复合组合物
文献发布时间:2023-06-19 09:44:49
技术领域
本发明涉及一种复合组合物,更具体而言,涉及一种含有具有氢生成能力的硅颗粒和/或其聚集体(凝集体,凝聚体)的复合组合物、以及含有该复合组合物的药品、氢供给材料、饲料、补充剂、食品添加物、健康食品和调配物。
背景技术
氢的应用被广泛进行,并且也存在许多备受期待的用途。作为其中的一个例子,能够看到涉及因氧化应激(oxidant stress)而引起的对象或成长的发表等。例如,在包括人类在内的动物体内中存在活性氧,上述活性氧源自:在体内通过代谢而在细胞内的线粒体中、和在紫外线照射下在皮下产生或从肺摄入的氧。虽然活性氧是生命维持所必需的,但另一方面,已知活性氧会氧化构成活体的细胞使其受损。特别是活性氧中氧化力最强的羟基自由基(hydroxyl radical)被认为会引起各种的疾病:如癌症、脑中风、心肌梗塞、糖尿病其它生活习惯病、皮肤的老化或皮肤炎等皮肤病症。因此,优选使得并未被用于对活体而言有益反应的剩余活性氧,特别是羟基自由基尽可能地不存在于体内。
如果仅处理活体内的氧化应激,则在体内生成的羟基自由基会通过与一些物质发生反应而消除。作为使羟基自由基消除的物质的一个例子,一般而言,推定为:多酚、维生素C、α-生育酚(α-tocopherol)或谷胱甘肽(glutathione)等在活体内具有的抗氧化物质。但是这些物质不仅会消除羟基自由基,就连过氧化氢等在体内具有功能的活性氧也会被消除,因此具有造成免疫力降低等危害(副作用)的可能性。另外,已知氢也能够消除羟基自由基。然而,因为氢在活性氧中仅会与羟基自由基发生反应,所以不会造成如上述的危害(副作用)。因此,提出了一种含有消除体内的羟基自由基的氢的富氢水(水素水,HydrogenWater)的生成装置的方案(例如,专利文献1)。
然而,氢在25℃时在水的溶解度是1.6ppm的极低的饱和溶解度,并且富氢水中的氢容易扩散至空气中。因此,为了将用于消除羟基自由基所需要的量的氢摄入到体内,需要较高地保持富氢水的溶解氢浓度。因此,在摄取富氢水的方法中,不可能将用于与体内的羟基自由基反应的足够量的氢摄入到体内。因此,为了使氢容易地摄入到体内,提出了一种含有氢和界面活性剂的含氢组合物的方案(专利文献2),但随着在体内的时间愈长,也不能够保持较高的氢浓度。羟基自由基的寿命短,并且在活体内会源源不绝地生成,因此,为了使其消除则需要源源不绝地将高浓度的氢供给到体内。
本发明人鉴于上述背景,公开了以硅细颗粒作为主成分,并且具有较高的氢生成能力的能够经口摄取的固体制剂(专利文献3)。
现有的技术文献
专利文献
专利文献1:日本专利第5514140号公报
专利文献2:日本专利特开2015-113331号公报
专利文献3:国际公开WO2017/130709号公报
专利文献4:国际公开WO2018/037752号公报
专利文献5:国际公开WO2018/037818号公报
专利文献6:国际公开WO2018/037819号公报
非专利文献
非专利文献1:松田等人,“通过硅纳米粒子的水分解与氢浓度,第62届应用物理学会春季学术演讲会演讲预印本,2015年,12-031
然而,假设即使摄取了富氢水,保持在25℃的状态下1升富氢水中所含有的氢量是以气体换算最大不超过18mL。另外,因为氢是最小的分子,轻且扩散速度快,所以不可能完全地保存于收容该富氢水的容器内。如果以在活体内的利用的示例作为一个例子进行说明,则在胃内富氢水中的氢大多会被气化。因此,足够量的氢无法被摄入到体内,从而有引起吞气症(aerophagia)(所谓的“打嗝”)的问题。因此,根据已知信息,不可能通过富氢水的摄取,长时间在胃内较高地保持氢浓度以及在连续而长时间断断续续地进行多次摄取。另一方面,在摄取通过表面活性剂内包氢的含氢组合物的情况下,不可能通过长时间地摄取含氢组合物而将足够量的氢摄入到体内。此外,这也会产生在胃内氢被释放的上述的问题。进一步地,在公知的方法中,假设是在皮肤上使用的情况下,因为是在空气中使用,所以会以在体内以上地非常快的氢的扩散速度在短时间扩散并飞散到空气中,因此极难或者不可能从皮肤进行吸收。此外,虽然在此针对活体内的状况进行了说明,但即使作为其它、工业用、植物用,或能源用,在实用化上也存在着各种的问题。
发明内容
本发明解决上述技术课题的至少一种,并且能够做出以下重大贡献:进一步增强具有低氧化硅(silicon sub-oxide)的硅细颗粒(微细颗粒fine particles)所有的氢的生成能力,即,使在体内或空间中长时间生成或以更高精度(准确度)提取大量的氢。此外,为了获得基于用途的任意的氢生成量,能够根据复合组合物的使用量、细颗粒的大小等任意地进行调节。
本发明人发现在至今为止的研究开发中并不是将氢气预先溶解于水等,而是找出在必要场所、部位生成氢的技术,为了大幅增加来自硅细颗粒的氢生成量,并且为了能够更长时间、进一步增强地或精度更高地进行提取,而重复进行了分析与检讨。至今,着眼于硅细颗粒所存在的环境(例如,就体内而言,肠液的pH值等、对人体不造成恶劣影响的氢生成的pH值),并且主要积累了用于使该环境最优化的研究。例如,在皮肤上等进行使用的情况下,能够通过并用适合的pH调节剂而实现相同的氢生成。另外,在上述的工业用、植物用或能源用等不同领域中,考虑适合其用途及情况的氢生成方法、氢的贮存方法以及氢的使用方法。因此,提供能够实现新颖、安全、并且有效的氢生成方法的复合组合物以及其使用的方法在产业上富有极大意义。
然而,在反复的研究开发中,着眼于硅细颗粒本身,更具体而言,着眼于硅细颗粒的表面、覆盖该表面的氧化硅膜的氧化状态和组成、氧化硅膜表面的物理性、化学性的表面构成状态以及该表面与该氧化硅膜的界面(表面)中的更加微观的物性或特征,并且本发明人发现:通过积极地活用该物性或特征,大幅地增加来自硅细颗粒的氢生成量,并且能够更长时间、进一步增强地或精度更高地提取氢生成能。另外,本发明人也同时发现:基于用途而需要的生成氢量能够根据复合组合物的调节方法、使用量、细颗粒的大小、pH值等而任意地进行调节。
本发明人针对通过实施特异的化学加工而制造的硅细颗粒的表面、覆盖该表面的氧化硅膜和/或该表面与该氧化硅膜的界面而从各种的观点进行分析和检讨。其结果明确了在制造当初的硅细颗粒上形成有特殊的状态的氧化硅膜。在进一步重复分析时,本发明人查明了以下观点:
(1)该氧化硅膜是被称为所谓的“低氧化硅”的大量含有在化学计量上与SiO
(2)存在具备硅颗粒和该氧化硅膜(含有低氧化硅和二氧化硅)的复合组合物;
(3)存在以构成该硅颗粒的硅细颗粒作为核,并且通过各种氧化硅膜(含有低氧化硅和二氧化硅)覆盖该硅细颗粒的表面的至少一部分而形成的复合组合物;以及
(4)该硅细颗粒的表面表示出亲水性,并且结合于该硅细颗粒的该表面的氢的浓度(SiH基的浓度)低、以及该氧化硅膜的表面具有大量OH基(即,SiOH基)。
本发明人发现了使用安全的硅,通过水(例如pH值为7以上)能够适当地选择任意的氢的生成速度、任意的生成气体量、任意的氢生成时间的技术、以及用于其的复合组合物。此外,如以下所示,通过详细地阐明含有低氧化硅的硅细颗粒的化学结构、分子结构、与反应对应的分子结构的变化,初次能够明确该复合组合物的详细的原子水平的状况的技术意义很大。
首先,低氧化硅大量含有硅悬挂键(silicon dangling bond)。该硅悬挂键被认为在氧化硅膜的能带隙(bandgap)内持有能级,化学物种经由其能级跳跃式地移动。因此,该硅悬挂键促进在对硅细颗粒进行氧化的化学物种(氢氧离子(OH-离子))的氧化硅膜中的扩散或迁移。另外,存在于硅与氧化硅膜的界面的硅悬挂键被认为会降低氢生成反应的活化能。
在此,本发明人得知了存在于氧化硅膜中的低氧化物(suboxide)起到了作为连锁反应介导活性中间体的作用。
根据本发明人等的至今为止的研究可知:由硅和水的反应的氢生成能够通过以下的化学反应式说明。
在化学反应式(1)中,硅和氢氧离子(OH-)反应生成SiO
[化1]
Si+2OH
2H
Si+2H
在此,化学反应式(1)并不是由一阶段的反应构成,而是由以下(4)至(7)所示的多阶段的反应构成的。
[化2]
在氢生成中,低氧化硅的量几乎不发生变化。这被认为是基于化学式(4)~(7)的反应并行而进行造成的。作为低氧化硅的Si
通过发生上述化学反应式(5)~(7),低氧化硅被氧化,并且使得在形成二氧化硅(SiO
