实施方式中的步骤S1001和S1003同样,因此省略说明。步骤S2003结束后,开始步骤S2005。
步骤S2005中,使试样的一部分与酰胺化反应溶液接触,进行酰胺化反应。可以通过分注等将包含步骤S2003中得到的试样的溶液分开,对于其一部分的溶液,使用与上述实施方式同样的酰胺化反应溶液在同样的条件下进行酰胺化反应。步骤S2005结束后,开始步骤S2007。
步骤S2007中,对于步骤S2003中得到的试样和步骤S2005中得到的试样,制备分析用试样。将使用供于酰胺化反应后的试样而制备的用于第一分析的分析用试样设为第一分析用试样,将使用供于酯化反应后的试样而制备的用于第二分析的分析用试样设为第二分析用试样。第一分析用试样和第二分析用试样与上述实施方式的分析用试样的制备方法同样通过公知的方法等而制备。步骤S2007结束后,开始步骤S2009。
步骤S2009中,对分析用试样进行分析。进行第一分析用试样的第一分析和第二分析用试样的第二分析。第一分析与第二分析优选通过相同的分析法来进行。例如,第一分析和第二分析都通过质谱分析来进行,或都通过色谱法来进行,或都通过LC/MS来进行。由此,能够根据第一分析所得到的数据与第二分析所得到的数据的比较来进行数据解析。例如通过质谱分析进行第一分析和第二分析的情况下,第二分析所得到的质谱图中的包含α2,3-唾液酸的糖链的峰在第一分析所得到的质谱图中偏移了与自酯化修饰向酰胺化修饰转化时的质量变化对应的m/z的量。如此,根据关于第一分析所得到的数据与第二分析所得到的数据的差异、以及伴随自酯化向酰胺化的转化的质量变化量的信息,能够进行鉴定峰等更详细的糖链的解析。步骤S2009结束后,处理结束。
(变形例2)
上述实施方式中,说明了将酯化的唾液酸通过酰胺化反应而酰胺化,连接方式特异性地进行唾液酸的修饰的例子。但是,也可以不特别以连接方式特异性的修饰为目的,而在将唾液酸酰胺化时使用上述酰胺化反应。
(变形例3)
发明人等出人意料地发现,在上述酰胺化反应中,根据酰胺化反应溶液的溶剂的组成,可以结构特异性地进行糖链中的唾液酸的酰胺化。这是指,在例如上述图1的流程图的步骤S1005或图3的流程图的步骤S2005中进行了酰胺化反应时,对于相同或不同连接方式的唾液酸,根据糖链中的唾液酸的位置或唾液酸附近的糖链的结构,将唾液酸选择性地酰胺化。此处,“唾液酸附近的糖链的结构”是指,关于唾液酸所直接键合的单糖与其它糖的键合的结构。
本变形例的酰胺化反应中,酰胺化反应溶液优选包含有机溶剂、更优选包含乙腈。酰胺化反应溶液的溶剂中的乙腈的浓度适宜调整,使得如上所述将唾液酸选择性地酰胺化。酰胺化反应溶液的溶剂更优选以重量/体积%计包含90%以上乙腈,进一步优选包含95%以上,更进一步优选包含98%以上。最优选酰胺化反应溶液的溶剂为乙腈,即实质上仅由乙腈组成。由此,能够将除α2,6-唾液酸以外的唾液酸、特别是α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸根据糖链的结构而选择性地酰胺化。尤其,可将在糖链的末端、特别是非还原末端存在的α2,3-唾液酸和α2,8-唾液酸选择性地酰胺化。关于本变形例的酰胺化反应中的其它条件,可以从与上述实施方式同样的条件中适宜选择。
更具体而言,使用包含有机溶剂、特别是乙腈的酰胺化反应溶液,将包含糖链的试样供于酰胺化反应,从而将以下的条件的唾液酸选择性地酰胺化。该条件是指,在除α2,6-唾液酸以外的唾液酸、特别是α2,3-或α2,8-唾液酸所直接键合的单糖的4位键合有另外的单糖。可以进行调整,使得不满足该条件的唾液酸不会通过包含乙腈等有机溶剂的酰胺化反应溶液而发生酰胺化。可以认为上述条件基于如下原理:唾液酸的内酯结构形成包含与该唾液酸相邻的单糖的4位的环状结构,因此,作为酰胺化反应的中间体等,该内酯结构产生影响。
图4A、4B和4C为示出本变形例的酰胺化反应中的选择性的一例的概念图。图4A和4B分别示出作为鞘糖脂的人双唾液酸神经节苷脂(Human Disialoganglioside)GD1a和GD1b的结构。图4A、4B和4C中,将发生酰胺化的可能性高的唾液酸用Neu5Ac(高)的符号表示,将发生酰胺化的可能性低的唾液酸用Neu5Ac(低)的符号表示,将发生酰胺化的可能性极低的唾液酸用Neu5Ac(极低)的符号表示。发生酰胺化的可能性高的唾液酸通过包含乙腈等有机溶剂或水等水系溶剂的酰胺化反应溶液而发生酰胺化。发生酰胺化的可能性低的氨基酸不会因包含乙腈等有机溶剂的酰胺化反应溶液而发生酰胺化,但会通过包含水等水系溶剂的酰胺化反应溶液而发生酰胺化。对于发生酰胺化的可能性极低的唾液酸,除非将酰胺化反应剂的浓度设定得高于将其它唾液酸酰胺化时的浓度,否则难以酰胺化。
