一种合成高纯度3-溴-N-苯基咔唑的方法
文献发布时间:2023-06-19 09:32:16
技术领域
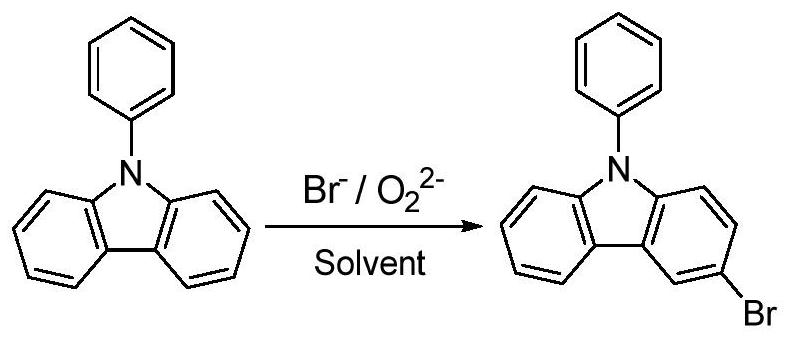
本发明属于一类N-苯基咔唑类衍生物的有机合成技术领域,具体涉及一种N-苯基咔唑氧化溴代反应合成3-溴-N-苯基咔唑的方法。
背景技术
有机电致发光二极管(Organic Light Emitting Diode,OLED)具有能耗低、亮度高、响应快、厚度薄、视角宽以及全固态、自发光、可挠曲等优良特性;可应用于更宽大的平板显示器和半导体,在固态照明与平板显示领域具有巨大的市场前景,是科研界和产业界的关注焦点和研究热点之一。OLED由有机半导体材料、发光材料以及电极层叠组成,有机电致发光材料对其性能影响最大。有机电致发光材料分为载流子传输材料和发光材料,其发光材料分为红、绿、蓝三种基元色光材料,作为主发光材料的磷光发光体材料的化学结构大都含有咔唑基团,而含有苯基咔唑基团的化合物在磷光主体材料的用途最为广泛。3-溴-N-苯基咔唑是一种合成含苯基咔唑基团化合物的重要中间体,可以合成N-苯基咔唑-3-硼酸,再经Suzuki 偶联、Buchwald-Harwig偶联反应等合成一系列含苯基咔唑基团的大分子的有机电致发光材料。例如,N-芳基咔唑类三苯胺结构材料具有优异的空穴传输性能、良好的热稳定性,作为一类重要空穴材料在有机电致发光材料中已经可以实际应用。
目前,合成3-溴-N-苯基咔唑的路线主要四种:
(一)中国专利申请号201110156767.X于2012年12月19日、中国专利申请号201310606035.5于2014年2月26日,均公开了一种N-苯基-3-溴咔唑的制备方法,以咔唑为原料,先经NBS溴代得到3-溴咔唑,再与卤苯经C-N偶联反应得到目标产物,合成过程如下所示:
(二)中国专利申请号201110382138.9于2012年6月13日,公开了一种高纯度3-溴-N- 苯基咔唑的制备方法,以咔唑为原料,先与卤苯经乌尔曼反应得到N-苯基咔唑,经N-溴代丁二酰亚胺(NBS)溴代得到目标产物,合成过程如下所示:
(三)以2,2'-二溴联苯为原料,与苯胺经偶联、关环、NBS溴代得到目标产物,如下所示;
(四)中国专利申请号201710375444.7于2017年12月8日,公开了一种高纯度3-溴-9- 苯基咔唑的制备方法,以对二溴苯为原料,硝化反应得到1,4-二溴-2-硝基苯,再与二苯胺经乌尔曼反应得到(4-溴-2-硝基苯基)二苯胺,后经还原、重氮化关环反应得到3-溴-N-苯基咔唑,路线如下所示。
以上合成3-溴-N-苯基咔唑的方法:路线(一)中咔唑的亲电溴代反应可能发生在C1、 C8、C3、C6,从而存在副产物较多弊端,另外3-溴咔唑进行乌尔曼反应时存在与另一分子 3-溴咔唑反应生成更为复杂的副产物的不足;路线(二)中N-苯基咔唑可以有效避免其他亲核试剂的干扰,同时因其苯环的空间位阻效应可有效降低C1、C8发生亲电取代的几率、提高目标产物3-溴-N-苯基咔唑的选择性,但N-苯基咔唑溴代反应使用NBS、溴素、二溴海因等溴代试剂,存在成本高、原子利用率低、污染重等缺点;路线(三)和(四)存在反应步骤复杂、条件苛刻、总反应收率低的不足,同时咔唑价格低、易得,以咔唑为原料合成3-溴 -N-苯基咔唑的方法是更为成熟、经济、简单的合成方法。因此,路线(二)是更为理想的合成3-溴-N-苯基咔唑的方法,但目前3-溴-N-苯基咔唑的制备过程复杂,N-苯基咔唑的溴代试剂存在成本高、污染大、原子利用率低、选择性差等不足,例如,NBS、二溴海因等作为该反应的亲电溴代试剂:①质子型溶剂条件下,N-苯基咔唑的溶解性和溶剂化率低,存在反应转化率和选择性差的缺陷;②非质子型极性溶剂或非质子型非极性溶剂条件下,因为N-苯基咔唑的溶解性和NBS的溶剂化率之间的矛盾,所以存在反应高选择性和高转化率不能兼得的缺陷;③溴源成本高、三废多;另外,单质溴具有强腐蚀性和强毒性,反应过程中仅有一半的溴被利用,另一半生成溴化氢而未被利用,存在溴原子利用率低、污染严重等不足。