因此,通过形成含有大量低氧化硅的氧化硅膜和/或该氧化硅膜与硅结晶层的界面,本发明人得到以下(X)和(Y)所示的见解。
(X)促进硅细颗粒与水分(特别是氢氧离子(OH
(Y)如上述的反应式(1)~(7)所示,利用OH
其结果是,本发明人通过制造上述的精心研究的硅细颗粒,实现了具备大量低氧化硅的硅细颗粒。
如上述,明确了用于形成使氢生成的优选的状态,上述状态为:硅细颗粒的氢生成能力更强,即,持续长时间地生成大量的氢气,或精度更高地进行提取。
另外,本发明人通过对上述的硅细颗粒实施进一步追加处理,不仅实现了具备大量低氧化硅的硅细颗粒,而且在宏观上观察时,也成功地使具备低氧化硅的硅细颗粒变成亲水性。具体而言,去除结合在硅细颗粒所具备的具有低氧化硅的该氧化硅膜表面的、由与硅原子反应而产生的氢原子,而实现了大量的氢氧基(OH基)结合在该氧化硅膜表面,换言之,通过实现大量的SiOH基,在宏观上观察时,具备含有低氧化硅的氧化硅膜的硅细颗粒变成为亲水性。其结果是,与水分的接触或反应被促进的硅细颗粒的的氢生成能力更强,即,持续长时间地生成大量的氢气,或能够精度更高地发挥。
如上所述,本发明人发现,通过对在具备低氧化硅的硅细颗粒的表面、覆盖该表面的氧化硅膜以及该表面与该氧化硅膜的界面的至少一部分进行研究,在微观上形成的物性或特征关系到硅细颗粒的氢生成能力更强,即,持续长时间地生成大量的氢气,或精度更高地进行提取。另外,本发明人进一步进行研究开发的结果同时也发现,包含具备至少一部分含有这样的低氧化硅的氧化硅膜的硅细颗粒的复合组合物能够作成口服以及外用的复合组合物。进一步地,本发明人还同时发现,基于用途所需的生成氢量、复合组合物的调节方法、复合组合物的使用量、构成复合组合物的细颗粒的大小或pH值等能够进行任意地调节。本发明是通过导入与至今不同的上述的各观点以及各研发而初次完成的创新。
本发明的一种复合组合物包含:硅细颗粒、覆盖该硅细颗粒的表面的至少一部分的低氧化硅(SiO
根据上述的复合组合物,覆盖硅细颗粒的表面的至少一部分的氧化硅膜包含上述的低氧化硅,因此硅细颗粒的氢生成能力更强,即,持续长时间地生成大量的氢气,或能够精度更高地进行提取。
而在本发明中,是不论结晶(不含有氧化硅膜)的直径大小,而以成为“直径”的测定对象的基本单位作为“微晶”来表示。
另外,本发明中的“硅细颗粒”的平均微晶直径在微米级以下,具体而言,是以微晶直径在1nm以上500μm以下的硅颗粒作为主要的颗粒。更狭义而言,本发明中的“硅细颗粒”的平均微晶直径是纳米级,具体而言,是以微晶直径在1nm以上50nm以下(更广义而言,是1nm以上500nm以下)的硅纳米颗粒作为主要的颗粒。另外,在本发明中“硅细颗粒”不仅是各硅细颗粒分散状态的物质,还包含多个硅细颗粒凝集而构成μm级(大约0.1μm以上500μm以下)的大小的聚集体的状态物质。此外,“硅细颗粒”的上述各数值范围仅为一个示例,因此并不限定其数值范围。基于“硅细颗粒”的用途、使用方法、所需功能等来选定适宜的微晶直径的直径。另外,只要“硅细颗粒”没有处于不产生其氢生成能力的状态,“硅细颗粒”能够在与其它物质混合后的状态下被使用。另外,本发明中的“氧化硅”是该低氧化硅与二氧化硅的混合组合物。
另外,本发明中的“含水液体”是指水或水溶液,包含例如动物(包括人)的消化道内液体。此外,“消化道内液体”表示小肠内液体以及大肠内液体。另外,自不用说“含水液体”的示例并不限于上述的示例。另外,本发明中的“pH调节剂”只要能够将pH值调节至超过7(代表性地,超过7.4)的碱性范围的药剂(以下,称为“碱性试剂”)即可,材料并没有特别限制。另外,还包含使用在动物(包括人)的皮肤上的。在将复合组合物作为活体内活性氧中和用药剂使用的情况下,优选地,使用被认定为药品(药典药品)、准药品(quasi drug,医药部外品,类药品)以及食品添加物的碱性试剂。如已述,复合组合物并不限定于动物(包括人)用。碱性试剂的示例能够采用:碳酸氢钠、碳酸钠、磷酸二氢钠、磷酸氢二钠、碳酸氢钾、碳酸钾、其它医药用、准药品用、食品用或化妆品用的pH调节剂。在其中最为通用的产品的碳酸氢钠作为医药用、准药品用或食品添加剂而广泛被应用的原因在于,兼备本发明所追求的pH调节功能以及在安全性、通用性优异的多个优点。另一方面,在工业用途上,并不限定于上述的pH调节剂,能够广泛地采用pH调节剂。在任意的pH调节剂,以不会被酸分解的方式作为优选的一个方式。特别优选的是,在经口摄取本发明的复合组合物的情况下,成为不会被胃酸分解或不易分解的形态。
发明效果
根据本发明的一种复合组组合物,覆盖硅细颗粒的表面的至少一部分的氧化硅膜包含上述的低氧化硅,因此能够让硅细颗粒的氢生成能力更强,或精度更高地进行提取。
附图说明
图1是使用X射线光电子能谱分析装置(XPS分析装置)测定第一实施方式的硅细颗粒的表面状态和第一实施方式的变形例(1)的硅细颗粒的表面状态时的Si 2p轨域的XPS谱图、以及测定使第一实施方式的变形例(1)的硅细颗粒接触约37℃的水而生成氢时的规定时间(反应时间)经过后的该表面的状态时的Si 2p轨域的XPS光谱。此外,虚线表示谱图在Si
图2是表示对于使第一实施方式的变形例(1)的硅细颗粒接触水而生成氢时的规定时间(反应时间)的、基于图1的测定结果计算出的、氧化硅膜的膜厚变化以及在其中含有的二氧化硅(SiO
图3是具有第一实施方式的粉碎工序后的低氧化硅的硅细颗粒的FT-IR谱图(上层),且是具有第一实施方式的改性工序后的低氧化硅的硅细颗粒的FT-IR谱图(下层)。
图4是表示关于构成第一实施方式的复合组合物的至少一部分的硅细颗粒的表面、覆盖该表面的含有低氧化硅的氧化硅膜和/或该表面与该氧化硅膜的界面的结构模型的概念图。
图5是表示利用碳酸氢钠和碳酸钠将第一实施方式的变形例(1)的一个示例中的硅细颗粒调节为pH10的水溶液与约36℃的水反应时的氢生成量与反应时间的关系的图表。
图6是表示利用碳酸氢钠将第三实施方式中的改性硅颗粒粉末调节为pH8.2的水溶液与约36℃的水反应时的氢生成量与反应时间的关系的图表。
图7是表示利用碳酸氢钠将第三实施方式的变形例(1)中的改性硅颗粒粉末调节为pH8.2的水溶液与约36℃的水反应时的氢生成量与反应时间的关系的图表。
图8是表示其它实施方式(3)中的、生成氢前的层状体与介质的积层结构的侧视图,(b)其它实施方式(3)中的、生成氢时的层状体与介质的积层结构的侧视图。
图9是表示其它实施方式(3)的变形例中的、层状体的结构体的侧视图。
符号说明
10a 层状体
20 基部
70 不透水性的膜
90b 介质
100 积层结构(叠层结构)
200 结构体
具体实施方式
参照附图并详细地说明本发明的实施方式。
[1]复合组合物及其制造方法
<第一实施方式>
本实施方式的复合组合物是具有氢生成能力的、含有硅细颗粒的复合组合物。此外,本实施方式的复合组合物是在覆盖上述的硅细颗粒的表面的至少一部分的氧化硅膜的中包含二氧化硅(SiO
另外,如后述,上述的氧化硅膜的膜厚根据生成氢而能够产生变化,但其范围为0.5nm以上20nm以下。此外,氧化硅膜中所含有的上述的低氧化硅的量也根据生成氢而能够产生变化,但后述的氢生成反应前的作为一个示例的优选的氧化硅膜中的该低氧化硅的组成比(硅原子数比)为10%以上。此外,虽然不需要特别设定上限,但若真要说的话,其上限为80%(即,80%以下)。
接着,对本实施方式的复合组合物的制造方法进行说明,并且对在其过程的数个工序中通过各种分析而测定或观察到的硅细颗粒的表面、覆盖该表面的氧化硅膜和/或该表面与该氧化硅膜的界面的状态进行说明。
在本实施方式的复合组合物中,使用通过乙醇等液体中利用珠磨机(bead mill)对例如市售的高纯度硅颗粒粉末(株式会社高纯度化学研究所制,粒度分布<ψ5μm(其中,代表性地,结晶粒径超过1μm的硅颗粒,纯度99.9%,i型5658硅>)进行粉碎处理而微细化的硅细颗粒作为硅颗粒。该硅细颗粒含有例如硅纳米颗粒和/或硅纳米颗粒的聚集体。此外,本实施方式并不限定于上述的作为该复合组合物的原料的硅颗粒粉末的大小、纯度、粉碎方法或分散溶剂。另外,在本实施方式以外的实施方式或变形例中所采用的示例也仅仅是一个例子,因此并不限定于该实施方式或该变形例。