GD1a糖链(图4A)包含:由葡萄糖(Glc)、2个半乳糖(Gal)和N-乙酰半乳糖胺(GalNAc)形成的直链、以及这2个Gal上分别键合的α2,3-唾液酸。对于连接方式为α2,3-,通过在唾液酸所键合的Gal的左下方记载该唾液酸而示意性示出(以下的各图中也同样)。本变形例的酰胺化反应中,在GD1a糖链中,在4位键合有GalNAc的Gal上键合的α2,3-唾液酸(Neu5Ac(高))选择性地发生酰胺化。此外,可以使未在4位键合单糖的Gal上键合的α2,3-唾液酸(Neu5Ac(低))不发生酰胺化。
GD1b糖链包含:由Glc、2个Gal和GalNAc形成的直链;在键合于Glc的Gal上键合的α2,3-唾液酸;以及键合于该α2,3-唾液酸的α2,8-唾液酸。对于连接方式为α2,8-,通过在唾液酸所键合的单糖的正下方记载该唾液酸而示意性示出(以下的各图也同样)。GD1a糖链与GD1b糖链为异构体的关系。本变形例的酰胺化反应中,GD1b糖链(图4B)中,在4位键合有Gal的Neu5Ac上键合的α2,3-唾液酸(Neu5Ac(高))选择性地发生酰胺化。此外,可以使未在4位键合单糖的Gal上键合的α2,3-唾液酸(Neu5Ac(低))不发生酰胺化。
图4C为示出A2GN1糖链的结构的概念图。A2GN1糖链具备:由N-乙酰基-D-葡萄糖胺(GlcNAc)和甘露糖(Man)形成的基本型的结构、以及2个侧链。2个侧链上分别键合有GlcNAc、半乳糖(Gal)和唾液酸(Neu5Ac)。对于连接方式为α2,6-,通过在唾液酸所键合的Gal的左上方记载该唾液酸而示意性示出(以下的各图也同样)。α2,6-唾液酸发生酰胺化的可能性极低,不容易通过酰胺化反应而酰胺化。
需要说明的是,本变形例除了图4A、4B和4C所示的糖链以外,也能够适用于包含糖脂型糖链和N型糖链等的各种糖链。
(变形例4)
上述变形例3中,也可以连接方式特异性且结构特异性地修饰氨基酸。本变形例中,α2,6-唾液酸通过酯化反应而酯化。除α2,6-唾液酸以外的唾液酸之中满足变形例3中记载的条件的唾液酸通过第一阶段的酰胺化而进行修饰。将发生该第一阶段的酰胺化的反应称为第一酰胺化反应,将第一酰胺化反应中的酰胺化反应溶液和酰胺化反应剂分别称为第一酰胺化反应溶液和第一酰胺化反应剂。除α2,6-唾液酸以外的唾液酸之中不满足变形例3中记载的条件的唾液酸通过第二阶段的酰胺化而进行修饰。将发生该第二阶段的酰胺化的反应称为第二酰胺化反应,将第二酰胺化反应中的酰胺化反应溶液和酰胺化反应剂分别称为第二酰胺化反应溶液和第二酰胺化反应剂。第一酰胺化反应剂和第二酰胺化反应剂以通过它们键合于唾液酸而生成的修饰体可利用质谱分析等分析进行区别并检测的方式来选择。
图5为示出本变形例的分析方法的流程的流程图。步骤S3001和步骤S3003与图1的流程图的步骤S1001和S1003同样,因此省略说明。通过酯化反应,至少α2,6-唾液酸发生酯化,其它唾液酸发生酯化(或内酯化)。步骤S3003结束后,开始步骤S3005。
步骤S3005中,使试样与第一酰胺化反应溶液接触,进行第一酰胺化反应。第一酰胺化反应溶液的溶剂优选包含乙腈等有机溶剂,特别优选实质上仅由乙腈等有机溶剂组成。第一酰胺化反应的结束后,优选从试样去除第一酰胺化反应溶液,其他与上述酯化反应后同样,可以适宜进行纯化、脱盐、增溶、糖链向固相载体的结合、或糖链自固相载体的游离等。通过第一酰胺化反应,α2,3-、α2,8-和α2,9-唾液酸、特别是α2,3-和α2,8-唾液酸之中,在非还原末端等存在的满足上述变形例3的条件的唾液酸通过第一酰胺化反应剂而进行修饰。步骤S3005结束后,开始步骤S3007。
步骤S3007中,使试样与第二酰胺化反应溶液接触,进行第二酰胺化反应。第二酰胺化反应溶液的溶剂包含水等水系溶剂。通过第二酰胺化反应,α2,3-、α2,8-和α2,9-唾液酸、特别是α2,3-和α2,8-唾液酸之中,满足上述变形例3的条件的唾液酸通过第二酰胺化反应剂而进行修饰。步骤S3007结束后,开始步骤S3009。步骤S3009和步骤S3011与上述图1的流程图的步骤S1007和S1009同样,因此省略说明。
通过本变形例的分析方法,能够连接方式特异性地将α2,6-唾液酸与除其以外的唾液酸进行区别并检测。除此之外,能够将α2,3-、α2,8-和α2,9-唾液酸根据该唾液酸存在于非还原末端等糖链的结构而进行区别并检测。
(方式)
本领域技术人员理解上述多个示例性的实施方式或其变形为以下的方式的具体例。