2012年,李猛等在《黑龙江科学》(2012年第3卷第1期第26-27页)发表了一篇名为“3-溴-9-苯基咔唑的合成”的合成3-溴-9-苯基咔唑的综述性文章,该文章总结了NBS、溴单质(Br
因此,寻求一种绿色、环保的制备方法,得到高纯度的3-溴-N-苯基咔唑是目前亟需解决的问题。
发明内容
1.要解决的问题
本发明旨在改进现有的以N-苯基咔唑为原料合成3-溴-N-苯基咔唑的方法,提供一种高原子利用率、后处理操作简单、适宜工业化生产、更为绿色经济环保、N-苯基咔唑氧化溴代反应合成3-溴-N-苯基咔唑的新方法。
2.技术方案
为了解决上述问题,本发明所采用的技术方案如下:
一种合成高纯度3-溴-N-苯基咔唑的方法,步骤为:将N-苯基咔唑溶于溶剂中后,于-20 ℃~10℃的低温下依次加入溴化剂和氧化剂,添加完毕后,保持反应温度为-20℃~10℃下反应至N-苯基咔唑的转化率达到≥99%停止反应,其中,添加溴化剂和氧化剂时保持反应体系温度变化在5℃以内;反应结束后,反应体系依次经过调pH至中性、洗涤、旋转蒸发、重结晶、真空干燥,得到高纯度3-溴-N-苯基咔唑。
具体地,一种合成高纯度3-溴-N-苯基咔唑的方法步骤如下:
在室温条件下,将N-苯基咔唑溶于有机溶剂中搅拌均匀,然后降低反应体系温度为-20 ℃~10℃,加入定量的溴化剂,待体系温度恒定后分批加入氧化剂,在-20℃~10℃条件下继续反应4~12小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去有机溶剂得到粗产品;然后采用重结晶的方法对粗产品多次提纯真空干燥得到高纯度的3-溴-N-苯基咔唑。
进一步地,所述溴化剂为含溴化合物,包括氢溴酸、溴化钾、溴化钠、溴化铵、溴化锂。
进一步地,所述溴化剂的质量分数为20%~50%。
进一步地,所述氧化剂为含有过氧根的化合物或过硫酸钾,优选双氧水、过氧化钠、过氧化镁。
进一步地,以含有过氧根的化合物作为氧化剂,N-苯基咔唑氧化溴代反应合成3-溴-N- 苯基咔唑的反应式如下所示:
进一步地,所述氧化剂的质量分数为20%~40%。
进一步地,所述溴化剂采用滴定的方式滴加,滴定速度为1~5mL/min,可以保证溴化剂添加过程中反应体系温度变化不大。
进一步地,所述氧化剂采用滴定的方式滴加,滴定速度为0.5~1.5mL/min,可以保证氧化剂添加过程中反应体系温度变化不大。
进一步地,所述溴化剂、氧化剂与N-苯基咔唑三者之间的摩尔比为 (1.00~1.20):(1.05~1.25):1。
进一步地,所述溶剂为正己烷、二氯苯、二氯甲烷、二氯乙烷、四氯化碳、三氯甲烷、氯苯、二硫化碳等中的一种或多种混合,均为水溶性差的溶剂,有利于产物提纯。
进一步地,由于上述反应产物纯度高,故采用重结晶便可实现提纯,所述重结晶过程中,采用正己烷、乙醇、甲醇、二甲苯、乙酸乙酯等中的一种或多种混合作为重结晶溶剂。
3.有益效果
相比于现有技术,本发明的有益效果为:
(1)本发明所使用的氧化溴代方法,具有高原子利用率、低原料成本、高反应选择性、制备周期短、制备操作简单、反应易控的特点;
(2)本发明具有较好的绿色、无毒、无害的优势,避免了NBS、溴素、二溴海因等溴代试剂对环境的污染和危害;
