用于病原微生物检测的超多重PCR建库试剂盒和检测方法
文献发布时间:2024-04-18 19:53:33
技术领域
本发明属于病原微生物检测技术领域,具体涉及用于病原微生物检测的超多重PCR建库试剂盒和检测方法。
背景技术
多重扩增(Multiplex polymerase chain reaction,MPCR)是在普通PCR的基础上,在同一个反应体系中加入不同的引物对,针对不同的模板或同一模板的不同区域进行特异性的扩增,从而得到多个目的片段,结合一定的检测手段进而实现同时对多个靶标进行诊断的技术。多重扩增具有高效率、高通量、低成本的特性而被深入研究,自1988年首次提出多重扩增这个概念以来,多重扩增已经应用于许多领域,包括基因突变与缺失、基因分型与定量、遗传检测、伴药诊断等。多重扩增所涵盖的范围广,能够同时对多个靶标进行检测,大幅提升检测效率的同时,还降低了检测成本。
随着科学技术的发展,多重扩增技术在扩增和检测方面已经取得了许多新的突破。在扩增方面,不再局限于在同一反应管中进行扩增,而是将不同的引物对和模板分散于相对独立的空间中进行扩增;在检测方面,不断有新的超多重检测技术的出现。
目前,应用多重扩增技术进行病原微生物检测是一个重要的研究方向,其关键是在低模板拷贝数的条件下实现超多重靶向扩增富集技术。结合二代测序或三代测序方法,多重PCR靶向高通量测序是一种潜在可以替代或解决病原感染检测问题的方法,此方法具有成本低、时效短、准确率高等优点。但是,因为多重PCR扩增的单个反应中,引物的数量往往达到几百甚至几万条,技术难度非常高;同时待检测的样本中病原微生物序列的丰度极低,进一步加大了多重PCR靶向富集的难度。近年也有报导利用多重PCR建库试剂盒进行病原微生物的检测,但大多只针对某一种类型样本的部分病原进行检测。除此之外,现有的病原微生物多重PCR法文库构建试剂盒因为在同一反应管中存在几百甚至几千条引物,而整个试剂盒的引物组及反应体系为整体分析优化的结果。因此,现有的病原微生物多重PCR建库试剂盒还存在以下的常见问题:(1)引物间相互干扰,形成大量二聚体或多聚体,降低了中靶产物的比例;(2)非特异扩增,产生大量非目标扩增子,影响后续测序;(3)扩增偏好性与稳定性差,无法进行准确的定量;(4)操作复杂、时间长、灵活性差;(5)部分临床样本病原微生物病原微生物载量极低,在常规体系下工作的多重扩增体系在低载量的条件下出现扩增异常。
发明内容
针对现有技术中的不足,本发明开发了一套可用于病原微生物检测的多重扩增技术体系,惊奇的解决了现有多重扩增技术中易出现引物二聚体或多聚体、多重扩增引物干扰、扩增效率差、扩增偏好大与中靶产物比例低的问题。
为了实现上述目的,本发明采用了以下技术方案。
本发明第一方面提供了一种用于病原微生物检测的超多重PCR建库试剂盒,所述试剂盒中的引物具有以下结构特征:
引物的3’末端具有相似性序列;
所述相似性序列由最末端的单一碱基限定区域和与所述单一碱基限定区域相邻的唯二碱基限定区域组成;
在所述单一碱基限定区域中仅含有一种类型的碱基,在所述唯二碱基限定区域中仅含有一种或两种类型的碱基;
任一引物中均不具有与所述相似性序列的反向互补可结合区域。
优选地,所述单一碱基限定区域为引物3’末端的最后一位碱基,所述唯二碱基限定区域为引物3’末端的倒数第2位和第3位碱基。
优选地,所述引物的5’端添加了具有发夹结构的通用序列。
具体地,所述的通用序列具有以下特征:
通用序列在F端和R端仅在中部存在特异性序列;
通用序列的5’端具有与其3’端结合的反向互补序列;
通用序列形成发夹结构的结合温度为55℃~65℃。
基于上述开发的具有特定序列结构的多重引物,本发明第二方面提供了一种用于病原微生物检测的方法,包括以下步骤:
S1、基于各病原微生物的靶向区域设计特异性引物;
S2、将通用序列添加至S1所述特异性引物的5’端;
S3、将待测核酸样品与S2所得引物置于反应体系中并进行扩增;
S4、以S3所得产物为模板,加入扩增引物进行扩增,且该扩增以所述通用序列为引物结合位点,所述扩增引物中含有标签序列、Index序列或Barcode序列;
S5、将S4所得产物与测序接头连接后进行测序,根据测序产生的数据分析待测核酸样品中检出的病原微生物信息。
其中,步骤S1中设计的特异性引物除了满足常规的引物设计要求外,还具有以下序列特征:
引物的3’末端具有相似性序列;
所述相似性序列由最末端的单一碱基限定区域和与所述单一碱基限定区域相邻的唯二碱基限定区域组成;
在所述单一碱基限定区域中仅含有一种类型的碱基,在所述唯二碱基限定区域中仅含有一种或两种类型的碱基;
任一引物中均不具有与该引物自身的相似性序列的反向互补可结合区域。
优选地,步骤S2所述的通用序列具有二级结构(发夹结构),且其具有以下特征:
通用序列在F端和R端仅在中部存在特异性序列;
通用序列的5’端具有与其3’端结合的反向互补序列;
通用序列形成发夹结构的结合温度为55℃~65℃。
更加优选地,采用具有二级结构的通用引物时,可以向S3所得产物中加入核酸内切酶进行反应,待反应结束后用磁珠纯化,再将纯化所得产物按照S4进行扩增反应。
优选地,步骤S4所述扩增引物的引物结合序列中去除了通用序列5’端会形成二级结构的序列。
本发明的有益效果为:本发明通过具有特定序列规则限制的多重特异性引物设计、并结合通用序列特殊二级结构限制,同时结合特殊的异常扩增片段消化,有效解决多重扩增中易出现引物二聚体或多聚体、多重扩增引物干扰、扩增效率差、扩增偏好大与中靶产物比例低的问题;可以在低至2copies/反应的多重扩增体系下实现500-20000重的扩增,且扩增靶标的覆盖率>80%;可以控制扩增循环数在30个循环以内,经纯化后的扩增产物总量>50ng,且在仅经过30个循环以内即可实现低拷贝条件先病原微生物的检出;最终测序数据中的中靶产物占比>80%。该多重体系可用于特异性扩增富集来源于血液、肺泡灌洗液、脑脊液、胸腹水等临床样本中的病原微生物,在4-6小时完成病原微生物的检测,相比常规的mNGS检测技术的24h,将检测时间缩短至20%,可以在病原微生物的检测应用中实现快速即时检测。
附图说明
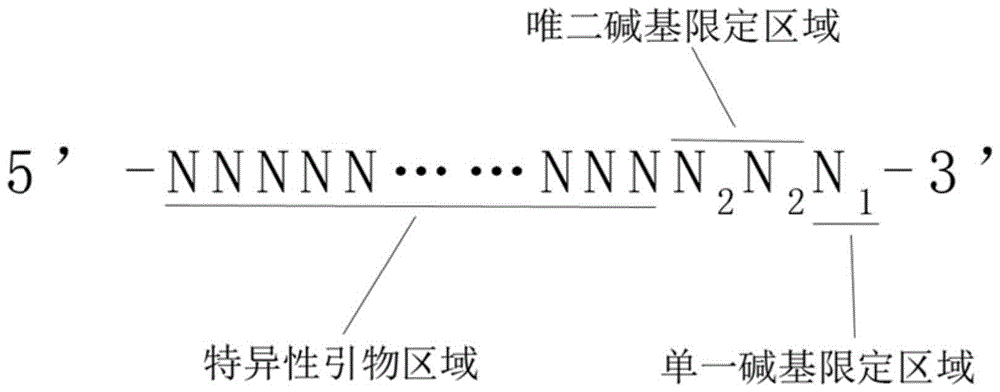
图1为本发明提供的特异性引物的结构示意图;
图2为本发明提供的通用引物及其单链产物二级结构的示例图;
图3为本发明提供的检测方法中采用核酸内切酶消化扩增产物中非完全匹配的二聚体的示意图;
图4为本发明提供的检测方法中第二轮扩增引物的二级结构示例图;
图5为采用本发明提供的检测方法在无人源基质和有人源基质条件下的病原检出线性图;
图6不同通用序列构建文库检测峰图。
具体实施方式
为了使本发明所要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。
本发明开发了一套具有特定序列规则限制的多重特异性引物设计、并结合通用序列特殊二级结构限制,同时结合特殊的异常扩增片段消化,有效的解决了现有多重扩增技术中的一些问题。
首先,本发明在基于检测靶标进行特异性引物设计时,除常规的引物二级结构、GC含量、自由能等要求外,本发明设计的特异性引物仅在5’端具有良好的特异性,而在3’端具有一定的相似性,且在引物其他区域不得具有与该相似性序列的反向互补可结合区域。
