用于检测基因组中拷贝数变异的方法和装置
文献发布时间:2023-06-19 11:21:00
相关申请
本申请根据35U.S.C.§119(e)要求2018年09月14日提交的,发明名称为“用于检测基因组中拷贝数变异的方法和装置”的美国临时申请序列号62/731,738的权益。
背景技术
拷贝数变异(CNV)是一种基因组部分被复制或缺失的现象,并且可能影响基因组中的大量碱基对。CNV可能会导致人类的微缺失和微复制综合征,以及其他遗传性病症,如自闭症谱系病症。
常规分子细胞遗传学方法,如染色体微阵列分析(CMA)和荧光原位杂交(FISH),是临床实验室检测染色体畸变的标准测定。然而,新一代测序(NGS)技术使全基因组测序(WGS)更易于使用,并且需要计算方法来分析基于WGS的测定。
发明内容
一些实施方式涉及一种用于检测遗传序列中的拷贝数变异(CNV)的方法,所述方法包括使用处理器执行以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
一些实施方式涉及至少一种非暂时性计算机可读存储介质,其上存储计算机可读指令,当由处理器执行时,所述计算机可读指令使处理器执行一种检测遗传序列中的CNV的方法。所述方法包括扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
一些实施方式涉及一种用于检测遗传序列中的CNV的系统,所述系统包括可操作地连接至计算机可读存储器的至少一个处理器。所述计算机可读存储器包含指令,当由所述至少一个处理器执行时,所述指令使所述至少一个处理器执行包含以下步骤的方法:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
在一些实施方式中,所述遗传序列是部分基因组序列。在一些实施方式中,所述遗传序列是全基因组序列(WGS)。
在一些实施方式中,所述方法包括将所述遗传序列与参照基因组进行比对。
在一些实施方式中,识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
在一些实施方式中,所述方法还包括计算所述遗传序列的读取深度。
在一些实施方式中,所述方法还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
在一些实施方式中,计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
在一些实施方式中,将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
在一些实施方式中,将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
在一些实施方式中,所述多个位元中的每个位元包含50个碱基对。
在一些实施方式中,所述方法还包括将所述多个位元中的一个或多个位元合并。
在一些实施方式中,过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
在一些实施方式中,通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
一些实施方式涉及一种诊断由至少一个致病性CNV引起的病症的方法。所述方法包括使用处理器执行以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述WGS的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。所述方法还包括确定所识别的至少一个CNV是至少一个致病性CNV;和基于所确定的至少一个致病性CNV来诊断病症。
一些实施方式涉及一种治疗由至少一个致病性CNV引起的病症的方法。所述方法包括使用处理器执行以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述WGS的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述WGS中的至少一个CNV。所述方法还包括:确定所识别的至少一个CNV是至少一个致病性CNV;基于所述至少一个致病性CNV来诊断病症;和实施治疗以减轻所诊断的病症的一种或多种症状。
在一些实施方式中,所述病症是以下选择之一:自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
在一些实施方式中,所述遗传序列是部分基因组序列。在一些实施方式中,所述遗传序列是WGS。
在一些实施方式中,所述方法包括将所述遗传序列与参照基因组进行比对。
在一些实施方式中,识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
在一些实施方式中,所述方法还包括计算所述遗传序列的读取深度。
在一些实施方式中,所述方法还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
在一些实施方式中,计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
在一些实施方式中,将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
在一些实施方式中,将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
在一些实施方式中,所述多个位元中的每个位元包含50个碱基对。
在一些实施方式中,所述方法还包括将所述多个位元中的一个或多个位元合并。
在一些实施方式中,过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
在一些实施方式中,通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
附图说明
将参考以下附图描述各个方面和实施方式。应当理解,附图不一定按比例绘制。在附图中,在各个附图中示出的每个相同或几乎相同的组件由相似的数字表示。为了清楚期间,并非每个组件都可以在每个附图中标记。
图1A示意性地描述了根据本文所述技术的一些实施方式的数据管线的说明性框图;
图1B示意性地描述了根据本文所述技术的一些实施方式的将聚类算法用于遗传序列的说明性应用;
图1C示意性地描述了根据本文所述技术的一些实施方式的将图1A的数据管线用于遗传序列的说明性应用;
图2是描述根据本文所述技术的一些实施方式的识别遗传序列中的至少一个拷贝数变异(CNV)的过程的流程图;
图3是描述根据本文所述技术的一些实施方式的诊断由遗传序列中的至少一个CNV引起的病症的过程的流程图;
图4是描述根据本文所述技术的一些实施方式的治疗由遗传序列中的至少一个CNV引起的病症的过程的流程图;
图5A和图5B显示了根据本文所述技术的一些实施方式的,由Coriell研究所进行的染色体微阵列(CMA),由杰克逊实验室进行的CMA和通过JAX-CNV算法分析的全基因组序列(WGS)鉴定的针对31个样品的检测到的CNV缺失和重复的比较;
图6A显示了根据本文所述技术的一些实施方式的,作为CNV尺寸的函数以及针对CNV缺失和CNV重复两者,由杰克逊实验室在31个样品上进行的由JAX-CNV检测的独特CNV的数量以及由JAX-CNV和CMA两者检测的CNV的数量;