(第1项)一个方式的分析用试样的制备方法,其为由包含糖链的试样制备分析用试样的方法,所述制备方法具备如下操作:进行酯化反应,所述酯化反应将前述糖链所包含的唾液酸的至少一部分供于除内酯化以外的酯化;使前述试样与酰胺化反应溶液接触,进行酰胺化反应,所述酰胺化反应溶液包含与通过前述酯化而进行了修饰的唾液酸发生反应的选自由氨、胺、肼、肼衍生物和羟胺以及它们的盐组成的组中的至少一种化合物,所述酰胺化反应将通过前述酯化而进行了修饰的唾液酸的酯化修饰转化为酰胺化修饰。由此,为了分析糖链所包含的唾液酸,能够通过新机理修饰唾液酸。
(第2项)另一个方式的分析用试样的制备方法中,第1项的方式的分析用试样的制备方法中,具备通过使前述试样与酯化反应溶液接触而进行前述酯化反应的操作,前述酯化反应溶液包含醇和酯化剂中的至少一者。由此,能够将糖链所包含的唾液酸更可靠地酯化。
(第3项)另一个方式的分析用试样的制备方法中,第2项的方式的分析用试样的制备方法中,前述酯化反应溶液包含酯化剂,前述酯化剂为三氮烯衍生物。由此,能够将糖链所包含的唾液酸效率良好地酯化。
(第4项)另一个方式的分析用试样的制备方法中,第2项或第3项的方式的分析用试样的制备方法中,前述酰胺化反应仅通过在用于从前述试样去除前述酯化反应溶液的操作后使前述试样与前述酰胺化反应溶液接触来进行。由此,能够简便地进行分析用试样的制备。
(第5项)另一个方式的分析用试样的制备方法中,第2项~第4项中任一方式的分析用试样的制备方法中,前述酰胺化反应溶液不包含脱水缩合剂。由此,能够更简便地制备酰胺化反应溶液。
(第6项)另一个方式的分析用试样的制备方法中,第1项~第5项中任一方式的分析用试样的制备方法中,在使前述试样与前述酰胺化反应溶液接触后,不进行使前述试样与脱水缩合剂反应的操作。由此,能够减少分析用试样的制备时的工序,能够更简便地进行该制备。
(第7项)另一个方式的分析用试样的制备方法中,第1项~第6项中任一方式的分析用试样的制备方法中,为了进行前述酰胺化反应而使前述试样与前述酰胺化反应溶液接触的时间短于30分钟。由此,能够在更短时间内高效地制备分析用试样。
(第8项)另一个方式的分析用试样的制备方法中,第1项~第7项中任一方式的分析用试样的制备方法中,在前述酰胺化反应前,不进行用于使通过前述酯化反应而生成的内酯结构裂解的操作。由此,能够将容易形成内酯的α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸等更可靠地酰胺化。
(第9项)另一个方式的分析用试样的制备方法中,第1项~第8项中任一方式的分析用试样的制备方法中,前述化合物为伯胺。由此,能够根据上述化合物的反应性,更可靠地修饰唾液酸。
(第10项)另一个方式的分析用试样的制备方法中,第9项的方式的分析用试样的制备方法中,与前述伯胺的氨基键合的碳原子上直接键合有1个以下的碳原子。由此,能够根据上述化合物的反应性,进一步可靠地修饰唾液酸。
(第11项)另一个方式的分析用试样的制备方法中,第1项~第10项中任一方式的分析用试样的制备方法中,前述化合物包含烷基。由此,能够根据上述化合物的反应性,更加可靠地修饰唾液酸。
(第12项)另一个方式的分析用试样的制备方法中,第1项~第10项中任一方式的分析用试样的制备方法中,前述酰胺化反应溶液的pH为7.7以上。由此,在碱性条件下酰胺化反应变得容易发生,能够更可靠地修饰唾液酸。
(第13项)另一个方式的分析用试样的制备方法中,第2项~第5项中任一方式的分析用试样的制备方法中,在使前述试样与前述酯化反应溶液接触时,将未通过前述酯化而修饰的唾液酸的至少一部分内酯化,在使前述试样与前述酰胺化反应溶液接触时,将通过前述内酯化而进行了修饰的唾液酸的内酯结构转化为酰胺化修饰。由此,通过酯化反应发生了内酯化的情况下,也能够与发生了酯化的情况同样地将唾液酸酰胺化。
(第14项)另一个方式的分析用试样的制备方法中,第1项~第13项中任一方式的分析用试样的制备方法中,在前述酰胺化反应中,将选自由α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸组成的组中的至少一种唾液酸酰胺化。由此,能够通过新机理将α2,3-唾液酸、α2,8-唾液酸或α2,9-唾液酸酰胺化。
(第15项)另一个方式的分析用试样的制备方法中,第1项~第14项中任一方式的分析用试样的制备方法中,在前述酰胺化反应中,调节前述酰胺化反应溶液中的前述化合物的浓度,使得不会发生通过前述酯化而进行了修饰的α2,6-唾液酸的酯化修饰向前述酰胺化修饰的转化。由此,能够将α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸与α2,6-唾液酸进行区别并解析。
(第16项)另一个方式的分析用试样的制备方法中,第1项~第15项中任一方式的分析用试样的制备方法中,前述酯化反应和前述酰胺化反应的至少一者在前述试样结合或吸附于固相载体的状态下进行。由此,纯化等操作变得简易,能够效率良好地制备分析用试样。