具体可参考图1:该特异性引物的结构分为特异性引物区域、唯二碱基限定区域与单一碱基限定区域,单一碱基限定区域限定在同一扩增体系中的所有引物仅包含一种类型的碱基,唯二碱基限定区域限定在同一扩增体系中的所有引物仅包含单一碱基和/或另一种碱基,同时限定任一引物的特异性引物区域不存在可以和该引物的唯二碱基限定区域+唯一碱基限定区域反向互补的区域。
进一步地,单一碱基限定区域为引物3’末端的最后一个碱基,唯二碱基限定区域为引物’末端的倒数第2位和第3位碱基时,其具体序列特征介绍如下:
①同一扩增体系中的所有引物的3’末端的最后一个碱基相同,具体为A、T、C、G、U或类似修饰碱基其中的一种,如为A;
②同一扩增体系中的所有引物的3’末端的倒数第2位和第3位仅包含A、T、C、G、U或类似修饰碱基其中的两种,如A、T两种;
此时,同一扩增体系的所有引物的3’末端相似,且具体只有四种可能形式:AAA、ATA、TAA、TTA;
③同一扩增体系中的任一引物的特异性引物区域(即图1中的NNNNN……NNN序列)中不存在与该引物3’末端3位碱基序列互补的区域,如一引物的3’末端为AAA,那么任一引物的特异性引物区域均不能有TTT这种序列。
本发明通过以上设计,可以使特异性引物3’末端的3个碱基不会结合形成二级结构,从而减少多重体系中引物的复杂性,并降低引物末端结合后出现延伸反应而导致的非特异性扩增。
其次,本发明在通用引物设计部分,在通用引物的5’端额外加上可以与其3’端结合的反向互补序列,使其可形成特殊发夹结构的二聚体,具体要求为:①通用序列的F端和R端具有极高的相似性,仅在引物的中间部分存在特异性序列;②通用引物的5端与3端可结合形成引物二聚体,其形成二聚体的结合温度在55℃~65℃之间。
本发明设计的通用引物可以确保在进行第一轮的特异性引物扩增时,额外加入的通用引物序列可以优先结合形成封闭的二聚体结构,避免引入的通用序列与多重扩增引物互相干扰而影响最终的扩增效果;同时,由于通用序列F端和R端的相似性,即使有二聚体或小片段的非特异性扩增,其单链序列由于序列互补,会形成自身封闭结合的反向互补二级结构,避免后续的扩增时继续放大。
本发明提供的通用引物的一个具体示例如图2所示:在该具体示例中,通用序列F和R在二级结构结合的区域具有相同的序列,在环区域具有不同的序列,当进行扩增反应退火时,通用引物会优先结合为颈环结构,封闭通用引物区域,避免该区域对多重引物产生影响;当通用序列F和R产生非特异性扩增时,其扩增产物也极易产生二级结构而避免其进一步的扩增放大。
基于本发明设计的通用引物和特异性引物,再将通用引物添加至特异性引物的5’端后即可用于病原微生物检测中的检测,该检测方法的步骤优选为:
(1)、将通用序列添加至特异性引物的5’端;可以理解的是,通用序列F端添加至所有上游引物的5’端,通用序列R端添加至所有下游引物的5’端;
(2)将步骤(1)所得引物与待测核酸样品及其他扩增所需试剂配制成反应体系,在PCR仪上进行扩增反应;
(3)向步骤(2)所得多重扩增产物中加入浓度梯度优化的核酸内切酶,可以特异性的消化反应体系中的非完全匹配的二聚体序列,进一步提高反应体系的稳定性;
(4)以步骤(3)所得产物为模板,进行第二轮加Barcode或Index的扩增反应;
(5)将S4所得产物与测序接头连接后进行测序,根据测序产生的数据分析待测核酸样品中检出的病原微生物信息。
在上述检测方法中,步骤(3)中的内切酶可以是具有识别并切割不完全配对DNA、十字型结构DNA、Holiday结构或DNA分叉点、异源性DNA功能的内切酶,如T4内切酶或T7内切酶。所述内切酶的消化形式见图3,核酸内切酶可以特异性消化未结合的单链区域。