图6B显示了根据本文所述技术的一些实施方式,针对每个基因突变,由杰克逊实验室在31个样品上进行的由JAX-CNV检测的独特CNV的数量以及由JAX-CNV和CMA两者检测的CNV的数量;
图7A显示了从顶部到底部和针对共计31个样品的CNV检测,针对降低的覆盖率值,由Coriell研究所进行CMA,由杰克逊实验室进行CMA以及通过JAX-CNV进行WGS分析;
图7B显示了根据本文所述技术的一些实施方式,作为覆盖率的函数以及针对CNV缺失,由杰克逊实验室在31个样品上进行的JAX-CNV与CMA之间的一致性;
图7C显示了根据本文所述技术的一些实施方式,作为覆盖率的函数以及针对CMV重复,由杰克逊实验室在31个样品上进行的JAX-CNV与CMA之间的一致性;
图8示意性地描述了根据本文所述技术的一些实施方式可以在其上实现本公开内容的任何方面的说明性计算装置X。
具体实施方式
拷贝数变异(CNV)是重复的基因组部分,群体中的不同个体表现出不同数量的重复基因组物质。CNV占人类基因组的4.8%至9.5%,并且CNV被认为在人类进化、基因组多样性和疾病易感性中起关键作用。然而,个体之间CNV的变化可能会导致微缺失和微复制综合征,并伴有诸如发育和/或智力障碍的症状。这些综合征可以包括但不限于自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
已将不同技术用于CNV检测的研究和临床实验室,包括荧光原位杂交(FISH)、基于PCR的测定、染色体微阵列(CMA),以及最近的新一代测序(NGS)。目前将CMA作为患有无法解释的发育迟缓或智力障碍、自闭症谱系病症和先天性异常患者的一线诊断测试。然而,CMA的执行成本可能很高,并且分辨率受到阵列期间使用的探针数量的限制。
在过去十年中,NGS技术的进步带来了DNA测序通量、速度和成本空前的提高。这些改进使全基因组测序(WGS)具有精确检测多种类型遗传变异的能力,从而可广泛用于研究和临床诊断。此外,随着NGS的进步,生物信息学工具的迅速发展,使NGS结果分析在临床实验室中变得可行。尽管已经开发了几种基于WGS的CNV调用算法,但是由于假阳性率和假阳性率通常很高(例如高于5%),因而在临床环境中很难检测出真正的致病性CNV,因此没有一种算法可广泛用于临床环境。
发明人已经认识到并意识到,临床环境中缺乏用于从NGS结果中准确有效地检测CNV的耐用的计算方法。因此,本文提供了用于检测遗传序列中的CNV的系统和方法,包括部分遗传序列(PGS)或完整遗传序列(WGS)。
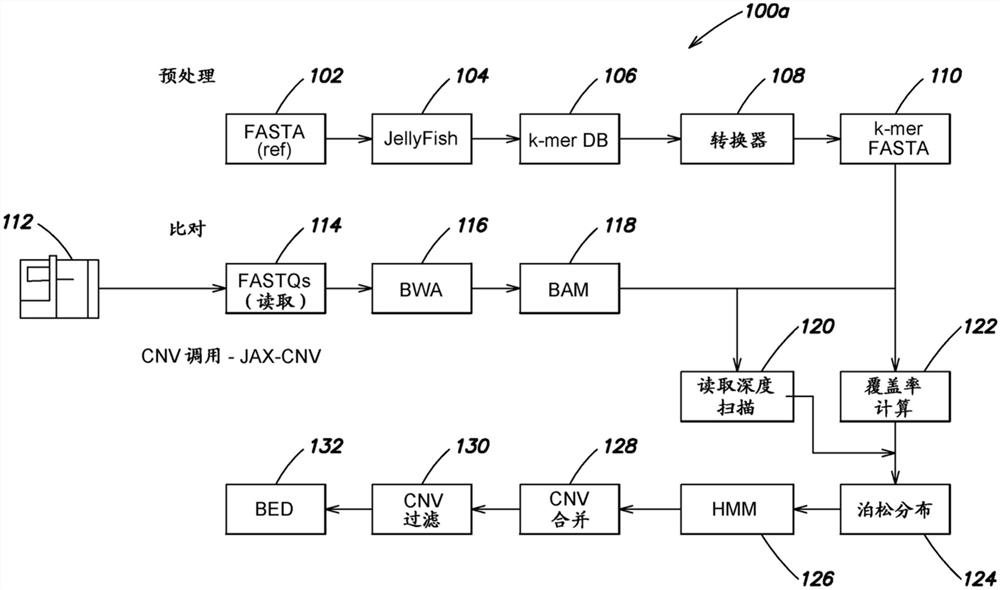
图1A显示了根据本文所述技术的一些实施方式的被配置为从遗传序列调用CNV的数据管线100的示意图。在一些实施方式中,数据管线100可以通过硬件(例如使用ASIC、FPGA或任何其他适宜电路)、软件(例如通过使用计算机处理器执行软件)或者其任何适宜组合来实现。
参照基因组(例如GRCh19或GRCh38)的预处理可以在目标遗传序列中调用CNV之前进行。预处理可以在调用CNV的每个实例之前进行,或者每个参照基因组仅进行一次。参照基因组的预处理可以包括读取FASTA(“Fast-All”)文件格式的参照基因组文件102,其中遗传序列可以使用单字母代码以基于本文的格式表示。
在步骤104中,可以对参照基因组的遗传序列内的每个k-mer的计数进行计算。k-mer是长度为k的遗传序列的子串。例如,尽管可以使用任何适当的k值,但是k可以是25个碱基对(在本文中为“bp”)。可以通过诸如JELLYFISH(例如JELLYFISH v2.2.6)的算法进行计算。该算法可以以二进制形式输出k-mer数据库106(在本文中为“k-mer DB”),所述二进制形式包含每个k-mer串以及其在遗传序列中出现的次数。
在一些实施方式中,在步骤108中,k-mer DB 106可以转换成k-mer FASTA文件110。k-mer FASTA文件110可以包含在遗传序列中已出现的每个k-mer的次数的log
根据一些实施方式,在开始调用CNV的算法之前,可以获取和处理遗传序列数据。遗传序列数据可以从例如新一代测序系统112或任何其他适宜测序方法获得。遗传序列数据可以代表,例如,部分遗传序列(PGS)或整个基因组序列(WGS)。遗传序列数据可以在FASTQ文件114中获得。
在一些实施方式中,在步骤116中,可以针对质量控制对FASTQ文件进行检查和/或针对参照基因组进行比对。可以通过例如FASTQC(例如FASTQC v0.11.5,未显示)进行质量控制。可以通过序列比对算法,比如例如BWA-MEM(例如BWA-MEM v0.7.15)对遗传序列与参照基因组进行比对。可以使用例如SAMTOOLS通过序列坐标对步骤116的比对结果进行排序。可以通过步骤116的算法来生成包含二进制格式的序列比对数据的二进制文件118(例如BAMbook文件)。可以将二进制文件118输入到CNV调用例程(在本文中为“JAX-CNV”)。
根据本文所述的一些实施方式,接下来可以将参照基因组的预处理结果和遗传序列数据的比对结果发送至JAX-CNV。在步骤120中进行的JAX-CNV的第一步可以是读取深度计算(“覆盖率”计算),其中计算特定核苷酸在测序结果中出现的次数。可以基于染色体中一个或多个独特的遗传区域(例如20个独特遗传区域)来计算每个常染色体的读取深度。可以扫描k-mer FASTA文件110和/或BAM文件118以确定每个常染色体中的独特遗传区域。当在所述区域内的每个k-mer仅出现一次并且所述区域的尺寸大于20Kb(例如20,000个碱基对)时,可以认为遗传区域是独特的。每个常染色体的读取深度可被计算为针对每个独特区域的每个碱基对计算的读取深度的平均值。
在一些实施方式中,然后可以针对样品的整个序列计算读取深度。可以应用四分位数范围来过滤异常值读取深度值,并且可以基于所有常染色体读取深度的平均值来计算遗传序列的总读取深度。将每个染色体的读取深度与遗传序列的读取深度进行比较,可以检测到遗传序列中的非整倍性。
在一些实施方式中,然后,可以将BAM文件118分成包含相同数量碱基对的位元。在一些实施方式中,所述位元可以包含50个碱基对。然后,可以在步骤122中执行读取深度计算,以计算每个位元的读取深度。可以将读取深度进一步转换为从0%至180%的百分位数,其中50%代表基线读取深度。例如,如果遗传序列的读取深度是50,且位元的读取深度是100,则位元的百分位数将是100%(100*50%/50)。
根据本文所述的一些实施方式,在步骤124和126中,可以将具有读取深度的泊松分布的隐马尔可夫模型(HMM)应用于百分位数值。隐马尔可夫模型可以将每个位元的百分位数转换为五个CNV状态之一:CN=0(缺失),CN=1(缺失),CN=2(正常),CN=3(重复)和CN>3(重复)。
在一些实施方式中,如果将位元尺寸设定为较小值(例如50个碱基对),则在分配的CNV状态中可能会出现噪音。使用更大尺寸的位元可能会降低噪音,但也会降低对小CNV的灵敏度。因此,根据本文所述的一些实施方式,在步骤128中合并相邻CNV可以减少在CNV状态中的噪音。如果CNV状态的长度短于5Kb,则可以将该状态与相邻状态合并。该合并步骤可能会导致JAX-CNV的分辨率为5Kb。