(第17项)另一个方式的分析用试样的制备方法中,第1项~第16项中任一方式的分析用试样的制备方法中,前述酰胺化反应溶液的溶剂包含有机溶剂。由此,能够抑制α2,6-唾液酸的酰胺化,精度更良好地进行连接方式特异性的解析。另外,能够进行结构特异性的唾液酸的酰胺化。
(第18项)另一个方式的分析用试样的制备方法中,第17项的方式的分析用试样的制备方法中,前述有机溶剂为乙腈。由此,能够更可靠地进行结构特异性的唾液酸的酰胺化。
(第19项)另一个方式的分析用试样的制备方法中,第17项或第18项的方式的分析用试样的制备方法中,在前述酰胺化反应中,将选自由α2,3-唾液酸、α2,8-唾液酸和α2,9-唾液酸组成的组中的至少一种唾液酸根据前述至少一种唾液酸在前述糖链中的位置或前述糖链的结构而酰胺化。由此,能够得到关于唾液酸的糖链中的位置或糖链的结构的信息,或进行利用它们的分析。
(第20项)另一个方式的分析用试样的制备方法中,第19项的方式的分析用试样的制备方法中,其中,还具备如下操作:在使用包含前述有机溶剂的酰胺化反应溶液进行前述酰胺化反应后,使包含水系溶剂的酰胺化反应溶液与供于前述酰胺化反应的前述试样接触,进行在前述酰胺化反应中未酰胺化的唾液酸的酰胺化。由此,能够将连接方式或糖链中的位置不同的3种以上的唾液酸进行区别并检测。
(第21项)另一个方式的分析用试样的制备方法中,第1项~第20项中任一方式的分析用试样的制备方法中,具备:由供于前述酰胺化反应后的前述试样制备第一分析用试样;以及,由供于前述酯化反应后且供于前述酰胺化反应前的前述试样制备第二分析用试样。由此,通过第一分析用试样的分析与第二分析用试样的分析的比较,能够进行更详细的糖链的解析。
(第22项)一个方式的分析方法具备如下操作:通过第1项~第20项中任一方式的分析用试样的制备方法制备试样、以及对制备的前述试样进行分析。由此,能够利用基于新机理的修饰,对糖链所包含的唾液酸进行解析。
(第23项)另一个方式的分析方法具备如下操作:通过第21项的方式的分析用试样的制备方法制备前述第一分析用试样和前述第二分析用试样;进行所制备的前述第一分析用试样和前述第二分析用试样的分析;以及,根据前述第一分析用试样的前述分析所得到的数据与前述第二分析用试样的前述分析所得到的数据的差异,进行前述试样所包含的糖链的解析。由此,通过第一分析用试样的分析与第二分析用试样的分析的比较,能够进行更详细的糖链的解析。
(第24项)另一个方式的分析用试样的制备方法中,第22项或第23项的方式的分析用试样的制备方法中,前述分析通过质谱分析和色谱法中的至少一者进行。由此,即使试样中包含各种各样的物质,也能够将它们分离并解析。
(第25项)一个方式的分析用试样的制备用试剂盒用于第1项~第20项中任一方式的分析用试样的制备方法。由此,能够效率良好地制备分析用试样。
本发明不限定于上述实施方式的内容。在本发明的技术构思范围内想出的其它方式也包括在本发明的范围内。
实施例
以下示出本实施方式的实施例,但本发明不限定于下述实施例。需要说明的是,以下,%的记载在没有特别提及的情况下是指体积/体积%。
(实施例1~4)
实施例1~4中,在将N型糖链键合于载体的状态下进行酯化反应和酰胺化反应,将所得到的糖链试样通过质谱分析进行分析。
<包含α2,3-唾液酸的评价用糖链试样的制作>
按照编号顺序进行以下的1-3的步骤、制作包含2个α2,3-唾液酸的糖链A2glycan(33A2)。使用PNGase F,使A2 glycan(33A2)自α2,3-唾液酸糖肽(α2,3-Sialylglycopeptide(SGP)、伏见制药所)游离。游离的糖链用Stage Tip Carbon脱盐处理。Stage Tip Carbon为将Empore Disc Carbon(3M制)切出直径约1mm,装入到200μL的吸头(tip)而成的碳柱。
1.对于分注有以1nmol/μL的浓度包含α2,3-SGP的溶液20μL的管,添加10μL的PNGase F(SIGMA)0.25U/μL(2.5U/管)。
2.轻轻进行敲打和离心,在37℃下进行过夜(o/n)的孵育。
3.第二天,使用Stage Tip Carbon将糖链脱盐。
<包含α2,6-唾液酸的评价用糖链试样的制作>
将包含α2,6-唾液酸的A2GN1糖链(东京化成工业)再溶解于水,混合,制成评价用糖链试样。
<酯化反应和酰胺化反应>
按照编号顺序进行以下的1-8的步骤,将制作的上述评价用糖链试样供于酯化反应和酰胺化反应。酯化反应中使用1-甲基-3-对甲苯基三氮烯(MTT)和1-乙基-3-对甲苯基三氮烯(ETT)中的任一者,并且,酰胺化反应中的酰胺化反应剂在各实施例的结果处说明。