(3)本发明具有高收率、低制备成本的优点,具备产业化的可行性,拓展了3-溴-N-苯基咔唑类衍生物作为中间体在有机光电材料设计的合成方法;
(4)本发明溴化剂为含溴化合物,优选氢溴酸,避免引入其他非氢离子造成后处理工序复杂;溴化剂的质量分数选择在20%~50%范围内,便于控制反应滴加速度和高的溶剂稳定性,从而便于控制添加溴化剂溶液时二溴代产物的生成,其中,溴化剂的质量分数低于20%导致无机相与有机相比例过高影响溴代转化率,溴化剂的质量分数高于50%导致存在较高浓度的溴单质且酸性过强而造成反应选择性差;溴化剂滴定速度控制在1~5mL/min,可以保证溴化剂添加过程中反应体系温度变化不大,减少添加溴代试剂时二溴产物的生成;
(5)本发明氧化剂优选含有过氧根的化合物,特别选择双氧水优势为易于控制其浓度、添加速度、原子利用率高(唯一副产物为水)、便于操作和处理等,而过氧化钠、过氧化镁等在有机溶剂中存在爆炸或燃烧的危险、不易控制反应温度;氧化剂的质量分数为20%~40%的优势为便于控制滴加速度、反应选择性高,其中,氧化剂的质量分数低于20%导致反应速度慢、过氧化氢分解过多影响氧化效果,氧化剂的质量分数高于40%导致不易控制反应温度、在有机溶剂中存在爆炸危险;氧化剂滴定速度控制在0.5~1.5mL/min,可以保证氧化剂添加过程中反应体系温度变化不大,从而保证高的反应速度和高选择性;
(6)本发明的溶剂使用水溶性差的溶剂,是因为水溶性较差的溶剂便于回收重复使用,减少溶剂损失从而降低生产成本;
(7)本发明反应结束后调PH至中性,以中和过量的溴单质或氢溴酸,转化成溴化钾等溶于水的无机盐,在分液过程中从无机层除去,从而减少产物中的杂质的溴含量;
(8)本发明反应体系温度为-20℃~10℃,其中,温度高于10℃时反应活性高更有利于二溴代反应对单溴代反应的竞争,造成反应选择性差;温度低于-20℃时N-苯基咔唑会从溶剂中析出存在固液包夹的情况,造成二溴代产物增多降低反应的选择性;
(9)本发明溴化剂过量,保证反应转化率≥99%,减少原料的剩余;氧化剂过量的目的是弥补因分解或与杂质反应等因素而造成氧化剂不足,保证溴化试剂完全转化为活性溴分子;
(10)本发明采用N-苯基咔唑作为反应物原料,N-苯基咔唑是由咔唑的9位N-H上引入苯基得到的,苯基的引入增加了N对咔唑C1、C3、C6、C8位的供电性,大大降低了咔唑溴化的选择性;由于苯基的空间位阻作用,咔唑C1、C8位的溴化相对困难;但是N-苯基咔唑 C3、C6位的选择性相对于咔唑更加困难,同时N-苯基也会参与溴化反应,故而N-苯基咔唑高选择性溴化获得3-溴-N-苯基咔唑更加困难,不容易控制溴化进程;N-苯基咔唑由于N-H 被苯基取代而溶解性明显好于咔唑;但其二溴代产物3,6-二溴-N-苯基咔唑的溶解性较差,如果溴源过量太多会造成更多的3,6-二溴-N-苯基咔唑的生成,很难得到高纯度的3-溴-N-苯基咔唑;因此严格控制溴源选择、溴源当量、反应温度等条件是获得解决N-苯基咔唑溴代选择性和获得高纯度3-溴-N-苯基咔唑的关键。
附图说明
图1为本发明合成3-溴-N-苯基咔唑反应式示意图;
图2为本发明实施例1中3-溴-N-苯基咔唑的核磁共振氢谱图;
图3为本发明实施例1中3-溴-N-苯基咔唑的核磁共振碳谱图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面结合实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本说明上述发明内容的技术均为本发明的范围。
实施例1