在一些情况下,CNV状态合并可能会合并包含太多不同状态的区域。为防止这种情况,如果将区域的原始状态分配为小于序列合并区域长度的80%,则将停止CNV状态合并并恢复原始状态和遗传区域。在识别出复杂区域并停止合并后,然后可以按其各自的序列长度对CNV状态进行排序。从最长到最短,每个CNV状态都可以在上游和下游扫描其他状态以进行进一步合并。
根据本文所述的一些实施方式,然后,在步骤130中,可以通过过滤CNV状态来产生候选CNV。可以将每个CNV状态区域分成10个等长的位元。可以为每个位元分配一个唯一性值,所述唯一性值对应于独特的(例如在遗传序列内仅出现一次)位元中k-mer的数量。如果位元的唯一性值低于阈值(例如,如果独特的k-mer的百分位数低于60%,尽管可以使用任何适宜阈值),则位元可以被顺序地过滤。
在一些实施方式中,可以在过滤之后应用聚类算法(未显示)以进一步聚类候选CNV片段。例如,如结合图1B进一步描述的,可以应用具有噪音的基于密度的空间聚类(DBSCAN)算法131。可以基于剩余候选CNV片段134在遗传序列内的位置进行分类。然后,可以基于以下两个条件将CNV片段134分为不同的原始簇135:a)任何两个连续CNV片段134之间的距离包括少于3,000,000个碱基对;或b)位于原始簇区域中的所有片段的类型(例如删除、重复)相同。接下来,对于每个原始聚类135,每个连续片段对f
为了克服具有小而稀疏片段的原始聚类的聚类偏倚,t可以将连续片段对的距离设置为d>3,而将不连续片段对的距离设置为d
对于仅有两个CNV片段的原始聚类(表示为f
图1C显示了根据本文所述技术的一些实施方式的JAX-CNV管线140的替代示意图,JAX-CNV管线140被配置为从遗传序列数据调用CNV。图1C可以显示出图1A的数据管线100步骤所应用的变换,以输入遗传序列数据。在一些实施方式中,JAX-CNV管线140可以通过硬件(例如使用ASIC、FPGA或任何其他适宜电路)、软件(例如通过使用计算机处理器执行软件)或者其任何适宜组合来实现。图1C的水平轴表示从遗传序列的第一个碱基对到最后一个碱基对的遗传序列的长度。
在一些实施方式中,如在步骤142中所示,然后,可以将BAMbook文件118划分为包含相同数量的碱基对的位元,并且可以计算每个位元的读取深度。如在步骤144中所示,可以将每个位元的读取深度进一步转换为从0%至180%的百分位数,其中50%代表基线读取深度。例如,如果遗传序列的读取深度是50,且位元的读取深度是100,则位元的百分位数将是100%(100*50%/50)。步骤142和144可以对应于图1A的步骤122。
接下来,在一些实施方式中,如在步骤146中所示,可以将具有读取深度的泊松分布的隐马尔可夫模型应用于百分位数值。隐马尔可夫模型可以将每个位元的百分位数转换为五个CNV状态之一:CN=0(缺失),CN=1(缺失),CN=2(正常),CN=3(重复)和CN>3(重复)。步骤146可以对应于图1A的步骤124和126。
在一些实施方式中,在步骤142中,如果将位元尺寸设定为较小值(例如50个碱基对),则在分配的CNV状态中可能会出现噪音。使用更大位元尺寸可能会降低噪音,但也可能会降低对小CNV的灵敏度。因此,根据本文所述的一些实施方式,在步骤148、150、152、154和156中合并相邻的CNV可以减轻在CNV状态中的噪音。步骤148、150、152、154和156可以对应于图1A的步骤128的一些或全部。在步骤148中,如果CNV状态的长度短于5Kb,则可以将该状态与相邻状态合并。
在一些情况下,如在步骤150中所示,CNV状态合并可能会合并包含太多不同状态的区域。如在步骤152中所示,为防止这种情况,如果将区域的原始状态分配为小于序列合并区域长度的80%,则将停止CNV状态合并并恢复原始状态和遗传区域。如在步骤154中所示,在识别出复杂区域并停止合并后,然后可以按其各自的序列长度对CNV状态进行排序。如在步骤156中所示,从最长到最短,每个CNV状态都可以在上游和下游扫描其他状态以进行进一步合并。如结合图1B所描述的,在CNV状态合并期间可以应用应用聚类算法的附加步骤。
根据本文所述的一些实施方式,在步骤158中,然后可以通过过滤CNV状态产生候选CNV。步骤158可以对应于图1A的步骤130的一些或全部。可以将每个CNV状态区域分成10个等长的位元。可以为每个位元分配一个唯一性值,所述唯一性值对应于独特的(例如在遗传序列内仅出现一次)位元中k-mer的数量。如果位元的唯一性值低于阈值(例如,如果独特的k-mer的百分位数低于60%,尽管可以使用任何适宜阈值),则位元可以被顺序地过滤。
图2是描述根据本文所述技术的一些实施方式的识别遗传序列中的至少一个CNV的过程200的流程图。在一些实施方式中,过程200的部分或全部可以通过硬件(例如使用ASIC、FPGA或任何其他适宜电路)、软件(例如通过使用计算机处理器执行软件)或者其任何适宜组合来实现。
根据本文所述的一些实施方式,在步骤202中,可以扫描待分析的遗传序列以鉴定至少一个常染色体中的至少一个独特的遗传序列。如结合图1A所描述的,步骤202可以对应于步骤120。当在所述区域内的每个k-mer仅出现一次并且所述区域的尺寸大于20Kb(例如20,000个碱基对)时,可以认为遗传区域是独特的。
根据本文所述的一些实施方式,在步骤204中,可以将遗传序列分成多个位元。在一些实施方式中,所述位元可以包含50个碱基对。在一些实施方式中,所述位元可以包含25个碱基对、50个碱基对或100个碱基对。在一些实施方式中,如果将位元尺寸设定为较小值(例如50个碱基对),则在随后步骤中在分配的CNV状态中可能会出现噪音。使用更大位元尺寸可能会降低噪音,但也可能会降低对小CNV的灵敏度。位元尺寸的选择可能取决于所需的灵敏度以及可接受的噪音水平。
根据本文所述的一些实施方式,在步骤206中,可以针对每个位元计算CNV状态。步骤206可以对应于如结合图1A所述的步骤124和126和/或如结合图1C所述的步骤146。根据本文所述的一些实施方式,可以将具有读取深度的泊松分布的隐马尔可夫模型(HMM)应用于每个位元的读取深度值得百分位数表示。隐马尔可夫模型可以将每个位元的百分位数转换为五个CNV状态之一:CN=0(缺失),CN=1(缺失),CN=2(正常),CN=3(重复)和CN>3(重复)。
根据本文所述的一些实施方式,在步骤208中,可以对CNV状态进行过滤以识别遗传序列中的至少一个CNV。步骤208可以对应于如结合图1A所述的步骤130和/或如结合图1C所述的步骤158。可以将每个CNV状态区域分成10个等长的位元。可以为每个位元分配一个唯一性值,所述唯一性值对应于独特的(例如在遗传序列内仅出现一次)位元中k-mer的数量。如果位元的唯一性值低于阈值(例如,如果独特的k-mer的百分位数低于60%,尽管可以使用任何适宜阈值),则位元可以被顺序地过滤。然后,可以基于过滤的CNV状态生成候选CNV。
图3是描述根据本文所述技术的一些实施方式的诊断由遗传序列中的至少一个CNV引起的病症的过程300的流程图。在一些实施方式中,过程300的部分或全部可以通过硬件(例如使用ASIC、FPGA或任何其他适宜电路)、软件(例如通过使用计算机处理器执行软件)或者其任何适宜组合来实现。