1.将制作的评价用糖链试样结合于酰肼珠(BlotGlyco;住友电木)。根据BlotGlyco的规程进行结合。
2.将使溶剂为DMSO且包含500mM的MTT或ETT的溶液加入到100μL珠中,在60℃下使其反应1小时(酯化反应)。
3.用甲醇200μL将珠清洗3次。
4.将酰胺化反应溶液100μL加入到珠中并搅拌后,通过离心去除酰胺化反应溶液。
5.用甲醇200μL将珠清洗3次。
6.使糖链试样从珠游离。根据Blotglyco的规程,进行游离。
7.将游离的糖链试样通过减压离心浓缩进行干固。
8.使用Stage Tip Carbon将糖链脱盐。
<质谱分析>
将脱盐并干固的糖链再溶解于20μL的水。将通过再溶解所得到的溶液0.5μL滴加到700μmμFocus plate(Hudson Surface Technology)。作为基质,添加包含溶解于50%乙腈(ACN)的5mg/mL 2,5-二羟基苯甲酸(DHB)的基质溶液(包含5mM的NaCl),在常温常压下风干后,添加乙醇0.2μL使其重结晶,得到质谱分析用试样。然后,使用MALDI-四极离子阱(Quadrupole Ion Trap)-飞行时间型(Time of Flight)质谱仪(MALDI-QIT-TOF-MS)(AXIMA-Resonance,Shimadzu/Kratos)以正离子模式进行测定。
<结果>
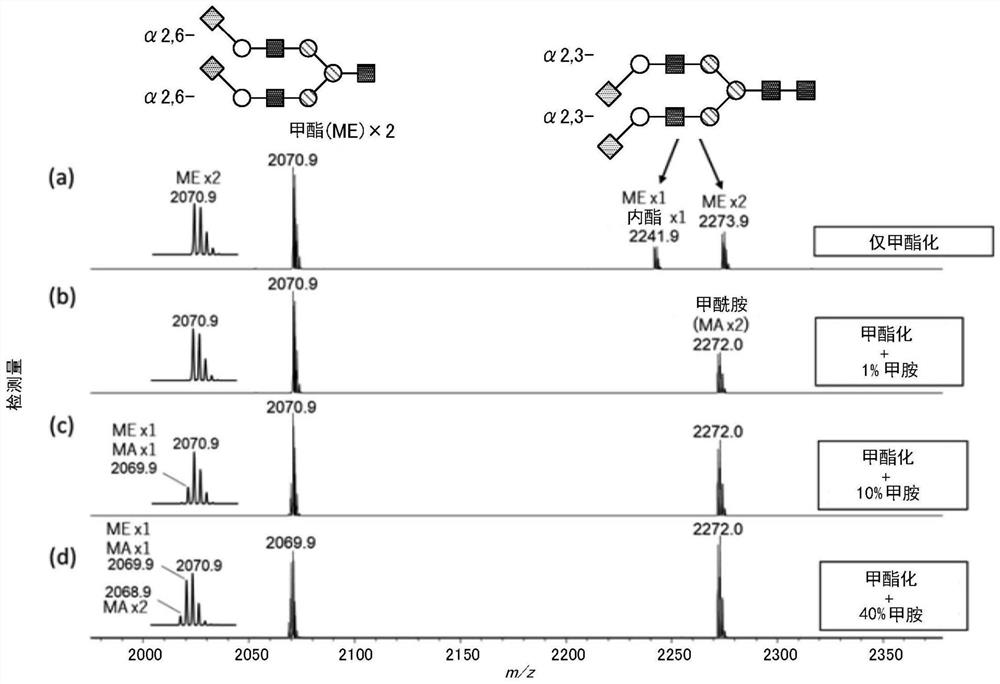
图6为示出本实施例中使用的包含α2,3-唾液酸的糖链试样(以下称为α2,3-糖链试样)的结构的概念图。α2,3-糖链试样具备由N-乙酰基-D-葡萄糖胺(GlcNAc)和甘露糖(Man)形成的基本型的结构、以及2个侧链。2个侧链上分别键合有GlcNAc、半乳糖(Gal)和唾液酸(Neu5Ac)。
本实施例中使用的包含α2,6-唾液酸的糖链试样(以下称为α2,6-糖链试样)为上述图4C所示的A2GN1糖链。α2,6-糖链试样与α2,3-糖链试样相比,除了唾液酸的连接方式之外,不同之处还在于少了一个GlcNAc。
(实施例1)
图7的质谱图(a)为在将糖链结合于固相载体的状态下对唾液酸进行甲酯化修饰反应后,使糖链游离而不进行酰胺化反应,从而得到的试样的质谱图。质谱图在横轴示出检测的离子的m/z、在纵轴示出检测的离子的检测信号的强度,以下的各图也同样。m/z2070.9的峰为糖链中的2处的唾液酸都进行了甲酯化的α2,6-糖链试样的峰。与此相对,关于α2,3-糖链试样,观测到2处的唾液酸进行了甲酯化的峰(m/z 2273.9)、以及2处的唾液酸之中的一者进行了甲酯化且另一者进行了内酯化的峰(m/z 2241.9)这两者。
图7的质谱图(b)、(c)和(d)为在将糖链结合于固相载体的状态下对唾液酸进行甲酯化修饰反应后,分别使用以重量/体积%计1%(b)、10%(c)和40%(d)甲胺溶液进行酰胺化反应而得到的试样的质谱图。供于甲酯化反应后,供于酰胺化反应的试样中,不仅进行了内酯化的α2,3-唾液酸转化为甲酰胺,而且进行了甲酯化的α2,3-唾液酸也转化为甲酰胺。令人吃惊地,虽然同为酯结构,但α2,6-唾液酸几乎不酰胺化,保持甲酯的状态不变。用10%以上的浓甲胺处理时,α2,6-唾液酸也稍微甲酰胺化,但其反应速度慢,与效率极良好地转化为酰胺的α2,3-唾液酸相对照,足以将它们彼此进行区别。本实施例显示出:利用上述实施方式中的酰胺化反应溶液能够将由α2,3-唾液酸形成的酯选择性地酰胺化,示出了本方法对于唾液酸的连接方式的识别是有效的。
(实施例2)
图8为示出在将糖链结合于固相载体的状态下对唾液酸进行甲酯化修饰反应后,分别使用以重量/体积%计5.6%氨水(a)、10%甲胺溶液(b)、17.5%乙胺溶液(c)、16.7%二甲胺溶液(d)和20%三甲胺溶液(e)进行酰胺化反应而得到的试样的质谱图的图。使用(a)的氨的情况下,未检测到图7的(a)所示的m/z2273.9、m/z 2241.9的峰,而是汇集成与糖链中的2个唾液酸进行了酰胺化的糖链对应的m/z 2243.9的峰。因此,可知进行了α2,3-唾液酸的结构特异性的酰胺化。(b)与图7同样。可知使用(c)的乙胺的情况下也没有问题,连接方式特异性地进行了乙酰胺化(m/z 2300.0)。另一方面,使用二甲胺(d)或三甲胺(e)的情况下,包含α2,3-唾液酸的糖链的峰消失。可以推测是因为,这些是难以发生内酯的氨解(参照非专利文献2)的胺,因此促进水解,结果恢复为原本的羧酸(-COOH)结构,电离效率降低,变得在正离子模式下检测不到。
(实施例3)