作为具体的示例,进行:使用珠磨装置(AIMEX株式会社制:RMH型卧式连续式ReadyMill),将200g的高纯度硅颗粒粉末(株式会社高纯度化学研究所制,粒度分布<ψ5μm,纯度99.9%以上)作为硅颗粒分散于4000mL的99%以上的乙醇、异丙醇(IPA)或甲醇等醇类(其中,在本实施方式中为乙醇)与少量的水(例如,为0.1wt%以上10wt%以下,更优选为超过1wt%且2wt%以下)的混合溶液中,加入ψ0.5μm的氧化锆(zirconia)制的磨珠(容量2900mL),在空气中室温下以转速2500rpm进行4小时粉碎而微细化的粉碎工序。此外,从提高最终被制造的硅细颗粒以及含有该硅细颗粒的复合组合物的安全性(例如,对人体的安全性)的精度的观点出发,采用乙醇(例如99.5wt%)作为该混合溶液所含有的醇类是一种优选的方式。
此外,作为上述的粉碎工序的其它一个示例,例如,在基于珠磨装置的粉碎时间为1小时的情况下,确认了能够获得以平均粒径100nm以下的硅纳米颗粒、包含该硅纳米颗粒的大小为40nm~0.5μm的硅细颗粒、和/或该硅纳米颗粒以及包含该硅纳米颗粒的硅细颗粒的聚集体为主的颗粒的硅颗粒。
此外,通过本发明人的更进一步的研究能够发现,除了上述的溶剂以外,通过使用进一步含有少量的水的溶剂,在粉碎处理中或在其后,会形成含有更多的低氧化物的氧化硅膜。该事实对进一步提高基于硅细颗粒的氢生成能力具有重大的贡献。另外,发明人还发现,通过本实施方式的粉碎工序,硅细颗粒的形状更接近大致球状或大致圆盘状。此外,粉碎方式并不限定于珠磨粉碎法。例如,也能够适当地采用高压碰撞法等其它粉碎方法。此外,主要溶剂的种类也并不限定于乙醇。例如主要溶剂的种类能够以异丙醇等其它醇类、和/或丙酮、乙腈、THF、醋酸酯等醇类以外的溶剂来代替上述的乙醇,或与该乙醇并用。
进一步补充说明,如在后详述那样地,本发明人通过FT-IR分析来分析硅细颗粒(包含硅纳米颗粒)时,得到了有趣的发现。具体而言,认为在制造硅细颗粒的过程(上述的粉碎工序)中,醇(代表性地是乙醇)中所包含的水和/或空气中的水蒸气与该硅细颗粒反应的结果是,会发生若干氢生成反应。已知在硅与水的初期反应中,水分子部分地解离为H和OH,吸附。在粉碎工序中与水发生若干反应的结果是,氢原子会结合到该硅细颗粒与氧化硅膜的界面的硅原子上,因此认为在FT-IR分析中确认了Si-H
其后,利用减压蒸发装置将包含硅细颗粒(含有硅纳米颗粒)的、在珠磨粉碎装置中通过磨珠分离的该混合溶液加热至40℃,蒸发该混合溶液,由此能够获得干燥的硅细颗粒(含有硅纳米颗粒)。能够将干燥的该硅细颗粒放入真空容器内或氮置换的容器内而保存。此外,被粉碎而得到的硅纳米颗粒的分离方法和干燥方法并不限定于在本实施方式中所公开的方法。例如,也能够采用在生产其它颗粒时所采用的公知的分离方法和/或干燥方法。
通过上述方法得到的硅细颗粒(含有硅纳米颗粒)的一个示例主要是由微晶直径为1nm以上500nm以下的硅纳米颗粒作为主成分而组成的。更具体而言,通过X射线衍射装置(株式会社Rigaku制,SmartLab)对硅纳米颗粒进行测定的结果,作为一个示例,获得以下的数值。在体积分布中,硅的微晶的模式直径为6.6nm,中径为14.0nm,平均微晶直径为20.3nm。此外,上述的结果仅仅是作为基于一个示例的粉碎工序等的结果,因此本实施方式并不限定于上述的数值。
在使用SEM(扫描电子显微镜)观察该硅细颗粒的一个示例时,该硅细颗粒是一部分凝集,且为大约0.1μm以上的略大的不定形的形状。另外,在使用TEM(透射电子显微镜)观察凝集的各个该硅细颗粒的聚集体时,包含于该观察视野的大多数的微晶的微晶直径为约2nm以上40nm以下。此外,上述的结果仅仅是作为基于一个示例的粉碎工序等的结果,因此本实施方式并不限定于上述的数值。
根据通过经过上述的粉碎工序而制造的硅细颗粒,如后述地,覆盖该硅细颗粒的表面的至少一部分的氧化硅膜富含的低氧化硅,因此能够让硅细颗粒的氢生成能力更强,或精度更高地进行提取。更具体而言,通过采用该硅细颗粒,能够实现例如从生成开始持续20小时以上的长时间的较高的氢生成速度。
<第一实施方式的变形例(1)>
通过进一步地使在上述的第一实施方式中制造的硅细颗粒的表面接触过氧化氢水,而进行该表面的改性的改性工序也是优选的一个方式。通过该改性工序,在宏观上观察时,含有硅纳米颗粒的硅细颗粒能够使硅细颗粒变换为亲水性。此外,使硅细颗粒的表面接触过氧化氢水的方法并没有限定。例如,能够使该硅细颗粒浸渍在收容于公知的容器的3wt%的过氧化氢水(例如为约10℃~约80℃,在更低成本的观点上,为约20℃~约50℃)中,进行改性工序。另外,作为过氧化氢水的代替,通过使该硅细颗粒浸渍在臭氧水和/或过碳酸钠中也能够得到同样的改性。或者,通过使该硅细颗粒与由过氧化氢水、臭氧水以及过碳酸钠的组中选出的至少一种接触也能够得到同样的改性。
具体而言,通过进行上述的改性工序,能够去除吸附在含有硅细颗粒所具备的低氧化硅的氧化硅膜的表面的氢原子,并且能够使氢氧基(OH基)(即,SiOH基)大量存在于该氧化硅膜的表面。其结果是,在宏观上观察时,因为能够将具有低氧化硅的硅细颗粒变成亲水性,所以被精度更高地促进与水分的接触或反应的具有低氧化硅的硅细颗粒能够更加增强氢生成能力,或精度更高地发挥。因此,具有低氧化硅的硅细颗粒(即,复合组合物的示例)能够发挥作为氢供给材料的功能。另外,如上所述,从由低成本且实现安全处理的观点出发,使用室温左右的过氧化氢水来进行的改性工序也是优选的。另外,从能够使用更安全且安心(例如,对人体的影响更少)的材料来生成氢的观点出发,在本实施方式的改质工序中采用过氧化氢水与乙醇同样地,是优选的一个方式。
在使用SEM(扫描电子显微镜)观察本变形例的具有低氧化硅的硅细颗粒的一个示例时,该硅细颗粒是一部分凝集,并且为0.1μm左右以上的略大的不定形的形状。另外,在使用TEM(透射电子显微镜)观察凝集的各个该硅细颗粒的聚集体时,包含于该观察视野内的大部分微晶的微晶直径为2nm以上40nm以下。
经详细调查使本变形例的具有低氧化硅的硅细颗粒与水接触时的氢的生成速度的结果是,如后所述,确认了相较于使第一实施方式的具有低氧化硅的硅细颗粒与水接触时的氢的生成速度,加速了约10倍以上。
而在本实施方式的改性工序中,为了对硅细颗粒的表面进行改性而采用了过氧化氢水,但如上述,实现第一实施方式的变形例(1)的改性工序的材料并不限定于过氧化氢水。若举出不同于上述的示例,使用过碳酸钠来代替过氧化氢水也是能够采用的一个方式。过碳酸钠通过与水反应而生成过氧化氢水,因此能够达成与第一实施方式的变形例(1)的效果相同的效果。
<第一实施方式与第一实施方式的变形例(1)的比较分析>
以下,表示利用各种分析法对第一实施方式的具有低氧化硅的硅细颗粒的表面(包含该表面上的氧化硅。以下相同)的状态、以及第一实施方式的变形例(1)的具有低氧化硅的硅细颗粒的表面(包含该表面上的氧化硅。以下相同)的状态进行测定和考察的结果。
[XPS分析结果]
本发明人利用X射线光电子能谱分析装置(XPS分析装置)(株式会社岛津制作所制,型式:KRATOS AXIS 165)而对上述的两个实施方式的硅细颗粒(复合组合物的一个示例)的表面进行分析。
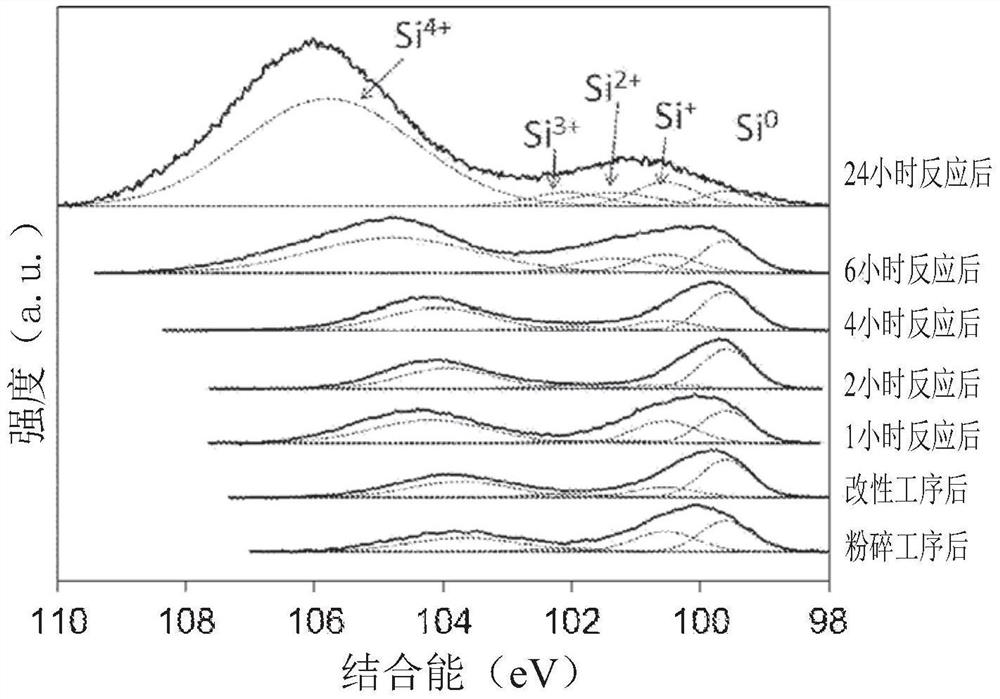
图1是使用辐射源(radiation source)为Mg Kα的XPS分析装置,在上述第一实施方式和第一实施方式的变形例(1)的具有低氧化硅的硅细颗粒与pH7且36℃的水持续反应的途中,测定各硅细颗粒的表面状态时的Si 2p轨域的XPS谱图,以及测定使第一实施方式的变形例(1)的硅细颗粒接触水而生成氢时的预定时间(反应时间)经过后的该表面状态时的Si 2p轨域的XPS谱图。
此外,图1的虚线表示谱图在Si
另外,在该分析中测定对象为:刚刚进行完粉碎处理后(图中的粉碎工序后)、刚刚进行完基于过氧化氢水的改性工序后(图中的改性工序后)、从经过改性工序的硅细颗粒与超纯水接触开始经过1小时反应后、从经过改性工序的硅细颗粒与超纯水接触开始经过2小时反应经过后、从经过改性工序的硅细颗粒与超纯水接触开始经过4小时反应后、从经过改性工序的硅细颗粒与超纯水接触开始经过6小时反应后、以及从经过改性工序的硅细颗粒与超纯水接触开始经过24小时后。此外,图1的刚刚进行完粉碎处理后(图中的粉碎工序后)的数据也能够作为上述第一实施方式的具有低氧化硅的硅细颗粒的结果而采用。