鉴于本发明提供的具有特殊二级结构限制的通用序列,步骤(4)的扩增反应中所用引物应满足以下要求:引物设计上会特异的去除5端可结合形成二聚体的核苷酸序列,以便在进行第二轮Barcode或Index扩增时,引物自身不会形成二级结构,同时由于该引物的结合区域还包括通用引物的特异性区域(即通用序列F和R中的特异性序列),其退火结合温度会高于通用引物形成二聚体的温度,在进行扩增时会优先结合引物进行扩增。
图4为第二轮扩增引物形成的二级结构示例图,通用引物结合区域与第一轮扩增的通用引物部分类似,差异部分是在引物的5’端进行了碱基替换,使得该引物不会形成二级结构,同时又保留了与通用引物结合而延续扩增的功能。
根据本发明提供的特异性引物、通用引物以及第二轮扩增所用引物的设计方法,本发明又提供了以下病原微生物件测的实施案例。在下述实施例中,若无特殊说明,均为常规方法;所述试剂和材料,若无特殊说明,均可从商业途径获得。
实施例1
本例构建了病原微生物多重检测体系,具体包括以下过程:
(1)用于检测细菌、真菌和病毒的特异性靶向引物的设计。
①选择的靶向区域是物种特异性的,仅在该靶标范围存在时可以进行扩增;
②基于选择的靶区域设计特异性引物,引物3’端最后一位限定为T碱基,末端倒数第2位、第3位限定为A或T碱基,即末三位有AAT、ATT、TAT、TTT这4种情况;
③引物的其他区域不包含可与末端3位碱基反向互补的序列,即不存在ATT、AAT、ATA、AAA这4个碱基序列;
④设计的引物仅能扩增靶标,且在GC含量、二级结构等特征方面满足常规的引物设计要求,具体为:引物长度18-25bp,GC含量40%-60%,退火温度55~62℃,优选60℃。
基于上述条件,本例共设计了59个靶标的80对特异性引物。可以理解的是,在具体实验时可以根据实验目的选择一种或多种组合一起进行多重扩增使用。
(2)通用引物序列的设计。
本实施例基于二代MGI平台的测序通用序列,设计可形成特异性二级结构的通用序列,具体设计的通用序列如下:
F端:GTCGGCTTGGCCTCCGACTT;
R端:GTCGGCTACGATCCGACTT。
F端序列与R端序列在引物的5’端与3’端一致,在引物的中间区域具有特异性序列。
(3)将步骤(2)中的通用序列额外添加至步骤(1)所得特异性引物的5’端(所得80对引物的序列见序列表,引物可以在引物合成公司进行合成),组成了可以用于进行特异性靶向富集的引物,即该引物包括两个部分:一是位于5’端的通用序列,该序列的引入是由于在第二轮扩增时,是以通用序列为引物结合位点的,可以避免扩增循环数过高造成的扩增偏好;二是位于3’端的特异性引物,该引物特异性的与病原微生物的靶标区域结合,可以特异性富集这部分待测序的靶标区域。
(4)采用步骤(3)所得引物进行第一轮扩增反应,该反应的通用配制体系如表1。
表1
将配制好的反应体系混匀后,瞬时离心,在PCR仪上进行扩增反应:
第一轮扩增需消耗约60min。
(5)使用核酸内切酶消化去除第一轮多重扩增过程产生的二聚体或多聚体,具体的反应体系配制如表2。
表2
将配制好的反应体系混匀后,瞬时离心,在PCR仪上37℃反应5min。反应完成后立即使用磁珠进行纯化。
(6)第二轮带标签序列PCR扩增反应。
第二轮扩增引物的引物结合序列与通用序列在3’端相同,但会特异的去除5’端会形成二级结构的引物区域,本实施例采用的引物具体为:
F端引物:GGTGCTGXXXXXXTCCACCATGGCTTGGCCTCCGACTT;
R端引物:GGTGCTGXXXXXXTCCACCATCGGCTACGATCCGACTT;
其中,XXXXXX部分为标签序列、Index序列或Barcode序列。
第二轮的加标签序列扩增的反应体系配制如表3。
表3
将配制好的反应体系混匀后,瞬时离心,在PCR仪上进行扩增反应:
第二轮扩增需消耗约30min。
(7)进行测序接头的连接。