图9为在将糖链结合于固相载体的状态下对唾液酸进行甲酯化修饰反应后,分别使用以水(a)、甲醇(b)、二甲基亚砜(c)和乙腈(d)作为溶剂的5或6%(重量/体积%)的甲胺溶液进行酰胺化反应而得到的试样的质谱图的图。显示出:即使是有机溶剂也没有问题,对α2,3-唾液酸进行了特异性酰胺化。与使溶剂为水的情况相比,在有机溶剂条件下进行时,更抑制了α2,6-唾液酸酯的酰胺化。
(实施例4)
图10的上部(a)为使用MTT对唾液酸进行甲酯化修饰反应后,使用17.5%(重量/体积%)乙胺溶液进行乙酰胺化反应而得到的试样的质谱图。图10的下部(b)为示出使用ETT对唾液酸进行乙酯化修饰反应后,使用10%(重量/体积%)甲胺溶液进行甲酰胺化反应而得到的试样的质谱图的图。可知即使改变酯与胺的组合也没有问题,将酯化的α2,3-唾液酸选择性地转化为酰胺。
(实施例5)
实施例5中,在将糖脂型糖链结合于载体的状态下进行酯化反应和酰胺化反应,将所得到的糖链试样通过质谱分析进行分析。
<评价用糖链试样的制作>
作为试样,使用作为鞘糖脂的人双唾液酸神经节苷脂(HumanDisialoganglioside)GD1a和GD1b(HyTest)。在包含0.2%Tritonx100的50mM乙酸钠缓冲液(pH5.5)45μL中溶解上述糖脂,在60℃下放置20分钟后,添加Endoglycoceramidase I(参考以下的文献从放线菌纯化而得到;Ishibashi Y,Nakasone T,Kiyohara M,Horibata Y,Sakaguchi K,Hijikata A,Ichinose S,Omori A,Yasui Y,Imamura A,Ishida H,Kiso M,Okino N,and Ito M.“A novel endoglycoceramidase hydrolyzesoligogalactosylceramides to produce galactooligosaccharides and ceramides,”Journal of Biological Chemistry,2007年,Volume 282,pp.11386-11396)5μL,在37℃下进行16小时糖链脱离反应。
<酯化反应和酰胺化反应>
按照编号顺序进行以下的1-7的步骤,将制作的上述评价用糖链试样供于酯化反应和酰胺化反应。
1.将制作的评价用糖链试样结合于固相载体(BlotGlyco)。根据BlotGlyco的规程,进行结合。
2.在使溶剂为DMSO且包含500mM的MTT的溶液中添加珠,进行酯化反应。
3.用甲醇200μL将固相载体清洗3次。
4.用甲胺溶液200μL将固相载体清洗3次,从而进行酰胺化反应。
5.用水200μL将固相载体清洗3次。
6.根据BlotGlyco的规程,使糖链的还原末端与MALDI用高灵敏度标签aoWR反应。
7.使用HILIC板(Waters)从试样去除过量试剂。
<质谱分析>
以DHB作为基质,使用Ultraflex II TOF/TOF-MS(Bruker)以正离子模式进行测定。
<结果>
本实施例中使用的GD1a糖链和GD1b糖链的结构分别示于上述图4A和4B。
图11为示出在将GD1a糖链作为试样进行甲酯化修饰反应后,未进行酰胺化的试样(a)和使用10%(重量/体积%)甲胺溶液进行了酰胺化的试样(b)的质谱图,以及在将GD1b糖链作为试样进行甲酯化修饰反应后,未进行酰胺化的试样(c)和使用10%(重量/体积%)甲胺溶液进行了酰胺化的试样(d)的质谱图的图。
对于GD1a,进行利用MTT的甲酯化且未进行酰胺化时,观察到与2个α2,3-唾液酸进行了甲酯化的情况对应的峰(m/z 1748)和与其中一处进行了内酯化的情况对应的峰(m/z1716)。甲酯化后,进行利用甲胺溶液的酰胺化反应时,汇集成甲酰胺化体。
对于GD1b,进行利用MTT的甲酯化且未进行酰胺化时,主要观察到发生了甲酯化和内酯化的糖链的峰(m/z 1716)。甲酯化后,进行利用甲胺溶液的酰胺化反应时,汇集成甲酰胺化体。这显示出:上述实施方式中的唾液酸连接方式特异性的唾液酸的自酯向酰胺的转化(酯酰胺转化)不仅对α2,3-唾液酸有效,而且对α2,8-唾液酸也有效。
(实施例6)
上述实施例中,在结合于固相载体的状态下进行酰胺化反应。本实施例中,将源自标准血清的N型糖链作为试样,在吸附于固相载体的状态下进行酰胺化反应,将得到的糖链试样通过质谱分析进行分析。
<评价用糖链试样的制作>
将市售的血清还原烷基化后,进行胰蛋白酶消化和利用PNGase F的糖链的游离,制备N型糖链。
<酯化反应>
按照编号顺序进行以下的1-5的步骤,将源自血清中糖蛋白的N型糖链供于酯化反应,用aoWR标记还原末端。
1.将制备的N型糖链结合于珠(BlotGlyco)。结合根据BlotGlyco的规程来进行。
2.将溶解于DMSO的500mM的MTT溶液加入到100μL珠中,在60℃下使其反应1小时。