另外,图2是表示对于使第一实施方式的变形例(1)的具有低氧化硅的硅细颗粒持续接触pH7且36℃的水而生成氢时的预定时间(反应时间)的、基于图1的测定结果而算出的二氧化硅(SiO
在此,如以下所示,上述的各膜厚的计算是基于Si 2p轨域的波峰的积分强度比并且使用以下所示的方法而算出的。
<膜厚的计算方法>
Si 2p轨域的XPS谱图被分离为基于在Si
[数1]
I(oxide)=I(Si
在此,I(Si
[数2]
在此,N是硅原子的数密度,σ是光电离截面(photoionization cross-section),λ是光电子的平均自由行程,下标文字的oxide和Si表示氧化硅膜和细颗粒的值。(σ
氧化硅膜中的硅原子的数密度N
[数3]
在此,N(SiO
另外,二氧化硅膜的膜厚(t
[数4]
在此,N
[数5]
<各分析结果的解析>
如图1所示,观察到起因于硅细颗粒的Si
比较图1中的、刚刚进行完粉碎处理后(相当于第一实施方式的具有低氧化硅的硅细颗粒)的结果与第一实施方式的变形例(1)的刚刚进行完基于过氧化氢水的改性工序后(图1中的改性工序后)的结果,观察到基于二氧化硅(SiO
另外,如图1所示,确认了当氢生成的反应时间愈长,基于二氧化硅的波峰强度会增加,因此二氧化硅膜的膜厚随着反应时间的经过而增加。在该分析中,在氢生成反应完成后所测定的二氧化硅膜的膜厚为10.5nm。值得一提的是,10.5nm厚的氧化硅膜是在36℃的非常低的温度条件下形成的。此厚膜化的实现可以说是由以下原因而产生的代表性的效果之一,所述原因为:如上所述,特别是第一实施方式的变形例(1)的硅细颗粒富含的低氧化硅、在氧化硅膜上存在有丰富的OH基(即,SiOH基)和/或因为表面上几乎不存在氢原子所以变成亲水性。
另外,本发明人从图1的XPS谱图求出第一实施方式的变形例(1)的刚刚进行完基于过氧化氢水的改性工序后(图1中的改性工序后)的氧化硅膜的膜中的、二氧化硅膜的膜厚和低氧化硅的膜厚。其结果是,形成于硅细颗粒的表面上的二氧化硅膜的膜厚为1.7nm,低氧化硅的膜厚为1.0nm。
因此,上述的计算结果证明了形成于硅细颗粒的表面的约2.7nm的氧化硅膜含有大量的低氧化物。低氧化硅包含大量的硅悬挂键。硅悬挂键被认为在能带隙内持有能级,经由该能级而氧化硅细颗粒的化学物种(氢氧离子(OH
通过本发明人的研究和分析得知:在该氧化硅膜中的、该二氧化硅的第一硅原子数(A
[数学式6]
另外,从特别能够让硅细颗粒的氢生成能力更强,或进度更高地进行提取观点出发可以说:该氧化硅膜中的该低氧化硅的组成比(硅原子数比)不论粉碎处理物、粉碎处理物的基于过氧化氢等的表面处理物皆为10%以上。另外,特别是,该组成比并没有上限,但若真要说的话,其上限为80%(即,为80%以下)。另外,上述的数值范围为20%以上70%以下是更优选的一个方式。
如上所述,第一实施方式的硅细颗粒和第一实施方式的变形例(1)的硅细颗粒都是通过形成大量含有的低氧化硅的氧化硅膜、和/或大量含有低氧化硅的该氧化硅膜与硅结晶层的界面而促进硅细颗粒与氢氧离子(OH-离子)的反应。其结果是,通过采用第一实施方式的具有低氧化硅的硅细颗粒和第一实施方式的变形例(1)的具有低氧化硅的硅细颗粒,能够让硅细颗粒的氢生成能力更强,即,持续长时间地生成大量的氢气,或精度更高地进行提取。
另一方面,如图2所示,确定了在氢生成反应的途中的阶段观察到低氧化硅的膜厚为大致恒定,另一方面,仅二氧化硅膜厚增加。这证明了已述的化学反应式(4)~(7)所示的各化学反应是在实质上同时进行的。由该结果也示出了低氧化硅作为介于连锁反应的活性中间体而发挥功能。
[FT-IR分析结果]
本发明人进一步利用傅立叶变换红外光谱仪(日本分光株式会社制,型式:FT/IR-6200)对具有第一实施方式中的刚刚进行完粉碎处理后的低氧化硅的硅细颗粒的表面、以及第一实施方式的变形例(1)中的刚刚进行完基于过氧化氢水的改性工序后的具有低氧化硅的硅细颗粒的表面进行分析。图3是该粉碎工序后的具有低氧化硅的硅细颗粒的FT-IR谱图,且是该改性工序后的具有低氧化硅的硅细颗粒的FT-IR谱图。此外,下层的谱图是为了使图容易观看,因此相较于上层以四倍强度的方式呈现。
通过该FT-IR分析,使用下述的方法计算出第一实施方式的刚刚进行完粉碎处理后的具有低氧化硅的硅细颗粒的表面的OH基(即,SiOH基)的表面浓度为1.5×10
首先,由Si 2p轨域的XPS谱图求出氧化硅膜的膜厚。假设是在由Si 2p轨域的XPS谱图求出氧化硅膜的膜厚的情况下所使用的相同圆筒型的结构模型。
在该模型中,氧化硅的体积V为下式(8)。
[数7]
V=π(R+t)
另一方面,氧化硅膜的表面积S为下式(9)。
[数8]
S=π(R+t)
氧化硅膜的LO波峰的面积强度I(LO)与氧化硅膜的体积和氧化硅膜中的硅原子的原子密度N(oxide)、以及O-Si-O逆对称伸缩振动(LO声子(phonon))的振子强度σ(LO)成正例。另一方面,存在于表面的OH基的O-H伸缩振动波峰的面积强度I(OH)与氧化硅膜的表面积、OH基的表面浓度c(OH)、以及O-H伸缩振动的振子强度σ(OH)成正例。因此,LO声子的波峰强度与O-H伸缩振动的波峰强度的比例(I(OF)/I(LO))能够通过下式(10)而求出。
[数9]
利用式(9)所计算的、第一实施方式的刚刚进行完粉碎处理后的具有低氧化硅的硅细颗粒的表面的OH基的表面浓度在过氧化氢处理前为2×10
如图3所示,Si-H伸缩振动轨域的FT-IR谱图被分离成以下六支波峰:在2240cm
如图3所示,可知在粉碎工序后的具有低氧化硅的硅细颗粒中,硅细颗粒的表面的至少一部分由薄的氧化硅膜所覆盖,并且氢原子结合到该氧化硅膜的表面上。其结果是,使该氧化硅膜的表面在宏观上观察呈现出疏水性。因此,存在于该氧化硅膜的表面的H-SiO
在第一实施方式的变形例(1)的刚刚进行完基于过氧化氢水的改质工序后的具有低氧化硅的硅细颗粒中,基于H-SiO
已知在粉碎工序后的具有低氧化硅的硅细颗粒中被观察到的在2252cm
由上述可知,存在于含有低氧化硅的该氧化硅膜的表面的氢氧基(OH基)的浓度为5×10
根据上述的各个分析结果,本发明人认为:硅细颗粒的表面、覆盖该表面的氧化硅膜和/或该表面与该氧化硅膜的界面的各个状态在上述的各个化学反应中遵循以下的结构模式而发生变化。
图4是表示关于构成本实施方式的复合组合物的至少一部分的具有低氧化硅的硅细颗粒的表面、覆盖该表面的含有低氧化硅的氧化硅膜和/或该表面与该氧化硅膜的界面的结构模型的概念图。此外,(a)~(d)分别表示以下的状态。
(a)粉碎工序后
(b)改性工序后
(c)与pH7的水接触,而氢生成反应进行时(反应时间约6小时以上)
(d)氢生成反应完成时
如图4所示,首先,在粉碎工序后,硅细颗粒被2.5nm左右的氧化硅膜所覆盖。另外,该氧化硅膜的表面上存在H-SiO
其后,通过进行改性工序,该氧化硅膜的表面发生剧烈地变化。通过改性工序,大量的H-SiO
进一步地,在接触水而进行氢生成反应时(图4(c)),由该硅细颗粒生成低氧化硅的反应速度与由低氧化硅生成二氧化硅的反应速度大致相等。其结果是,低氧化硅大致恒定,另一方面,二氧化硅的量(膜厚)增加。例如,在二氧化硅膜的膜厚变为约15nm时,氢生成反应停止(图4(d))。此外,图4所记载的15nm仅为一个示例,本实施方式并不限定于该数值。另外,通过本发明人的分析可知,经过本实施方式的粉碎工序和改性工序的具有低氧化硅的硅细颗粒从基于与水接触而开始生成氢到经过168小时(7天)后的该氧化硅膜(包含二氧化硅和低氧化硅)的膜厚为3nm以上20nm以下(代表性地,为15nm以下)。因此,从氢生成开始到经过168小时(7天)后的该氧化硅膜的膜厚只要在上述的数值范围就能够确认为是精度更高的本实施方式的硅细颗粒。硅细颗粒与水的反应并不限定于此条件。作为其它的条件,可举出:例如,图5所示的在pH10、36℃的反应条件。
如上所述,通过进行本实施方式的粉碎工序和改性工序,能够实现去除吸附在具有低氧化硅的硅细颗粒所具备的氧化硅膜的表面的氢原子,并使大量的氢氧基(OH基)存在于含有低氧化硅的该氧化硅膜的表面的状态。其结果是,在宏观上观察时,硅细颗粒能够变为亲水性,因此与水分的接触和反应被更高精度地促进的硅细颗粒能够让氢生成能力更强,即,使大量的氢在体内长时间地生成,或精度更高地发挥。另外,如上所述,从在实现低成本且安全处理的观点的出发,使用室温左右的过氧化氢水来进行改性工序也是优选的。
<第一实施方式的变形例(2)>