将第二轮扩增产物使用常规的纯化磁珠进行1X纯化,具体流程参考纯化磁珠的使用说明书,推荐使用诺维赞或翊圣的纯化磁珠。纯化完成后进行Qubit浓度测定,将准备同批进行测序的样本等量混合,混合后的总量在50~500ng即可进行后续连接反应。且连接体系的配制如表4。
表4
将配制好的反应体系混匀后,瞬时离心,20℃连接反应10min,反应结束后即连接上测序接头。连接产物按照Nanopore官方的测序流程进行上机反应。连接与上机反应耗时约0.5h。
(8)测序与数据分析。
测序产生的数据在明德病原微生物分析软件上进行同步分析,测序时间达到1~2h即可以检出主要的病原微生物。
实施例2
本例对临床血液感染样本进行检测,先使用明德自动化提取仪的磁珠法进行核酸的提取。提取方法参照试剂盒说明书和仪器使用说明书。再根据实施例1设计的引物和检测方法进行建库、测序和分析。具体的检出性能如表5所示。
表5
在不进行人源基因组DNA去除的情况下,可以在低至2copies/反应的多重扩增体系下实现616重的扩增,且引物的工作率可以达到91%,扩增靶标的检出率达到93%;可以控制扩增循环数在30个循环以内,经纯化后的扩增产物总量>50ng,且在仅经过30个循环以内即可实现低拷贝条件下病原微生物的检出;最终测序数据中的中靶产物占比>80%。
实施例3
对于多重扩增,已有的试剂或方法并不能进行相对定量,且由于扩增偏好和引物干扰问题,测序的病原体序列数并不能反应临床样本中的病原微生物含量。
根据实施例1设计的引物和检测方法,本例对测序检出序列数进行了线性测试。本例采用的待测样品为2~100copies/mL的病原体梯度样品,检测结果如图5所示,表明:在2~100copies/mL的检测条件下,无论是否存在人源基质,本发明的方法均表现出良好的线性关系,R
实施例4
本例对比了本发明试验流程与PacBio流程、Nanopore流程中通用序列的性能。
在进行两步法PCR扩增添加Barcode和Index时,PacBio与Nanopore官方均推荐有适用于自身的通用序列,其开发的各类试剂也均是基于该通用序列进行开发,在设计原则上该通用序列是完全不会形成二级结构的。其中,Nanopore官方使用的通用序列为:
F端:TTTCTGTTGGTGCTGATATTGC,
R端:ACTTGCCTGTCGCTCTATCTTC。
PacBio官方推荐的通用序列为:
F端:GCAGTCGAACATGTAGCTGACTCAGGTCAC,
R端:TGGATCACTTGTGCAAGCATCACATCGTAG。
在相同的实验条件对PacBio、Nanopore和按照本发明设计的能形成二级结构的通用序列的多重扩增性能进行比较,发现PacBio与Nanopore的通用序列在扩增时均会存在二聚体或多聚体的残留,而采用本发明的方法可以完全避免二聚体的干扰。
图5为不同通用序列构建文库检测峰图,可以明显看到,Nanopore通用序列(左)和PacBio通用序列(中)构建的文库在检出时出现了明显的二聚体峰,而本发明(由)构建的文库完全无二聚体峰的干扰。
综上,本发明开发的用于病原微生物检测的多重扩增技术体系,惊奇的解决了多重扩增中引物二聚体与引物互相干扰的问题,在不进行人源基因组DNA去除的情况下,可以在低至2copies/反应的多重扩增体系下实现超多重的扩增,且扩增靶标的覆盖率>80%;可以控制扩增循环数在30个循环以内,经纯化后的扩增产物总量>50ng,且在仅经过30个循环以内即可实现低拷贝条件先病原微生物的检出;最终测序数据中的中靶产物占比>80%;还能基于该方法获得的测序数据进行相对定量。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。