根据本文所述的一些实施方式,在步骤302中,可以扫描待分析的遗传序列以鉴定至少一个常染色体中的至少一个独特的遗传区域。步骤302可以对应于如结合图1A所述的步骤120和/或如结合图2所述的步骤202。当在所述区域内的每个k-mer仅出现一次并且所述区域的尺寸大于20Kb(例如20,000个碱基对)时,可以认为遗传区域是独特的。
根据本文所述的一些实施方式,在步骤304中,可以将遗传序列分成多个位元。如结合图2所述的,步骤304可以对应于步骤204。在一些实施方式中,所述位元可以包含50个碱基对。在一些实施方式中,所述位元可以包含25个碱基对、50个碱基对或100个碱基对。在一些实施方式中,如果将位元尺寸设定为较小值(例如50个碱基对),则在随后步骤中在分配的CNV状态中可能会出现噪音。使用更大位元尺寸可能会降低噪音,但也可能会降低对小CNV的灵敏度。位元尺寸的选择可能取决于所需的灵敏度以及可接受的噪音水平。
根据本文所述的一些实施方式,在步骤306中,可以针对每个位元计算CNV状态。步骤306可以对应于如结合图1A所述的步骤124和126,如结合图1C所述的步骤146和/或如图2所述的步骤206。根据本文所述的一些实施方式,可以将具有读取深度的泊松分布的隐马尔可夫模型(HMM)应用于每个位元的读取深度值得百分位数表示。隐马尔可夫模型可以将每个位元的百分位数转换为五个CNV状态之一:CN=0(缺失),CN=1(缺失),CN=2(正常),CN=3(重复)和CN>3(重复)。
根据本文所述的一些实施方式,在步骤308中,可以对CNV状态进行过滤以识别遗传序列中的至少一个CNV。步骤308可以对应于如结合图1A所述的步骤130,如结合图1C所述的步骤158和/或如结合图2所述的步骤208。可以将每个CNV状态区域分成10个等长的位元。可以为每个位元分配一个唯一性值,所述唯一性值对应于独特的(例如在遗传序列内仅出现一次)位元中k-mer的数量。如果位元的唯一性值低于阈值(例如,如果独特的k-mer的百分位数低于60%,尽管可以使用任何适宜阈值),则位元可以被顺序地过滤。然后,可以基于过滤的CNV状态生成候选CNV。
根据本文所述的一些实施方式,在步骤310中,可以确定所识别的候选CNV是否包含致病性CNV。致病性CNV可以包含与针对众所周知的复制和/或缺失疾病或者在本领域中是有据可查的基因组坐标重叠的CNV。致病性CNV可以例如与诸如但不限于以下的病症相关:自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
在一些实施方式中,确定所识别的候选CNV是否由致病性CNV构成可以包括对JAX-CNV输出的候选CNV进行人工审核的过程。在一些实施方式中,确定所识别的候选CNV是否包含致病性CNV可以是使用计算系统(例如结合图9所述的计算系统900)的部分或完全自动化过程。
根据本文所述的一些实施方式,在步骤312中,可以基于确定所识别的候选CNV包含致病性CNV来诊断病症。病症可以是被诊断为以下中的任何一种的病症:例如,自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
图4是描述根据本文所述技术的一些实施方式的治疗由遗传序列中的至少一个CNV引起的病症的过程400的流程图。在一些实施方式中,过程400的部分或全部可以通过硬件(例如使用ASIC、FPGA或任何其他适宜电路)、软件(例如通过使用计算机处理器执行软件)或者其任何适宜组合来实现。
根据本文所述的一些实施方式,在步骤402中,可以扫描待分析的遗传序列以鉴定至少一个常染色体中的至少一个独特的遗传区域。步骤402可以对应于如结合图1A所述的步骤120,如结合图2所述的步骤202和/或如结合图3所述的步骤302。当在所述区域内的每个k-mer仅出现一次并且所述区域的尺寸大于20Kb(例如20,000个碱基对)时,可以认为遗传区域是独特的。
根据本文所述的一些实施方式,在步骤404中,可以将遗传序列分成多个位元。步骤404可以对应于如结合图2所述的步骤204和/或如结合图3所述的步骤304。在一些实施方式中,所述位元可以包含50个碱基对。在一些实施方式中,所述位元可以包含25个碱基对、50个碱基对或100个碱基对。在一些实施方式中,如果将位元尺寸设定为较小值(例如50个碱基对),则在随后步骤中在分配的CNV状态中可能会出现噪音。使用更大位元尺寸可能会降低噪音,但也可能会降低对小CNV的灵敏度。位元尺寸的选择可能取决于所需的灵敏度以及可接受的噪音水平。
根据本文所述的一些实施方式,在步骤406中,可以针对每个位元计算CNV状态。步骤406可以对应于如结合图1A所述的步骤124和126,如结合图1C所述的步骤146,如结合图2所述的步骤206和/或如结合图3所述的步骤306。根据本文所述的一些实施方式,可以将具有读取深度的泊松分布的隐马尔可夫模型(HMM)应用于每个位元的读取深度值得百分位数表示。隐马尔可夫模型可以将每个位元的百分位数转换为五个CNV状态之一:CN=0(缺失),CN=1(缺失),CN=2(正常),CN=3(重复)和CN>3(重复)。
根据本文所述的一些实施方式,在步骤408中,可以对CNV状态进行过滤以识别遗传序列中的至少一个CNV。步骤408可以对应于如结合图1A所述的步骤130,如结合图1C所述的步骤158,如结合图2所述的步骤208和/或如结合图3所述的步骤308。可以将每个CNV状态区域分成10个等长的位元。可以为每个位元分配一个唯一性值,所述唯一性值对应于独特的(例如在遗传序列内仅出现一次)位元中k-mer的数量。如果位元的唯一性值低于阈值(例如,如果独特的k-mer的百分位数低于60%,尽管可以使用任何适宜阈值),则位元可以被顺序地过滤。然后,可以基于过滤的CNV状态生成候选CNV。
根据本文所述的一些实施方式,在步骤410中,可以确定所识别的候选CNV是否包含致病性CNV。如结合图3所述的,步骤410可以对应于步骤310。致病性CNV可以包含与针对众所周知的复制和/或缺失疾病或者在本领域中是有据可查的基因组坐标重叠的CNV。致病性CNV可以例如与诸如但不限于以下的病症相关:自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
在一些实施方式中,确定所识别的候选CNV是否由致病性CNV构成可以包括对JAX-CNV输出的候选CNV进行人工审核的过程。在一些实施方式中,确定所识别的候选CNV是否包含致病性CNV可以是使用计算系统(例如结合图9所述的计算系统900)的部分或完全自动化过程。
根据本文所述的一些实施方式,在步骤412中,可以基于确定所识别的候选CNV是否由致病性CNV组成来诊断病症。如结合图3所述的,步骤412可以对应于步骤312。病症可以是被诊断为以下中的任何一种的病症:例如,自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
根据本文所述的一些实施方式,在步骤414中,可以实施治疗以减轻与步骤412诊断的病症相关的一种或多种症状。治疗可以包括遗传咨询、职业治疗、言语治疗、物理治疗和/或心血管药物或外科手术中的一种或多种。