而在第一实施方式和第一实施方式的变形例(1)中,通过使该硅细颗粒浸渍于室温左右的过氧化氢水中而进行改性工序,但改性工序的方法并不限定于第一实施方式的变形例(1)所公开的方法。例如,作为浸渍在过氧化氢水中的代替,利用公知的球磨机,并通过该球磨机而使过氧化氢水与该硅细颗粒接触也是能够采用的其它的一个方式。
在本变形例(2)中,例如使用球磨机,在空气中,与第一实施方式同样地,将市售的高纯度硅颗粒粉末作为硅颗粒(粒度分布<ψ1mm(但代表性地,结晶粒径超过100μm的硅颗粒),纯度99.9%)在乙醇与水的混合溶液中进行基于使用了直径5mm的不锈钢球的球磨法的粉碎处理。其结果是,通过在空气中分离球与一次硅颗粒,能够获得平均粒径为3μm的硅颗粒。
此外,通过使用第一实施方式中的珠磨装置而进一步对根据本变形例(2)而得到的硅颗粒进行粉碎的粉碎工序,得到与第一实施方式相同的具有低氧化硅的硅细颗粒也是能够采用的其它的一个方式。
在上述的“其它的一个方式”中,在体积分布中主要是由微晶直径为5nm以上500nm以下的具有低氧化硅的硅细颗粒作为主成分而组成的。更具体而言,通过X射线衍射装置测定具有低氧化硅的硅细颗粒的结果得到了以下的数值。在体积分布中,硅的微晶的模式直径为9.3nm,中径为26.1nm,平均微晶直径为41.5nm。使用SEM观察该硅细颗粒时,具有低氧化硅的硅细颗粒的一部分凝集,形成了0.5μm至5μm的略大的不定形的聚集体。另外,在使用TEM观察各个硅细颗粒时,其大多的微晶直径为约5nm以上50nm以下。
<第一实施方式的变形例(3)>
另外,将5mg的第一实施方式、第一实施方式的变形例(1)或第一实施方式的变形例(2)的硅细颗粒与约500mg的碳酸氢钠粉末(和光纯药株式会社制,纯度为99.5%)进行混合。混炼(kneading,捏揉)该混合物,并使用打锭法,从而能够获得作为直径约8mm、高度约4mm的圆柱型的块状体的锭剂。此外,锭剂仅为块状制剂的一个示例。此外,将稳定的具有低氧化硅的硅细颗粒和碳酸氢钠等pH调节剂分别在酸性下稳定且碱性下溶解的纳米胶囊(nanocapsule)、微胶囊(microcapsule)、通常的胶囊,或进行涂层(coating)是优选的一个方式。通过采用上述方式,能够避免在酸性条件的水分存在下的反应,而促进在碱性且水分存在下溶解而硅细颗粒与水的反应。
<第一实施方式的变形例(4)>
另外,在第一实施方式和第一实施方式的变形例(1)中,在使用了珠磨机的粉碎工序中,采用了乙醇与少量的水(0.1wt%~2wt%)的混合溶液,但第一实施方式不限定于该混合溶液。例如,作为乙醇代替,即使是在采用2-丙醇的情况下,或者采用已述的各种溶剂的情况下,都能够达到与第一实施方式或第一实施方式的变形例(1)的效果的相同的效果。
<第二实施方式>
本实施方式的复合组合物的特征之一的点在于,作为第一实施方式的粉碎工序中所采用的乙醇和少量的水的代替,通过使用酸性的溶液(代表性地,pH值为3~6)进行粉碎工序而制造。此外,能够将与第一实施方式重复的说明予以省略。
本实施方式的复合组合物的原料是例如使用喷射磨机(jet mill)法对市售的纯度为99.99%以上的i型多结晶硅进行粉碎,并经过300μm的筛而得到的高纯度硅颗粒粉末(粒径300μm以下)。此外,本实施方式并不限定于作为上述的该复合组合物的原料的硅颗粒粉末的大小、纯度、粉碎方法或分散溶剂。
作为具体的示例,使用珠磨装置(AIMEX株式会社制:RMH型卧式连续式ReadyMill)。在该珠磨装置中,进行将2.1g的上述的高纯度硅颗粒粉末作为硅颗粒分散于78mL的pH值被调整为3~5的酸性溶液(本实施方式为盐酸(HCl水溶液))中后,加入ψ0.5μm的氧化锆制的磨珠(容积2900mL),在空气中,将该珠磨装置的冷却水的温度设定至6℃的状态,以转速2500rpm进行75分钟的粉碎而微细化的粉碎工序。此外,用于形成该酸性的溶液的pH调整用的溶液并不限于盐酸。例如,从能够提高最终被制造的硅细颗粒(包含硅纳米颗粒和/或硅纳米颗粒的聚集体)和含有该硅细颗粒的复合组合物的安全性(例如对人体的安全性)的精度的观点出发,pH值为5或6也是优选的一个方式。
确认了即使是在采用本实施方式的复合组合物的制造方法的情况下,也能够达成通过第一实施方式的复合组合物及其制造方法而达成的效果的至少一部分的效果。
[XPS分析结果]
本发明人利用X射线光电子能谱分析装置(XPS分析装置)(株式会社岛津制作所制,型式:KRATOS AXIS 165)而对本实施方式的硅细颗粒(复合组合物)的一个示例的表面进行分析。此外,对于作为该分析的对象的硅细颗粒,在上述粉碎工序中所使用的酸性的溶液的pH值为5.0。对观察到的XPS谱图进行解析的结果是,使用pH5.0的盐酸(HCl水溶液)并进行基于珠磨法的粉碎而形成的硅细颗粒是具有1.6nm的二氧化硅膜、1.0nm低氧化硅以及2.6nm的氧化硅膜。
<第三实施方式>
本实施方式的复合组合物的特征在于,并不进行第一或第二的各实施方式中的粉碎工序而形成的点。此外,能够将与第一实施方式或第二实施方式,或这些的各个变形例重复的说明予以省略。
本实施方式的复合组合物的原料是第二实施方式的高纯度硅颗粒粉末(粒径300μm以下)。此外,本实施方式并不限定于作为上述该复合组合物的原料的硅颗粒粉末的大小、纯度、粉碎方法或分散溶剂。
在本实施方式中并不进行该粉碎工序,但与第一实施方式的变形例(1)相同地,进行通过使该硅颗粒粉末的表面接触过氧化氢水而进行该表面的改性的改性工序。具体而言,通过使浓度例如3wt%的过氧化氢水接触该硅颗粒粉末30分钟,而形成本实施方式的改性硅颗粒粉末(复合组合物的一个示例)。
其后,使用碳酸氢钠并添加78mL的调制到pH8.2的36℃的水(水溶液)来制作水溶液,并将该改性硅颗粒粉末分散于该水溶液中。此外,在本实施方式中,使用氢浓度计对通过使该改性硅颗粒粉末浸渍于该水溶液中生成的氢气的量进行测定。图6是表示本实施方式中的氢气的生成量与反应时间的关系的图表。
<第三实施方式的变形例(1)>
另外,作为本实施方式的变形例之一,对除了将在形成第三实施方式的该改性硅颗粒粉末过程中所采用的300μm的筛变换成45μm的筛以外,进行与第三实施方式相同的处理的示例(变形例)进行说明。
在本实施方式中,形成了粒径45μm以下的改性硅颗粒粉末(复合组合物的一个示例)。图7是表示本变形例的氢气的生成量与反应时间的关系的图表。
如上所述,在第三实施方式及其变形例中,也能够确认在相对短时间内生成可以说是充分的量的氢。因此,即使在不进行第一或第二的各实施方式的粉碎工序而形成复合组合物的情况下,也能够达成第一实施方式或第二实施方式的效果的至少一部分。
<其它的实施方式(1)>
此外,上述的各实施方式或其变形例的复合组合物例如能够作为制剂而活用。另外,其活用例并不限定于锭剂。例如,作为锭剂的代替,即使采用使粉状的该复合组合物内包于胶囊内的胶囊剂的情况下,也能够达成与上述的效果相同的效果。该复合组合物在不是块状而是表面积较大的粉状时能够生成更多的氢,但通过作成锭剂或胶囊剂,能够使经口摄取变得容易。另外,通过作成锭剂或胶囊剂,在胃内保持为一定程度的块状,另一方面,在通过胃后会进行崩解而成为粉状。因此,能够在想要抑制氢生成反应的胃内,减少该复合组合物暴露在胃液和/或胃的内容物的表面积,而在想要促进氢生成反应的小肠和/或大肠中使暴露在含水液体的表面积变多。
另外,该复合组合物也可以是颗粒的制剂。相较于锭剂或胶囊剂,颗粒的制剂在经口摄取后在更早的阶段变成为粉状。但是,因为胃液的pH值低(约1.5),所以即使在到达胃而立刻变成粉状时也几乎不会生成氢,而使其在通过胃后的水存在条件下生成氢。
该复合组合物也可以是粉剂。粉剂是例如含有健康食品的食品构成成分,例如在将该复合组合物用作食品添加物的情况下较为方便。在用作食品添加物的情况下,能够将微晶直径在1nm以上10μm以下,或1nm以上500nm以下(更狭义地为1nm以上100nm以下)的硅颗粒混合作为该复合组合物来使用。优选地,硅细颗粒含有1质量%以上。原本硅细颗粒的含量并没有设定上限,但考虑入口质量(eating quality),优选为40质量%以下。
另外,能够适用于锭剂的覆盖层的示例为作为覆盖锭剂的最外层的涂层剂的、公知的胃难溶性肠溶性的材料。另外,能够适用于胶囊剂的覆盖层的示例为内包该复合组合物的、由公知的胃难溶性肠溶性材料所制造的胶囊本身。