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二氯甲烷,搅拌至完全溶解,然后降低反应体系温度至-10℃;逐滴加入24.3g氢溴酸(40%,0.12mol,17.6mL),待反应温度恒定在-10℃下再逐滴加入13.6g双氧水(30%,0.12mol,12.2mL),其中,溴化剂的滴加速度为3.0mL/min,氧化剂的滴加速度为0.5mL/min,滴加过程可以严格控制反应温度在-10℃以下;在温度为-10℃的条件下反应6小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二氯甲烷得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
如图2所示为本实施例3-溴-N-苯基咔唑的核磁共振氢谱,其中
如图3所示为本实施例3-溴-N-苯基咔唑的核磁共振碳谱,其中
实施例2
在室温条件下,向装有搅拌器、温度计的1000mL四口圆底烧瓶中加入243.3g N-苯基咔唑(99%,1.00mol)、500mL二氯甲烷,搅拌至完全溶解,然后降低反应体系温度至-10℃;逐滴加入212.4g氢溴酸(40%,1.05mol,153.9mL),待反应温度恒定在-10℃下再逐滴加入119.0g双氧水(30%,1.05mol,107.2mL),其中,溴化剂的滴加速度为3.0mL/min,氧化剂的滴加速度为0.5mL/min,滴加过程可以严格控制反应温度在-10℃以下;在温度为-10 ℃的条件下反应6小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二氯甲烷得到粗产品,将粗产品用500mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
实施例3
本实施例与实施例1基本相同,其不同之处在于:添加40.5g氢溴酸(20%,0.10mol, 35.0mL),添加17.9g双氧水(20%,0.105mol,16.7mL)。
本实施例反应结束后HPLC检测N-苯基咔唑转化率未能达到99%,3-溴-N-苯基咔唑24.5 g(HPLC检测含量为≥99%),收率76.0%;添加完氢溴酸和双氧水的反应体系过程中,无机相与有机相体积比接近1:1,生成的溴分子浓度过低造成N-苯基咔唑无法完全转化为3-溴 -N-苯基咔唑而影响反应的转化率和收率。
实施例4
本实施例与实施例1基本相同,其不同之处在于:添加19.4g氢溴酸(50%,0.12mol, 12.8mL),添加10.6g双氧水(40%,0.125mol,9.2mL)。
本实施例反应结束后HPLC检测N-苯基咔唑完全转化,3-溴-N-苯基咔唑23.4g(HPLC 检测含量为≥99%),收率72.6%;添加氢溴酸和双氧水过程中,反应体系温度不易控制,过量的溴源体系造成生成的3,6-二溴-N-苯基咔唑的增多,而3,6-二溴-N-苯基咔唑在大部分有机溶剂中溶剂性比较差,通过重结晶等手段不易与3-溴-N-苯基咔唑分离,虽有高转化率但低选择性、低收率等不足。
实施例5
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL正己烷,搅拌至完全溶解,然后降低反应体系温度至-20℃;逐滴加入35.7g溴化钾水溶液(40%,0.12mol),待反应温度恒定在-20℃下再逐滴加入108.1 g过硫酸钾水溶液(30%,0.12mol),其中,溴化剂的滴加速度为3.0mL/min,氧化剂的滴加速度为0.5mL/min,滴加过程可以严格控制反应温度在-20℃以下;在温度为-20℃的条件下反应12小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去正己烷得到粗产品,将粗产品用100*3mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