发明人进一步认识并意识到,CNV检测的常规方法已经达到某些临床基准。因此,发明人已针对准确度和灵敏度在来自Coriell研究所的与各种体质障碍(即,DiGeorge综合征、Williams综合征、猫叫综合征、Smith-Magenis综合征、Wolf-Hirschhorn综合征、Miller-Dieker无脑回畸形综合征、法洛氏四联综合征、1p缺失综合征和Angelman综合征)相关的31个样品(如表1中所示)中检测了JAX-CNV。Coriell研究所报告,测试样品中共计存在45个CNV(25个缺失和20个重复,尺寸范围从101千碱基(Kb)至94兆碱基(Mb)),其为JAX-CNV的灵敏度分析设定了初始基线。
在45个Coriell登记的CNV中,有41个被鉴定为是致病性的。通过Illumina配对末端测序对这些样品进行WGS,读取长度为2x150bp,读取深度约为40。将BWA-MEM用于针对GRCh38人参照基因组(chr1-22,X、Y和M)的比对,然后应用JAX-CNV进行CNV调用。如表1中所示,JAX-CNV从WGS数据库中准确地检测了全部45个Coriell登记的CNV,其中“O”表示通过所述方法在不同读取深度下检测到的CNV。“*”表示CNV在检测方法之间不是50%相互重叠,而是在人工审核中被回收的。阴影单元格表示未调用CNV。
在杰克逊实验室CLIA认证实验室的标准操作规程(在本文中为“JAX-GM”)之后,通过临床验证的Affymetrix CytoScan HD平台(Affymetrix,Santa Clara,CA)对这31个待测样品进行进一步评估,以检测染色体失衡。与其他一些临床实验室一样,JAX-GM的临床实验室为使用CMA进行临床CNV检测提供了更高分辨率(即,低于50Kb)。CNV微阵列分析是由在JAX-GM的细胞遗传学实验室使用Affymetrix Cytoscan HD平台进行的。该阵列包括2,696,550个探针,其中包括743,304个SNP探针和1,953,246个非多态性拷贝数探针。RefSeq基因的平均探针间隔为880bp,代表96%的基因。按照生产厂商的方案进行DNA标记、玻片杂交、洗涤和扫描。CEL文件是通过Affymetrix GeneChip Command Console软件从扫描的阵列图像文件生成的,并导入到Affymetrix染色体分析套件(ChAS v3.3)软件中。拷贝数数据文件(CYCHP文件)是使用Affymetrix CytoScan HD阵列版本NA36(hg38)作为参照生成的。使用下述过滤标准分析数据:大于50Kb,至少有50个连续标记物。
JAX-GM临床验证的CMA平台报告了共计105个CNV(每个样品0-9个CNV)。由于阵列上的探针覆盖范围有限,CMA平台未能检测到六个Coriell登记的CNV,包括四个缺失(101.5Kb-119 Kb)和两个重复(118Kb-148.8Kb)(表1),因为至少需要50个阵列探针才能确保CMA平台进行可靠且高质量的CNV调用。结果,JAX-CNV能够识别出Coriell报告的全部45个染色体畸变,而JAX-GM CMA却错过了其中的6个(JAX-GM CMA平台的假阴性率为13.33%)。
表1
根据本文所述技术的一些实施方式,图5A和图5B显示了表1的总结,比较了通过由Coriell研究所进行的CMA,通过由杰克逊实验室进行的CAM鉴定的以及通过JAX-CNV算法的全基因组序列(WGS)分析针对31个样品检测的CNV缺失(图5A)和重复(图5B)的比较。由Coriell研究所进行的CMA由内圈表示,由JAX-GM进行的CMA由中圈表示,以及由JAX-CNV进行的分析由外圈表示,划分代表围绕圈的圆周排列的单个染色体。
由于Affymetrix CytoScan HD是在JAX-GM的经过临床验证的平台,因而,在理想情况下,在该平台识别的所有CNV应被JAX-CNV检测,以显示出使用JAX-CNV作为一线诊断测定进行WGS的潜力。在JAX-GM的CMA平台的CNV尺寸截止值≥50Kb。通过此标准,JAX-GM CMA平台从31个测试样品中识别出112个CNV,其中包括45个Coriell登记CNV中的39个。在这112个CNV中,有4个缺失和3个重复是边界质量调用,因此随后通过ddPCR测定进行了验证。设计了针对这七个区域的ddPCR测定,但由于该基因组区域的复杂性,在16p13处(chr16:14961449-15030399)获得了69Kb的增益。
根据Bio-Rad QX200
利用CMA平台,通过ddPCR确认了其余6个畸变(4个缺失和2个重复)为假阳性。最有趣的假阳性CNV是位于共同复制区域的在6p25的缺失。1000基因组计划3,25包括2,504个样品,在26个研究人群中显示出这种重复的0.99等位基因频率。因此,该“缺失”实际上可能是正常的两个副本数量的结果,但由于参照样品带有重复而显示为缺失。因此,将105个CNV(61个缺失和44个重复)用于与下文所述的JAX-CNV进行比较。
当应用50%相互重叠来评价CNV调用时,JAX-CNV成功地从WGS数据(图3)中识别了全部105个CNV(65个被识别为致病性的)。值得注意的是,有两个缺失(GM11428和GM14164)和四个重复(GM03997、GM09687、GM11428和GM13590)不符合CMA调用50%相互重叠的基准,但其仍位于具有更小或更大尺寸的相同区域中。图6A显示了根据本文所述技术的一些实施方式的,作为CNV尺寸的函数以及针对CNV缺失和CNV重复两者,由杰克逊实验室在表1中所述的31个样品上进行的由JAX-CNV检测的独特CNV的数量(浅灰)以及由JAX-CNV和CMA两者检测的CNV的数量(深灰)。图6B显示了根据本文所述技术的一些实施方式,针对每个基因突变,由杰克逊实验室在表1中所述的31个样品上进行的由JAX-CNV检测的独特CNV的数量(浅灰)以及由JAX-CNV和CMA两者检测的CNV的数量(深灰)。总体而言,与由JAX-GM进行的CMA相比JAX-CNV检测到多754个CNV,真对每个样品平均多10个CNV。检测到的280个CNV被认为具有致病性。超过半数的JAX-CNV独特调用小于100Kb,且89%小于300Kb。这可能是由于WGS和JAX-CNV提供了比基于阵列的技术更高分辨率的事实,基于阵列的技术受到使用的探针数量的限制。
尽管NGS的成本下降了,但是发明人已经认识并意识到,当将WGS视为临床诊断中的一线测定时,其价格仍然高得令人望而却步。根据本文所述的一些实施方式,为了解决这个问题并证明JAX-CNV的能力,发明人对WGS数据的读取深度进行了下采样,并评估了JAX-CNV在这些较低读取深度上的敏感性。最初对这些样品进行了测序,读取深度范围从30x至48x。在对齐的BAMbook文件上,通过SAMBAMBA35执行了不同覆盖率的模拟。基于原始WGS数据产生了包括30x、20x、15x、10x和9x在内的一系列读取深度。然后,将JAX-CNV应用于具有不同读取深度的下采样WGS数据。
在45个Coriell登记的CNV中,有33个大于300Kb,这是CAP标准的截止尺寸。即使将读取深度降至9x,对于检测这些大于300Kb的CNV,JAX-CNV仍然保持100%的灵敏度。使用9x的读取深度可以显着降低WGS用于临床诊断的成本。