如上述,作为该复合组合物的活用例而优选的制剂的示例为:使便于经口摄取的充分的量的块状制剂,即锭剂或粉状的该复合组合物(能够包括成为聚集体状态的物质)内包于胶囊的胶囊剂。此外,在采用锭剂的情况下也可以进一步含有崩解剂。另外,崩解剂能够采用公知的材料。另外,更优选的崩解剂的优选的示例为有机酸,最优选的示例为柠檬酸。在此,有机酸还能够发挥使硅细颗粒或硅纳米颗粒形成为块状的结合剂的功能。此外,该复合组合物还能够作为例如添加在各食材上的粒状、片状和/或碎状的食用增味材料(代表性地,为“香松(Furikake)”)而活用。
<其它的实施方式(2)>
另外,上述各实施方式或其变形例的复合组合物例如通过使用使与该复合组合物接触的“介质”而能够经皮或经黏膜而将氢摄入体内(包括皮肤本身或黏膜本身)。此外,本实施方式的介质并不限定为特别的材料或商品。只要是生理上能够接受的介质就能够达成本实施方式的效果。因此,具备该复合组合物和接触该复合组合物的该介质的物质能够发挥作为氢供给材料的功能。
作为具体例之一,从在生活上增加人体的部位接触水(或含水液体)或含有该水(或含水液体)的介质(以下,也总称为“介质”)的机会的观点来说,优选的介质的示例是从由液状、胶状(gel,凝胶状)、乳霜状、糊状、乳液状以及慕斯状的组选出的至少一种。另外,其它优选的介质的示例是入浴水(优选地是碱性的入浴水)。因此,在本实施方式的一个示例中,以制造该入浴水作为介质的制造方法。
若进一步对入浴水作详细地说明,在一般的浴缸(包括钱汤的浴缸、公共浴室以及旅馆所设置的屋内或屋外的浴缸)内,代表性地,将自来水作为入浴水而加以储存。在储存该入浴水的前后,将上述的复合组合物配置或投入至该浴缸内,并通过进行使该复合组合物接触作为介质的入浴水的接触工序而生成氢(H
因此,能够经由作为在生理学上能够接受的介质的入浴水,使通过上述的接触工序而生成的氢(H
<其它的实施方式(3)>
另外,上述的各实施方式或其变形例的复合组合物也能够采用的其它的一个方式是例如:将该复合组合物形成层状的层状体,或在被形成为层状的基底材料中包含该复合组合物的层状体。因此,该层状体能够发挥作为氢供给材料的功能。
在本实施方式中,通过在将该复合组合物形成为层状的层状体,或在被形成为层状的基底材料中使用包含该复合组合物的层状体,能够形成层状体与介质的积层结构100。图8(a)是表示生成氢前的层状体与介质的积层结构100的侧视图,图8(b)是表示生成氢时的层状体与介质的积层结构100的侧视图。
如图8(a)和图8(b)所示,上述的积层结构100在基部20(例如,纤维、天然树脂、合成树脂、金属、半导体、陶瓷或玻璃)上或其上方至少具备层状体10a和介质90b。在此,介质90b的优选的一个示例是在生理学上能够接受并且从由液状、胶状、乳霜状、糊状、乳液状以及慕斯状的组中选出的至少一种的材料。另外,介质90b能够含有碳酸氢钠所代表的pH调节剂。此外,基部20具有伸缩性是优选的一个方式。另外,特别是即使在不具备基部20仍能够保持层状体10a与介质90b的积层结构100的情况下,不一定需要设置基部20。
如图8(a)所示,在生成氢前的阶段,为了不使层状体10a与介质90b接触,在层状体10a与介质90b之间设置不透水性的膜70。此外,能够将由公知的不透水性材料所形成的膜作为膜70而活用。例如,不透水性的膜70的材料是公知的聚乙烯等高分子。另外,作为其它的示例,使用国际公开第WO2011/036992号公报中公开的、具有水解性以及不透水性的片也是能够采用一个方式。
另一方面,如图8(b)所示,如果将膜70朝箭头方向抽出,会使层状体10a与介质90b的至少一部分直接接触。其结果是,例如通过与碳酸氢钠所代表的pH调节剂的相互作用,当层状体10a接触含有pH值为7以上(更优选为超过7、更进一步优选为超过7.4)的含水液体的介质90b时氢能够生成。
此外,在本实施方式中,通过将膜70朝箭头方向(纸张左边方向)抽出而以使层状体10a与介质90b直接接触的方式形成,但膜70的去除方法并没有特别限定。例如,在去除膜70的至少一部分时或溶解该膜的至少一部分时,使介质90b以与该复合组合物(在本实施方式中为层状体10a)接触的方式形成是能够采用的一个方式。此外,对于用于使膜70的至少一部分溶解的材料的示例,使用国际公开第WO2011/036992号公报中公开的、具有水解性以及不透水性的片也是能够采用的一个方式。
另外,作为其他的方式,在生成氢前的阶段中,作为层状体10a的代替,也能够采用由不透水性的膜70覆盖上述各实施方式或其变形例的复合组合物。只要通过使膜70抽取或溶解,从而使得该复合组合物与介质90b的至少一部分直接接触,则能够达到与层状体10a的情况相同的效果。
另外,例如,在介质是从由液状、胶状、乳霜状、糊状、乳液状以及慕斯状的组中选出的至少一种的情况下,也被认为无法维持如图8(b)所示的两层(层状体10a和介质90b)明确地被分离的状态。反而,在这样的情况下,从精度更高地促进氢生成的观点出发,层状体10a与介质90b的接触面积会变多,因此优选。此外,例如,为了附着在人的部位上,介质包含在生理学上能够接受的粘结剂也是能够采用的一个方式。
<其它的实施方式(3)的变形例>
作为上述的其它的实施方式(3)的一个变形例,将该复合组合物形成为层状的层状体能够采用单体,也能采用与基部20的层积构造。作为图9所示的一个示例的结构体200在基部20上具备有层状体10a。此外,在即使不具备基部20而仍能够保持层状体的形状的情况下,则不需要必须设置基部20。另外,从精度更高地避免与空气中的水分接触的观点出发,也可以设置为不透水性的膜70覆盖层状体10a。
如图9所示,例如,在层状体10a接触人的皮肤或黏膜后,通过让含有来自其皮肤或黏膜的水分的汗液或体液与层状体10a接触而生成氢是本实施方式的优选的一个方式。由这样的方式,与其它的实施方式(3)相同地,也能够实现人将氢摄入到体内。此外,作为汗液或体液的代替,水分也能够在使用层状体10a前(例如前一刻),将水(净水等)通过喷雾等而进行供给。
值得一提的是,在其它的实施方式(3)及其变形例中所采用的结构体或层积结构是在各种的“生活场面”中能够采用的结构。例如,能够采用(能够包含)该介质的代表性商品例为以下的(A)~(D)。
(A)从洗面奶、洗发水、沐浴露、液体洗手香皂以及液体沐浴香皂的组中选出的一种清洗剂。
(B)从化妆水(例如,含有玻尿酸)、美容液、乳液、乳剂、化妆用乳霜(例如,含有胶原蛋白)、粉底霜、皮肤面膜剂(包含具有凝胶(或胶状剂)的皮肤面膜剂)、刮胡膏、头发润丝精、护发乳、润发乳、头发化妆品、制汗剂以及防紫外线化妆品的组中选出的一种化妆用材料。
(C)从软膏以及泥罨剂(湿布剂,日本外用贴膏剂)的组中选出的一种治疗用材料。
(D)从吸水性树脂、吸水性无纺布、吸水性纤维、吸水毡(felt)以及吸水凝胶(或胶状剂)的组中选出的一种卫生材料。
在此,上述的“头发化妆品”包含:头发定型剂、发油、桩油、塑型(剂)、造型(剂)、吹发(剂)、梳发(剂)、固定发膏、发棒、发蜡、造型慕斯、发胶、发膏剂、护发霜、造型发泥、造型液、男用护发液、造型喷雾、整发水。另外,上述的“卫生材料”包含:卫生手套、头套、头带、床垫、床单、成人失禁用品、生理用品、衣物、伤口治疗用品(包括伤口敷料、胶布以及绷带)、包括成人用尿布及儿童用尿布的抛弃式尿布、纱布、袍服、卫生纸巾(包括湿巾、洗脸巾、贴布、湿纸巾及卫生棉)、脱脂绵、绵棒、OK绷、手术胶布。
<其它的实施方式(4)>
另外,上述的各实施方式及其变形例的复合组合物也能够作为例如饲育用(在本发明中,包括牧场中的饲养)的动物(包括狗、猫、马、羊、兔或鸡,鱼类除外。)、食用动物、该动物的毛或皮能够利用于衣物或皮制品(包括收纳包、各种箱盒或包)等动物(包括狐狸、熊、鹿、蛇或鳄鱼)、作为医疗而活用的动物,或包括养殖用的鱼类的鱼类等的饲料而使用。进一步地,也能够作为工业用药品或药剂使用。
另外,上述的各实施方式或其变形例的复合组合物也能够作为人类的补充剂或食品添加物来利用。而值得一提的是,因为上述的各实施方式或其变形例的复合组合物具有氢生成能力而能够发挥对各种食品或材料的防腐功能。例如,使包含蔬菜、水果、鲜鱼以及精肉的生鲜食品接触含有水的该复合组合物或浸渍了该复合组合物的水,由此能够使该生鲜食品长久保存。另外,使该复合组合物浸渍含有水(水分)的香水、乳液,或含有化妆水的各种化妆品或各种香妆品中,由此能够使该化妆品或该香妆品长久保存。