反应体系中钾离子和硫酸根的去除方法:反应结束中和至中性后通过多次萃取分液除去无机层,钾离子和硫酸根离子在无机相中分离除去。
本实施例反应过程中,以温度为-20℃、正己烷作为溶剂条件下,部分N-苯基咔唑以固体状态存在呈固液混合物,溴源体系加入后无机相与有机相体积比大于2:1造成溴分子浓度过低,严重影响反应的速度,同时大量的3,6-二溴-N-苯基咔唑生成,造成分离困难,3-溴-N- 苯基咔唑收率很不理想。
实施例6
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二硫化碳,搅拌至完全溶解,然后降低反应体系温度至10℃;逐滴加入30.9g溴化钠水溶液(40%,0.12mol),待反应温度恒定在10℃下再逐滴加入31.2 g过氧化钠水溶液(30%,0.12mol),其中,溴化剂的滴加速度为3.0mL/min,氧化剂的滴加速度为0.5mL/min,滴加过程可以严格控制反应温度在10℃以下;在温度为10℃的条件下反应6小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二硫化碳得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
反应体系中钠离子的去除方法:反应结束中和至中性通过多次萃取分液,钠离子在无机相中分液去除。
本实施例的合成反应,过氧化钠溶于水过程有氧气生成,有部分氧化剂以氧气的形式逃逸到空气,生成的活性溴分子减少,造成3-溴-N-苯基咔唑收率降低。
值得注意的是,溴化剂还可以选择其他含溴化合物,包括但不限于溴化铵、溴化锂;氧化剂还可以选择其他过氧化物,包括但不限于过氧化镁;只是盐离子处理方式有所不同。
对比例1
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL四氢呋喃,搅拌至完全溶解,然后降低反应体系温度至-10℃;逐滴加入24.3g氢溴酸(40%,0.12mol,17.6mL),待反应温度恒定在-10℃下再逐滴加入13.6g双氧水(30%,0.12mol,12.2mL),滴加过程严格控制反应温度在-10℃以下;在温度为-10℃的条件下反应6小时,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去四氢呋喃得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
对比例1使用四氢呋喃作为溶剂,N-苯基咔唑的溶解性好,Br
对比例2
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二氯甲烷,搅拌至完全溶解,逐滴加入24.3g氢溴酸(40%,0.12mol,17.6mL),待反应温度恒定后再逐滴加入13.6g双氧水(30%,0.12mol,12.2mL),室温条件下反应6小时停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二氯甲烷得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
对比例2采用二氯甲烷作为溶剂、室温条件下,无法控制N-苯基咔唑的溴代选择性,有超过25%的N-苯基咔唑生成3,6-二溴-N-苯基咔唑,N-苯基咔唑还有少量剩余(5%左右),造成分离困难,最后得到的HPLC检测含量为≥99%产品收率较低。