对于小于300Kb的其余12个CNV,JAX-CNV获得了可重复的结果,可将读取深度降至15x,或原始读取深度的31.25-50%(见表1)。在10x测序读取深度处,JAX-CNV无法识别两个重复,一个是在GM14164的染色体区域22q11.21的148.8Kb重复,另一个是在GM18828的染色体区域1q31的118Kb重复。通过JAX-GM CMA也未检测到这两个重复。在9x读取深度处,JAX-CNV识别所有缺失,包括JAX-GM CMA无法识别的4个调用;然而,JAX-CNV遗漏了7个重复,包括在GM03997的染色体区域5q35的130Kb重复,在GM09711的染色体区域2q13的140Kb重复,在GM13480的染色体区域9p24的107Kb重复,在GM13590的染色体区域9q13的120Kb重复,在GM13590的染色体区域17q11的101Kb重复,在GM14164的染色体区域22q11的148Kb重复,以及在GM18828的染色体区域1q31的118Kb重复。
为了更好地理解测序读取深度的影响,发明人将分析扩展到了由JAX-GM CMA调用的105个CNV。图7A显示了从顶部到底部和针对通过JAX-GM CMA调用的105个CNV,针对降低的读取深度的值,由Coriell研究所进行CMA,由JAX-GM进行CMA以及通过JAX-CNV进行WGS分析。对于所有105个CNV(61个缺失和44个重复),在20x读取深度处实现了100%一致性。然而,随着读取深度降低,方法之间的一致性降低。分别对于15x、10x和9x序列读取深度,JAX-CNV分别遗漏了1个CNV(重复)、4个CNV(1个缺失和3个重复)和15个CNV(1个缺失和14个重复)。
图7B显示了根据本文所述技术的一些实施方式,作为覆盖率的函数以及针对CNV缺失,由杰克逊实验室在31个样品上进行的JAX-CNV与CMA之间的一致性。图7C显示了根据本文所述技术的一些实施方式,作为覆盖率的函数以及针对CMV重复,由杰克逊实验室在31个样品上进行的JAX-CNV与CMA之间的一致性。遗漏的CNV长度的范围从79Kb至311Kb。因此,WGS上JAX-GM CMA和JAX-CNV之间的一致性为20x序列读取深度为100%,15x序列读取深度为99%,10x测序读取深度为96%,以及9x测序读取深度为87%。与具有15x或更低覆盖率的重复(图7C)相比,缺失(图7B)显示出更高的一致性。
图8示意性地描述了可以在其上实现本公开内容的任何方面的说明性计算机800。
在图8中所示的实施方式中,计算机800包括处理单元801,其具有一个或多个处理器,以及非暂时性计算机可读存储介质802,其可以包括例如易失性和/或非易失性存储器。存储器802可以存储一个或多个指令以对处理单元801进行编程以执行本文描述的任何功能。除了系统存储器802之外,计算机800还可以包括其他类型的非暂时性计算机可读介质,诸如存储器805(例如一个或多个磁盘驱动器)。存储器805还可以存储一个或多个应用程序和/或由应用程序使用的资源(例如软件库),其可以被加载到存储器1302中。
计算机800可以具有一个或多个输入设备和/或输出设备,如图8中所示的设备806和807。这些设备尤其可以用于呈现用户界面。可以用于提供用户界面的输出设备的实例包括用于视觉呈现输出的打印机或显示器,以及用于听觉呈现输出的扬声器或其他声音生成设备。可以用于用户界面的输入设备的实例包括键盘和定点设备,如鼠标、触摸板和数字化平板电脑。作为另一个实例,输入设备807可以包括用于捕获音频信号的麦克风,并且输出设备806可以包括用于视觉渲染的显示屏和/或用于听觉渲染所识别的文本的扬声器。作为另一个实例,输入设备807可以包括传感器(例如起搏器中的电极),以及输出设备806可以包括被配置为解释和/或渲染由传感器收集的信号的设备(例如,被配置为基于起搏器中电极收集的信号生成心电图的设备)。
如图8中所示,计算机800还可以包括一个或多个网络接口(例如网络接口810),以使得能够通过各种网络(例如网络820)进行通信。网络的实例包括局域网或广域网,如企业网络或因特网。此类网络可以基于任何适当的技术且可以根据任何适当的协议进行操作,并且可以包括无线网络、有限网络或光纤网络。此类网络可以包括模拟和/或数字网络。
此外,本技术可以体现为以下配置:
(1)一种用于检测遗传序列中的拷贝数变异(CNV)的方法,所述方法包括使用处理器执行以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
(2)根据(1)所述的方法,其中所述遗传序列是部分基因组序列。
(3)根据(1)所述的方法,其中所述遗传序列是全基因组序列(WGS)。
(4)根据(1)-(3)中任一项所述的方法,还包括将所述遗传序列与参照基因组进行比对。
(5)根据(1)-(4)中任一项所述的方法,其中识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
(6)根据(1)-(5)中任一项所述的方法,还包括计算所述遗传序列的读取深度。
(7)根据(1)-(6)中任一项所述的方法,还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
(8)根据(1)-(7)中任一项所述的方法,其中计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
(9)根据(1)-(8)中任一项所述的方法,其中将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
(10)根据(1)-(9)中任一项所述的方法,其中将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
(11)根据(1)-(10)中任一项所述的方法,其中其中所述多个位元中的每个位元包含50个碱基对。
(12)根据(1)-(11)中任一项所述的方法,其中还包括将所述多个位元中的一个或多个位元合并。
(13)根据(1)-(12)中任一项所述的方法,其中过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
(14)根据(13)所述的方法,其中通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
(15)至少一种非暂时性计算机可读存储介质,其上存储计算机可读指令,当由处理器执行时,所述计算机可读指令使处理器执行一种检测遗传序列中的拷贝数变异(CNV)的方法,所述方法包括以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
(16)根据(15)所述的至少一种非暂时性计算机可读存储介质,其中所述遗传序列是部分基因组序列。
(17)根据(15)所述的至少一种非暂时性计算机可读存储介质,其中所述遗传序列是全基因组序列(WGS)。
(18)根据(15)-(17)中任一项所述的至少一种非暂时性计算机可读存储介质,所述方法还包括将所述遗传序列与参照基因组进行比对。
(19)根据(15)-(18)中任一项所述的至少一种非暂时性计算机可读存储介质,其中识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
(20)根据(15)-(19)中任一项所述的至少一种非暂时性计算机可读存储介质,还包括计算所述遗传序列的读取深度。