<其它的实施方式(5)>
另外,上述的各实施方式或其变形例的复合组合物通过在自然的状态下凝集而能够构成μm级(例如,20μm左右)的直径大小的聚集体。通过该聚集体或结合剂的添加或压缩等,人为地使该复合组合物集合,由此能够形成作为能够被人的指头拎住程度的大小的块状的固体的制剂的调配物。该调配物也能够对植物(包括树木)适用。
具体而言,在本实施方式中,通过将该调配物埋入该植物被种植或野生(含有水分)的土中,将土作为含有含水液体的介质而活用。使该调配物接触作为介质的土而生成氢(H
另外,通过将该调配物导入或投入存在于自然或人工的积水(介质)中,而使该调配物与含水液体接触也能够作为本实施方式的其它的方式而采用。通过使该调配物与含水液体接触而生成氢(H
另外,假设,如果上述的积水的pH值是比弱酸性更高的pH值(例如,pH值为5以上),通过使用本实施方式的该调配物与碳酸氢钠的混合物,变成较高的pH值,从而能够满足作为容易生成氢(H
通过使用本实施方式的该调配物与碳酸氢钠的混合物,即使假设作为介质的该土或该积水为中性或弱酸性,通过将该调配物埋入或导入或投入作为介质的该土或该积水中,也会使得该复合组合物经过接触该介质的接触工序。其结果是,能够促进氢(H
因此,能够经由作为介质的该土或该积水,使通过上述的接触工序而生成的氢(H
另外,在本实施方式中,该复合组合物或该调配物并不限定于直接使用的情况。该复合组合物或该调配物包含于例如动物用药品、家畜或宠物用食品或动物用饲料,或植物用药品、植物用肥料或植物用堆肥等“基底材料”中的方式,或调配于该“基底材料”中的方式也是能够采用的优选的一个方式。例如,将例如0.1wt%~50wt%的该复合组合物或该调配物作为添加剂混入或混炼至该基底材料是代表性的一个示例。因此,上述的“基底材料”不仅是将该复合组合物混入植物用药品、植物用肥料或植物用堆肥的调配物,在本实施方式中也是广义上的“调配物”。因此,作为用于动物或植物例如经皮或经黏膜而将氢摄入体内的优选一个方法,能够采用上述的基底材料与介质接触。
<实施例>
以下,为了更详细地说明上述的各实施方式而列举实施例予以说明,但是上述的实施方式并不是通过这些示例来进行限定。
(实施例1-1)
首先,以200g的第一实施方式的具有低氧化硅的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料,加入反应容器,并添加500mL的浓度3wt%的过氧化氢水。一边搅拌一边设定至35℃,通过使硅细颗粒浸渍于过氧化氢水中30分钟,进行硅细颗粒的表面的改性工序。此后,利用无灰定量滤纸(GE Healthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对表面改性后的该硅细颗粒(即,第一实施方式的变形例(1)的复合组合物的一个示例)进行减压过滤,由此进行固液分离。此后,经过水洗,在使硅细颗粒分散在乙醇中后,通过离心分离进行固液分离。在40℃减压下,对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存在真空容器内或氮置换后的容器内。进行了改性工序的硅细颗粒的表面表示出亲水性。
(实施例1-2)
首先,以200g的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料,加入反应容器,并添加250mL的浓度10wt%的过氧化氢水。一边搅拌一边设定至20℃,通过使硅细颗粒浸渍于过氧化氢水中60分钟,进行硅细颗粒的表面的改性工序。此后,利用无灰定量滤纸(GE Healthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对表面改性后的该硅细颗粒(即,第一实施方式的变形例(1)的复合组合物的一个示例)进行减压过滤,由此进行固液分离。此后,经过水洗,在使硅细颗粒分散在乙醇中后,通过离心分离进行固液分离。在40℃减压下,对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存在真空容器内或氮置换后的容器内。进行了改性工序的硅细颗粒的表面表示出亲水性。
(实施例1-3)
首先,以200g的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料,加入反应容器,并添加500mL的浓度3wt%的过氧化氢水。一边搅拌一边设定至60℃,通过使硅细颗粒浸渍于过氧化氢水中30分钟,进行硅细颗粒的表面的改性工序。此后,利用无灰定量滤纸(GE Healthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对表面改性后的该硅细颗粒(即,第一实施方式的变形例(1)的复合组合物的一个示例)进行减压过滤,由此进行固液分离。此后,经过水洗,在使硅细颗粒分散在乙醇中后,通过离心分离进行固液分离。在40℃减压下,对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存在真空容器内或氮置换后的容器内。进行了改性工序的硅细颗粒的表面表示出亲水性。
(实施例2)
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将78mL的pH7且36℃的水添加至该原料(硅细颗粒)中而使该原料分散,由氢浓度计对一边搅拌一边生成的氢气进行测定。相继生成的氢气的量如表1所示。此外,也通过气相色谱质谱分析法(GC/MC)分析法鉴别氢气的生成量并进行定量评价。
另外,在该原料与水的反应完成后,对反应后的该硅细颗粒进行基于第一实施方式的XPS装置和FT-IR装置的分析。其结果表示:反应后的该硅细颗粒依然保持着氢生成能力。
[表1]
(实施例3)
将5mg的第一实施方式的变形例(1)的硅细颗粒(即,表面改性后的硅细颗粒)作为原料。将78mL的pH7且36℃的水添加至该原料(硅细颗粒)中而使该原料分散,由氢浓度计对生成的氢气进行测定。相继生成的氢气的量如表2所示,为表1的结果的十倍以上的值。此外,也通过GC/MC分析法鉴别(鉴定)氢气的生成量并进行定量评价。
此外,在该原料与水的反应完成后,对反应后的该硅细颗粒进行基于第一实施方式的XPS装置和FT-IR装置的分析。其结果表示:反应后的该硅细颗粒(即,表面改性后的硅细颗粒)依然保持着氢生成能力。
[表2]
(实施例4)
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将78mL的利用碳酸氢钠调制成pH8.3的36℃的水添加至该原料(硅细颗粒)中而使该原料分散,由氢浓度计对生成的氢气进行测定。相继生成的氢气的量如表3所示。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
另外,在该原料与水的反应完成后,对反应后的该硅细颗粒进行基于第一实施方式的XPS装置和FT-IR装置的分析。其结果表示:反应后的该硅细颗粒依然保持着氢生成能力。
[表3]
(实施例5)
将5mg的第一实施方式的变形例(1)的硅细颗粒(即,表面改性后的硅细颗粒)作为原料。将78mL的利用碳酸氢钠调制成pH8.3的36℃的水添加至该原料(硅细颗粒)中而使该原料分散,由氢浓度计对生成的氢气进行测定。相继生成的氢气的量如表4所示,为表3的结果的十五倍以上的值。该氢气生成量的増大是基于pH値成为碱性的效果。更具特色的是,相较于表3的结果,特别是在反应的初期阶段的氢气的生成量有明显提升。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
另外,在该原料与水的反应完成后,对反应后的该硅细颗粒进行基于第一实施方式的XPS装置和FT-IR装置的分析。其结果表示:反应后的该硅细颗粒(即,表面改性后的硅细颗粒)依然保持着氢生成能力。
[表4]
(实施例6)