对比例3
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二氯甲烷,搅拌至完全溶解,然后降低反应体系温度至-10℃;逐滴加入47.9g液溴(40%,0.12mol),待反应温度恒定在-10℃下其中,液溴的滴加速度为0.5mL/min,滴加过程可以严格控制反应温度在-10℃以下;在温度为-10℃的条件下反应6小时停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二氯甲烷得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P真=0.095MPa条件下烘干 3小时得到白色晶状的3-溴-N-苯基咔唑8.5g(HPLC检测含量为≥99%),收率26.4%。
本对比例采用二氯甲烷作为溶剂、-10℃条件下,质量分数为40%的液溴作为溴源,反应选择性较差,液溴仅有一半的溴被利用,N-苯基咔唑转化率仅为78%左右,溴原子利用率低、反应转化率低。
对比例4
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二氯甲烷,搅拌至完全溶解,然后降低反应体系温度至-10℃;平均分三次加入12.3g溴化钠固体(99%,0.12mol),待反应温度恒定在-10℃下再平均分五次加入9.4g过氧化钠固体(99%,0.12mol),添加过程可以严格控制反应温度在-10℃以下;在温度为-10℃的条件下反应,期间液相检测N-苯基咔唑的转化率达到≥99%停止反应。反应结束后向反应体系加入定量的碳酸钠饱和水溶液调节混合液的pH至中性,采用水洗分离法洗涤3次,旋转蒸发除去二氯甲烷得到粗产品,将粗产品用100mL乙醇重结晶三次得到白色固体,将白色固体转入真空干燥箱中60℃、P
对比例4采用二氯甲烷作为溶剂、-10℃条件下,溴化钠作溴化剂、过氧化钠作氧化剂,两者在二氯甲烷中溶解性较差,反应时间长,过氧化钠添加过程和溴化反应过程温度有大幅上升、不易控制,最终收率较低。
对比例5
在室温条件下,向装有搅拌器、温度计的250mL四口圆底烧瓶中加入24.3g N-苯基咔唑(99%,0.10mol)、50mL二氯甲烷,搅拌至完全溶解,然后降低反应体系温度至-10℃;逐滴加入24.3g氢溴酸(40%,0.12mol,17.6mL),其中,氢溴酸的滴加速度为3.0mL/min,滴加过程可以严格控制反应温度在-10℃以下;在温度为-10℃的条件下反应6小时,期间液相检测N-苯基咔唑的转化率。
对比例5采用二氯甲烷作为溶剂、-10℃条件下,氢溴酸作溴化剂、不添加氧化剂,仅有少量N-苯基咔唑溴代生成3-溴-N-苯基咔唑(7%左右),延长反应时间长不能提高反应转化率,证明氢溴酸部分氧化为溴分子,不添加氧化剂反应转化率无法达到≥99%。
本发明中氧化溴代反应合成3-溴-N-苯基咔唑的方法,作为有机合成反应绿色化发展趋势的具体事例,有效克服了传统方法中存在的溴原子利用率低、区域选择性较差、三废多及对生成设备要求高等缺点,为合成3-溴-N-苯基咔唑提供了一种廉价、环境友好、经济、溴原子利用率高、操作简单的溴化方法。
本发明所述内容并不仅限于本发明所述实施例内容。本文中应用了具体实施例对本发明结构及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
- 一种合成高纯度3-溴-N-苯基咔唑的方法
- 一种N-苯基-3-(4-溴苯基)咔唑的合成方法