(21)根据(15)-(20)中任一项所述的至少一种非暂时性计算机可读存储介质,所述方法还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
(22)根据(15)-(21)中任一项所述的至少一种非暂时性计算机可读存储介质,其中计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
(23)根据(15)-(22)中任一项所述的至少一种非暂时性计算机可读存储介质,其中将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
(24)根据(15)-(23)中任一项所述的至少一种非暂时性计算机可读存储介质,其中所述多个位元中的每个位元包含50个碱基对。
(25)根据(15)-(24)中任一项所述的至少一种非暂时性计算机可读存储介质,所述方法还包括将所述多个位元中的一个或多个位元合并。
(26)根据(15)-(25)中任一项所述的至少一种非暂时性计算机可读存储介质,其中过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
(27)根据(26)所述的至少一种非暂时性计算机可读存储介质,其中通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
(28)一种用于检测遗传序列中的拷贝数变异(CNV)的系统,所述系统包括:可操作地连接至计算机可读存储器的至少一个处理器,所述计算机可读存储器包含指令,当由所述至少一个处理器执行时,所述指令使所述至少一个处理器执行包含以下步骤的方法:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述遗传序列的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV。
(29)根据(28)所述的系统,其中所述遗传序列是部分基因组序列。
(30)根据(28)所述的系统,其中所述遗传序列是全基因组序列(WGS)。
(31)根据(28)-(30)中任一项所述的系统,还包括将所述遗传序列与参照基因组进行比对。
(32)根据(28)-(31)中任一项所述的系统,其中识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
(33)根据(28)-(32)中任一项所述的系统,还包括计算所述遗传序列的读取深度。
(34)根据(28)-(33)中任一项所述的系统,还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
(35)根据(28)-(34)中任一项所述的系统,其中计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
(36)根据(28)-(35)中任一项所述的系统,其中将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
(37)根据(28)-(36)中任一项所述的系统,其中将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
(38)根据(28)-(37)中任一项所述的系统,其中所述多个位元中的每个位元包含50个碱基对。
(39)根据(28)-(38)中任一项所述的系统,还包括将所述多个位元中的一个或多个位元合并。
(40)根据(28)-(39)中任一项所述的系统,其中过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
(41)根据(40)所述的系统,其中通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
(42)一种诊断由至少一个致病性拷贝数变异(CNV)引起的病症的方法,所述方法包括:使用处理器执行以下步骤:扫描遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述WGS的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述遗传序列中的至少一个CNV;确定所识别的至少一个CNV是至少一个致病性CNV;和基于所确定的至少一个致病性CNV来诊断病症。
(43)根据(42)所述的方法,其中所述病症是以下选择之一:自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
(44)根据(42)-(43)中任一项所述的方法,其中所述遗传序列是部分基因组序列。
(45)根据(42)-(44)中任一项所述的方法,其中所述遗传序列是全基因组序列(WGS)。
(46)根据(42)-(46)中任一项所述的方法,其中识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
(47)根据(42)-(46)中任一项所述的方法,还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
(48)根据(42)-(47)中任一项所述的方法,其中计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
(49)根据(42)-(48)中任一项所述的方法,其中将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
(50)根据(42)-(49)中任一项所述的方法,其中将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
(51)根据(42)-(50)中任一项所述的方法,其中所述多个位元中的每个位元包含50个碱基对。
(52)根据(42)-(51)中任一项所述的方法,还包括将所述多个位元中的一个或多个位元合并。
(53)根据(42)-(52)中任一项所述的方法,其中过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
(54)根据(53)所述的方法,其中通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
(55)一种治疗由至少一个致病性拷贝数变异(CNV)引起的病症的方法,所述方法包括:使用处理器执行以下步骤:扫描所述遗传序列以识别至少一个常染色体内的至少一个独特遗传区域;将所述遗传序列分成多个位元,所述多个位元中的每个位元包含所述WGS的多个碱基对;计算所述多个位元中的每个位元的CNV状态;和过滤所述CNV状态以识别所述WGS中的至少一个CNV;确定所识别的至少一个CNV是至少一个致病性CNV;基于所述至少一个致病性CNV来诊断病症;和实施治疗以减轻所诊断的病症的一种或多种症状。