将200g的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将250mL的稀释35%的过氧化氢得到的浓度10wt%的过氧化氢水添加至该原料(硅细颗粒)中而使该原料分散。在35℃的条件下,使硅细颗粒浸渍于过氧化氢水中30分钟,由此进行对硅细颗粒的表面进行改性的改性工序。此后,利用无灰定量滤纸(GEHealthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对表面改性后的该硅细颗粒(即,第一实施方式的变形例(1)的复合组合物的一个示例)进行减压过滤,由此进行固液分离。在使硅细颗粒分散于乙醇中后通过离心分离进行固液分离。在40℃减压下,对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存于真空容器内或氮置换后的容器内。进行了改性工序的硅细颗粒的表面表示出亲水性。
此后,利用XPS分析装置分析该硅细颗粒的表面的结果是,相对于氧化硅膜中的硅原子数(即,二氧化硅的硅原子数与低氧化硅的硅原子数的总计(100%))的低氧化硅中的硅原子数的比(即低氧化硅的组成比)为约60%。
(实施例7)
以下,对采用过碳酸钠作为第一实施方式的变形例(1)的过氧化氢水的代替的示例进行说明。
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将180mL的水添加至该原料(硅细颗粒)中,进一步添加1.8g的过碳酸钠以制成水溶液,并使该原料分散于该水溶液。在30℃的条件下,使硅细颗粒一边搅拌一边浸渍30分钟,由此进行对硅细颗粒的表面进行改性的改性工序。此后,利用无灰定量滤纸(GEHealthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对表面改性后的该硅细颗粒(即,第一实施方式的变形例(1)的复合组合物的一个示例)进行减压过滤,由此进行固液分离。此后,在通过水洗而去除附着在硅细颗粒的碳酸钠后,在使硅细颗粒分散于乙醇中后通过离心分离而进行固液分离。在40℃减压下对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存于氮置换后的容器内。进行了改性工序的硅细颗粒的表面表示出亲水性。
(实施例8)
将5mg的通过实施例(7)而得到的硅细颗粒(即,通过过碳酸钠而表面改性的硅细颗粒)作为原料。将78mL的pH7且36℃的水添加至该原料(硅细颗粒)中,并使该原料分散于该水中。在该示例中,利用氢浓度计对通过浸渍于该水溶液而生成的氢气的量进行测定。
确认了从氢生成时开始经过168小时(7天)后的该氧化硅膜(包含二氧化硅和低氧化硅)的膜厚为约13nm。因此,从氢生成时开始经过168小时(7天)后的该氧化硅膜的膜厚只要在已述的3nm以上20nm以下(代表性地为15nm以下)的数值范围,就能够精度更高地认为是第一实施方式的变形例(1)的硅细颗粒。
(实施例9)
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将78mL的利用碳酸氢钠和碳酸钠调制成pH10的36℃的水(水溶液)添加至该原料(硅细颗粒)中以制成水溶液,并使该原料分散于该水溶液中。在该示例中,使用氢浓度计对通过使该原料浸渍在该水溶液而生成的氢气的量进行测定。相继生成的氢气的量相比于使用pH7或pH8.3的水的情况大幅度地增加。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。另外,图5是表示该示例中的氢气的生成量与反应时间的关系的图表。
(实施例10)
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将78mL的利用苛性钠(氢氧化钠)调制成pH8.3的36℃的水(水溶液)添加至该原料(硅细颗粒)中以制成水溶液,并使该原料分散于该水溶液中。在该示例中,使用氢浓度计对通过使该原料浸渍在该水溶液而生成的氢气的量进行测定。相继生成的氢气的量相比于使用pH7的水的情况大幅度地增加。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
(实施例11)
将5mg的第一实施方式的变形例(1)的硅细颗粒(即,表面改性后的硅细颗粒)作为原料。制成将30mL的利用苛性钠调制成pH8.3的36.5℃的水(水溶液)至该原料(硅细颗粒)中,并添加到该水溶液中而使该原料分散。在该示例中,使用氢浓度计对通过使该原料浸渍在该水溶液而生成的氢气的量进行测定。相继生成的氢气的量相比于使用pH7的水的情况大幅度地增加。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
(实施例12)
将5mg的第一实施方式的硅细颗粒(即,第一实施方式的复合组合物的一个示例)作为原料。将78mL的利用苛性钠调制成pH8.3并预先加热至60℃的水(水溶液)一点一点地添加至该原料(硅细颗粒)中而使该原料分散于该水溶液中。在该示例中,使用氢浓度计对通过使该原料浸渍于该水溶液而生成的氢气的量进行测定。
在该示例中,确认了相较于例如表3所示的结果氢的生成明显地加快。另外,相继生成的氢的生成速度相较于使用了pH7的水的情况大幅度地增加。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
(实施例13)
以5mg的第一实施方式的变形例(1)的硅细颗粒(即,表面改性后的硅细颗粒)作为原料。将78mL的利用苛性钠调制成pH10并预先加热至60℃的水(水溶液)一点一点地添加至该原料(硅细颗粒)中而使该原料分散于该水溶液中。在该示例中,使用氢浓度计对通过使该原料浸渍于该水溶液而生成的氢气的量进行测定。
在该示例中,确认了相较于实施例11的情况氢的生成进一步加快。此外,也通过GC/MC分析法鉴别氢气的生成量并进行定量评价。
(实施例14)
利用无灰定量滤纸(GE Healthcare Japan株式会社制,级42,颗粒保持能力:2.5μm)对在第二实施方式中制造的复合组合物进行减压过滤,由此进行固液分离。在40℃减压下对固液分离后的表面改性后的硅细颗粒进行干燥处理。此后,干燥后的该硅细颗粒被保存于真空容器内或氮置换后的容器内。在第二实施方式中制造的复合组合物的表面表示出亲水性。
根据实施例14而得到的复合组合物的一个示例主要是由微晶直径为1nm以上500nm以下的硅纳米颗粒作为主成分而组成的。更具体而言,通过X射线衍射装置(株式会社Rigaku制,SmartLab)对硅纳米颗粒进行测定的结果,作为一个示例,平均微晶直径为28.0nm。此外,上述的平均微晶直径的结果仅仅是作为基于一个示例的粉碎工序等的结果,因此本实施例并不限定于上述的数值。
(实施例15)
以5mg的根据实施例14而得到的硅细颗粒(即,第二实施方式的复合组合物的一个示例)作为原料。将78mL的pH7且36℃的水添加至该原料(硅细颗粒)而使该原料分散,由氢浓度计对一边搅拌一边生成的氢气进行测定。相继生成的氢气的量如表5所示。此外,也通过气相层析质谱分析法(GC/MC)分析法鉴别氢气的生成量并进行定量评价。
[表5]
而在第一接触工序中使上述的各实施方式的硅细颗粒与pH值不足7的第一含水液体接触,在其后的第二接触工序中使上述的各实施方式的硅细颗粒与pH值为7以上的第二含水液体接触,从而能够在第二接触工序中生成氢。因此,上述各实施方式的硅细颗粒能够在与pH值为7以上的含水液体接触时具有显著的氢生成能力。
另外,上述各实施方式中的用于生成氢的第二含水液体的温度条件并没有限定。虽然是能够依赖于第二含水液体的pH,但只要第二含水液体的温度在80℃以下,便能够实现精度较高地促进氢生成。然而,第二含水液体的温度的上限原本并没有限定。例如,在将本实施方式的复合组合物作为工业药品使用的情况下,也可以超过50℃。但是,会产生温度愈高设备(包括容器)被要求较高的耐热性的在使用上需要注意的问题,因此,优选地,在作为工业药品使用的情况也在100℃以下。
产业上的可利用性
本发明的复合组合物能够被广泛地利用于各种产业,上述产业包含:活用氢的农业、农牧业、林业、水产业、宠物产业、或处理盆栽或鲜花的业种、医药(包括准药品)和医疗业界、食品业界、兽医业及树木医业,或处理工业用还原剂、防锈用途或工业药品的合成工序的产业,进一步地,还含有燃料电池等新兴的能源产业。
- 包含蜡组合物的木塑复合材料组合物、由其生产木塑复合材料的方法及蜡组合物作为用于生产木塑复合材料的润滑剂的用途
- 用于形成复合物的组合物、由此形成的复合物和用于口服的包含所述复合物的组合物