3.将珠用甲醇200μL清洗3次。
4.用aoWR试剂标记糖链,并回收。
5.使用HILIC板(Waters)去除aoWR试剂。
<酰胺化反应>
将上述酯化反应后的源自人血清糖蛋白的N型糖链用99%乙腈/1%乙酸稀释,制备成90%乙腈浓度。然后,将包含糖链试样的溶液应用于HILIC板(waters),通过从背面进行真空抽吸而使溶液在板的载体中通液,使糖链吸附到HILIC载体上。然后,为了进行酯酰胺转化,将包含5~40%(重量/体积%)的乙胺的乙腈作为酰胺化反应溶液添加到板上,通过从背面进行真空抽吸而使酰胺化反应溶液在板的载体中通液。然后,在板上添加95%乙腈/1%乙酸,同样从背面进行真空抽吸,从而清洗板载体。将该操作重复三次。最后将5%乙腈/1%乙酸添加到板上,从HILIC板将糖链洗脱。
<质谱分析>
将DHB作为基质,使用Ultraflex II TOF/TOF-MS(Bruker)以正离子模式进行测定。
<结果>
图12为示出本实施例中检测的源自人血清糖蛋白的N型糖链的结构的一例的概念图。图12所示的糖链试样具备由GlcNAc和Man形成的基本型的结构、以及3个侧链。3个侧链上分别键合有GlcNAc、Gal和唾液酸(Neu5Ac)。唾液酸的连接方式为α2,3-或α2,6-。后述的实施例7中也检测同样的糖链。
图13为示出本实施例中得到的质谱图中的、观察到图12所示的糖链的峰的部分的图。质谱图(a)为在进行甲酯化修饰反应后未进行酰胺化反应的试样的质谱图。质谱图(b)-(e)为在进行甲酯化修饰反应后,分别使用以重量/体积%计5%(b)、10%(c)、20%(d)和40%(e)乙胺溶液进行酰胺化反应而得到的试样的质谱图。
仅进行了甲酯化的谱(a)中,在m/z 3352.19观测到图12的三链三唾液酸糖链,在m/z 3498.32观测到在其上键合岩藻糖(用+Δ示意性示出岩藻糖向糖链上的键合)而成的岩藻糖基化三链三唾液酸糖链。这些由于全部的唾液酸都进行了甲酯化,因此无法进行α2,3-与α2,6-的区别。对于m/z 3352.19,也观测到+14Da的峰,但其相当于无差别地将羟基甲基化而得到的超甲基化峰。
与此相对,在HILIC板上使用乙胺进行了酯酰胺转化的情况下((b)-(e)),与其浓度相应地观察到峰的偏移。仅进行了甲酯化的三链三唾液酸糖链(m/z3352.19)为3个唾液酸都是α2,6-的糖链、与2个唾液酸是α2,6-且1个唾液酸是α2,3-的糖链的混合物,其存在比率约为1:9。通过酯酰胺转化而将α2,3-从甲酯转化为乙酰胺,因此质量增加了13Da的糖链的峰的强度增加。图13、图14和图15中,用箭头A13示意性示出与13Da对应的m/z的变化。另外,岩藻糖基化三链三唾液酸糖链以几乎100%的存在比率具有2个α2,6-、1个α2,3-,因此发生酯酰胺转化时,质量完全偏移13Da。分别还检测到+13Da的峰(m/z3379.44和3525.57),但这是α2,6-唾液酸酯也部分转化为乙酰胺而得到的超酰胺化峰。作为总结,可知图13(b-d)中,根据所使用的乙胺的浓度,α2,3-唾液酸特异性地发生了酯酰胺转化。该实验结果显示出:连接方式特异性酯酰胺转化也可在吸附于HILIC板的状态下发生。
图14为示出以仅酰胺化反应溶液的通液阶段不从背面进行真空抽吸而是自然落下的方式进行与图13同样的实验的情况下得到的试样的质谱图的图。由此,能够使反应时间从数秒增加至20~30分钟。由此,即使是低浓度的胺,也能够效率良好地发生酯酰胺转化。
(实施例7)
本实施例中,以源自标准血清的N型糖链作为试样,在液相中进行酰胺化反应,将得到的糖链试样通过质谱分析进行分析。评价用糖链试样的制作和酯化反应在与实施例6同样的条件下进行。
<酰胺化反应>
将通过上述方法制备的源自人血清糖蛋白的N型糖链分注于管内,用包含2.5~40%(重量/体积%)乙胺的乙腈溶液稀释至10倍,用涡旋混合器轻轻混合,从而进行酰胺化反应。然后,将包含试样的溶液应用于HILIC板(Waters),通过从背面进行真空抽吸,从而使该溶液在板的载体中通液,使糖链吸附到HILIC载体上。然后,将95%乙腈/1%乙酸加入到板上,同样从背面进行真空抽吸,从而清洗板载体。将该操作重复三次。最后将5%乙腈/1%乙酸添加到板上,将糖链从HILIC板洗脱。
<质谱分析>
将DHB作为基质,使用MALDI-QIT-TOF-MS(AXIMA-Resonance,Shimadzu/Kratos)以正离子模式进行测定。
图15为示出本实施例中得到的质谱图中的、观察到图12所示的糖链的峰的部分的图。质谱图(a)为在进行甲酯化修饰反应后未进行酰胺化反应的试样的质谱图。质谱图(b)-(f)为在进行甲酯化修饰反应后,分别使用以重量/体积%计2.5%(b)、5%(c)、10%(d)、20%(e)和40%(f)乙胺溶液进行酰胺化反应而得到的试样的质谱图。