(56)根据(55)所述的方法,其中所述病症是以下选择之一:自闭症谱系病症、癫痫、精神分裂症、TAR综合征、HNPP综合征、3q29微缺失综合征、Sotos综合征、8p23.1缺失综合征、Langer-Giedion综合征、WAGR综合征、Koolen-de Vries综合征、Beckwith-Wiedemann综合征、DiGeorge综合征、腓骨肌萎缩症、Miller-Dieker无脑回畸形综合征、Angelman综合征、Williams综合征、18p缺失综合征、猫叫综合征、Smith-Magenis综合征、1p缺失综合征、Prader-Willi综合征、De Grouchy综合征、Xp11.2重复综合征和Wolf-Hirschhorn综合征。
(57)根据(55)-(56)中任一项所述的方法,其中所述遗传序列是部分基因组序列。
(58)根据(55)-(56)中任一项所述的方法,其中所述遗传序列是全基因组序列(WGS)。
(59)根据(55)-(58)中任一项所述的方法,其中识别所述至少一个常染色体内的至少一个独特遗传区域包括:确定所述至少一个独特遗传区域的每个25k-mer在所述遗传序列内仅出现一次;和确定所述至少一个独特遗传区域包含大于20,000个碱基对。
(60)根据(55)-(59)中任一项所述的方法,还包括:基于所述至少一个独特遗传区域的读取深度来计算所述至少一个常染色体的读取深度;将所述至少一个常染色体的所述读取深度与所述遗传序列的所述读取深度进行比较;和基于比较的读取深度来确定所述遗传序列是否包含非整倍性。
(61)根据(55)-(60)中任一项所述的方法,其中计算所述多个位元中的每个位元的CNV状态包括:计算所述多个位元中的每个位元的读取深度;将所述多个位元中的每个位元的所述读取深度转换成百分位数;和将所述百分位数转换成CNV状态。
(62)根据(55)-(61)中任一项所述的方法,其中将所述读取深度转换成百分位数包括:用所述多个位元中的每个位元的所述读取深度除以所述多个碱基对中的碱基对数量并乘以所述遗传序列的所述读取深度。
(63)根据(55)-(62)中任一项所述的方法,其中将每个位元的所述百分位数转换成CNV状态包括应用具有所述遗传序列的读取深度的泊松分布的隐马尔可夫模型(HMM)。
(64)根据(55)-(63)中任一项所述的方法,其中所述多个位元中的每个位元包含50个碱基对。
(65)根据(55)-(64)中任一项所述的方法,还包括将所述多个位元中的一个或多个位元合并。
(66)根据(55)-(65)中任一项所述的方法,其中过滤所述CNV状态包括:将合并的位元划分成多个区域,每个区域包含相等数量的碱基对;为每个区域分配唯一性值;和滤除唯一性值低于阈值的区域。
(67)根据(66)所述的方法,其中通过确定所述区域中的唯一k-mer的数量来计算所述唯一性值。
因此,已经描述了该技术的至少一个实施方式的几个方面,应当意识到的是,本领域技术人员将容易想到各种改变、修改和改进。
此类改变、修改和改进旨在作为本公开内容的一部分,并且旨在落入本发明的精神和范围内。此外,尽管指示了本发明的优点,但是应当意识到的是,并非本文描述技术的每个实施方式都将包括所描述的每个优点。一些实施方式可以不实现在此描述为有利的任何特征,并且在一些情况下,可以实现所描述的特征中的一个或多个以实现进一步的实施方式。因此,前述描述和附图仅作为实例。
可以以多种方式中的任何一种来实现本文所述技术的上述实施方式。例如,实施方式可以使用硬件、软件或其组合来实现。当以软件实现时,软件代码可以在任何适宜处理器或处理器集合上执行,无论是在单台计算机中提供还是在多台计算机中分布。此类处理器可以以集成电路形式实现,在集成电路部件中具有一个或多个处理器,包括本领域公知的商业上可购买的集成电路部件,其名称诸如CPU芯片、GPU芯片、微处理器、微控制器或协处理器。或者,可以在诸如ASIC的定制电路中实现处理器,或者可以在通过配置可编程逻辑设备而得到的半定制电路中实现。作为又一个替代方案,处理器可以是较大电路或半导体设备的一部分,无论是市售的、半定制的还是定制的。作为特定实例,一些可商购的微处理器具有多个核,使得这些核中的一个或一个子集可以构成处理器。但是,嗯可以使用任何适当形式的电路来实现处理器。
而且,本文概述的各种方法或过程可以被编码为可在允许多种操作系统或平台中的任何一种的一个或多个处理器上执行的软件。可以使用多种适宜编程语言和/或编程工具中的任何一种来编写这种软件,包括脚本语言和/或脚本工具。在一些情况下,此类软件可以被编译为在框架或虚拟机上执行的可执行机器语言代码或中间代码。另外地或可替代地,可以解释这样的软件。
本文公开的技术可以体现为用一个或多个程序编码的非暂时性计算机可读介质(或多种计算机可读介质)(例如计算机存储器、一个或多个软盘、密纹声像盘、光盘、磁带、闪存、在现场可编程门阵列或其他半导体设备中的电路配置或者其他非暂时性、有形计算机存储介质),当在一个或多个处理器上执行所述程序时,将执行用于实现上文讨论的本公开内容的各种实施方式的方法。一种或多种计算机可读介质可以是可移动的,从而可以将存储在其上的一个或多个程序加载到一个或多个不同的计算机或其他处理器上,以实现如上所述的本公开内容的各个方面。
如本文所用,术语“程序”或“软件”指可以用来对一个或多个处理器进行编程以实现如上所述的本公开内容的各个方面的任何类型的计算机代码或计算机可执行指令的集合。此外,应当意识到的事,根据该实施方式的一个方面,当被执行时,执行本公开内容的方法的一个或多个计算机程序不需要驻留在单个计算机或处理器上,而是可以以模块化的方式分布在多个不同计算机或处理器中以实现本公开内容的各个方面。
计算机可执行指令可以是由一台或多台计算机或其他设备执行的多种形式,如程序模块。程序模块可以包括执行特定任务或实现特定抽象数据类型的例程、程序、对象、部件、数据结构等。在各种实施方式中,可以根据需要组合或分布程序模块的功能。
而且,数据结构可以以任何适宜形式存储在计算机可读介质中。为简化说明,可以显示数据结构具有通过数据结构中的位置而相关的字段。此类关系同样可以通过将字段的存储分配给传达字段之间关系的计算机可读介质中的位置来实现。然而,可以使用任何适宜机制来建立数据结构的字段中的信息之间的关系,包括通过使用指针、标签或在数据元素之间建立关系的其他机制。
本发明的各个方面可以单独地,组合地使用,或者以在前述实施方式中未具体讨论的各种排布来使用,因此本发明的应用不限于在前述说明书或附图中示出的前述组件的细节和排布。例如,在一个实施方式中描述的方面可以以任何方式与其他实施方式中描述的方面组合。
而且,本发明可以体现为一种方法,已经提供了一个实例。作为该方法的一部分执行的动作可以以任何适宜方式排序。因此,可以构造实施方式,其中以与所示出的顺序不同的顺序来执行动作,即使在说明性实施方式中被示为顺序动作,其也可以包括同时执行一些动作。
权利要求中修饰权利要求元素的诸如“第一”、“第二”、“第三”之类的序数术语本身并不意味着一个权利要求元素相对于另一个或时间上的任何优先级、优先权或顺序或者执行方法的动作的时间顺序。此类术语仅被用作标记,以将具有特定名称的一个权利要求元素与具有相同名称(但使用序数术语)的另一元素区分开。
而且,本文所使用的措词和术语是出于描述的目的,并且不应被视为限制。“包括(including)”、“包含(comprising)”、“具有(having)”、“含有(containing)”、“涉及(involving)”及其变体的使用意在涵盖其后列出的项目和其他项目。
- 用于检测基因组中拷贝数变异的方法和装置
- 一种基因组拷贝数变异的检测方法及包含该方法的装置