对于糖链试样,添加各浓度的乙胺后进行混合,直至用HILIC板开始纯化为止为一分钟左右,但确认到在使用2.5%左右的低浓度乙胺的情况下也没有问题地发生了酯酰胺转化。另外,可知在使用高浓度的胺的情况下也抑制了α2,6-唾液酸酯的酰胺化。本实施例中用HILIC载体进行了纯化,但酯酰胺转化在吸附于HILIC载体前以液相反应的形式进行,显示出:连接方式特异性的酯酰胺转化以液相反应的形式也没有问题地进行。
(实施例8)
实施例8中,在将由鞘糖脂(Glycosphingolipid;GSL)得到的GSL糖链结合于载体的状态下进行乙酯化反应,对于得到的糖链试样进行酰胺化反应,通过质谱分析进行分析。
<包含α2,3-唾液酸的糖链试样的制作和酯化反应>
通过与实施例5的方法同样的方法,制作包含总计2个α2,3-或α2,8-唾液酸的糖链(GD1a、GD1b)。游离的糖链通过Glycoblotting法键合于固相载体,用1-乙基-3-对甲苯基三氮烯(ETT)进行乙酯化,以aoWR标记糖链(100μL)的形式回收。对于A2GN1糖链,与上述实施例1同样地制作试样,与本实施例的上述记载同样地供于酯化反应。
<酰胺化反应和质谱分析>
将得到的aoWR标记糖链溶液25μL与250μL的胺溶液(浓度1mol/L、溶剂为乙腈)混合,使用HILIC板(Waters)从试样去除过量试剂。将DHB作为基质,使用Ultraflex II TOF/TOF-MS(Bruker)以正离子模式进行测定。对于A2GN1糖链,也同样地供于酰胺化反应和质谱分析。
<结果>
图16为示出将GD1a和A2GN1糖链作为试样进行乙酯化修饰反应后,未进行酰胺化反应的试样(a);以及使用分别包含1M(M为mol/L)甲胺、乙胺和丙胺的乙腈溶液进行酰胺化反应而得到的试样(b、c、d)的质谱图的图。对于GD1a,进行利用ETT的乙酯化且未进行酰胺化时,检测到与2个α2,3-唾液酸进行了甲酯化的情况对应的峰(m/z 1776)。以下的各图中,质谱图中示出的箭头将峰与产物的修饰体关联地显示。甲酯化后,进行利用甲胺溶液的酰胺化反应时,汇集成仅一个唾液酸进行了甲酰胺化的产物(m/z 1761)。同样地在使用乙胺、丙胺溶液的情况下也汇集成仅一个唾液酸进行了酰胺化的产物(m/z 1775、1789)。
图17示出将GD1b和A2GN1糖链作为试样进行乙酯化修饰反应后,未进行酰胺化反应的试样(e);以及使用分别包含1M(M为mol/L)甲胺、乙胺和丙胺的乙腈溶液进行酰胺化反应而得到的试样(f、g、h)的质谱图的图。对于GD1b,进行利用ETT的乙酯化且未进行酰胺化时,检测到与1个α2,3-唾液酸和1个α2,8-唾液酸进行了甲酯化的情况对应的峰(m/z1776)。甲酯化后,进行利用甲胺溶液的酰胺化反应时,汇集成仅一个唾液酸进行了甲酰胺化的产物(m/z 1761)。同样地在使用乙胺、丙胺溶液的情况下也汇集成仅一个唾液酸进行了酰胺化的产物(m/z 1775、1789)。在关于图16的(a)-(d)和图17(e)-(h)的全部反应中,作为内标糖链而添加的具有α2,6-唾液酸的A2GN1糖链以乙酯体的形式被检测。
图18为示出乙酯化后供于丙酰胺化的GD1a(图18的(a))和GD1b(图18的(b))(均m/z 1779)的MS/MS谱的图。可知在GD1a中,在m/z 1090检测到相当于乙酯化的GM3糖链的碎片,在GD1b中,仅α2,8-唾液酸发生了丙酰胺化。即,显示出在本条件下发生了位置特异性的α2,3-/α2,8-唾液酸的酯酰胺交换,显示出上述方法不仅对判别唾液酸连接方式是有效的,而且对键合位置的解析也是有效的。
(实施例9)
实施例9中,作为酰胺化反应溶液,使用以乙腈为溶剂的丙胺溶液,将丙胺溶液中的丙胺的浓度分别制备成0M-3M的范围的多个浓度。此外,通过与实施例8同样的方法进行GSL糖链的质谱分析。
<结果>
图19为示出分别使用各种浓度的丙胺溶液作为酰胺化反应溶液时得到的GD1a的质谱图的图。GD1a的情况下,使用0.125M的丙胺溶液时,几乎不进行酯酰胺交换,使用0.25M或高于0.5M的浓度时,仅一个存在于糖链末端的α2,3-唾液酸发生丙酰胺化。
图20为示出分别使用各种浓度的丙胺溶液作为酰胺化反应溶液时得到的GD1b的质谱图的图。GD1b的情况下,与丙胺浓度无关,仅有容易形成内酯的α2,8-唾液酸发生了丙酰胺化。其结果也启示了,不仅显示出存在于末端的α2,3-或α2,8-唾液酸酯的酰胺化最佳浓度,而且能够识别α2,8-唾液酸与α2,3-唾液酸。
- 分析用试样的制备方法、分析方法和分析用试样的制备用试剂盒
- 分析用试样的制备方法、分析方法和分析用试样的制备用试剂盒