一种宏蛋白质组挖掘方法及其在获取肠道微生物蛋白水解特征中的应用
文献发布时间:2023-06-19 10:55:46
技术领域
本发明涉及生物信息分析技术领域,更具体的,涉及一种宏蛋白质组挖掘方法及其在获取肠道微生物蛋白水解特征中的应用。
背景技术
肠道微生物生活在一个动态的环境中,面临着来自药物、饮食、微生物竞争和宿主内源化学成分的蛋白毒性和代谢压力。细菌已经进化出不同的调节策略以适应不断变化的环境,包括基因表达的改变、细胞分化和运动的变化,在这些调节策略中,蛋白水解起到了至关重要的作用,蛋白水解调控是影响所有生物的重要过程,细菌使用能量依赖的蛋白酶来降解错误折叠的蛋白,或者激活调节蛋白来对动态的肠道环境做出快速反应。微生物通过蛋白水解进行调节的功能非常广泛,例如应激反应、细胞生长分裂、生物膜形成、蛋白质的分泌。
炎症性肠病(IBD)是一种受遗传和环境因素影响的慢性炎症性疾病,主要包括克罗恩病(CD)和溃疡性结肠炎(UC)。已有报道证实IBD与肠道微生物失调有关。在IBD肠道微生物组研究中,宏基因组学和16S rRNA基因测序占绝大多数。然而,需要宏转录组学或宏蛋白质组学通过分别直接测量RNA和蛋白质来精确定位功能和代谢活动。此外,在蛋白质水平上还有重要的调节模式,例如蛋白质水解调控,这些调节模式无法通过RNA研究获得,但可以使用宏蛋白质组学进行研究。
然而,在IBD等复杂疾病状态下,肠道微生物蛋白质水解的特征变化尚未被研究,因此亟需一种能够在复杂疾病状态下掌握肠道微生物蛋白质水解特征的方法。
发明内容
本发明所要解决的技术问题是克服现有技术存在的上述问题,首先提供一种以半胰蛋白酶多肽为中心的宏蛋白质组挖掘方法,也提供一种比较蛋白质水解程度的方法。
本发明的第二个目的是提供上述方法在获取肠道微生物蛋白水解特征中的应用。
本发明的目的通过以下技术方案实现:
一种确定蛋白质水解程度的方法,包括以下步骤:
S1、获取样品的(宏)蛋白质组数据或公共数据库中发表的(宏)蛋白质组数据;
S2、利用大的宏蛋白数据库以及PEAKS DB软件执行第一次搜索,得到至少一个肽被鉴定出来的蛋白质;
S3、利用PEAKS DB软件、MaxQuant软件和pFind软件将组学数据与S2得到的蛋白质序列进行搜库鉴定,保留同时被PEAKS DB、MaxQuant和pFind三种软件同时鉴定的肽;
S4、区分出S3得到的肽中半胰蛋白酶多肽(Semi-tryptic peptide)和完全胰蛋白酶多肽(full tryptic peptide);
S5、以半胰蛋白酶多肽归一化后的相对丰度来确定蛋白质水解程度,其中,半胰蛋白酶多肽归一化的相对丰度是通过将半胰蛋白酶多肽的相对丰度归一化到完全胰蛋白酶多肽的相对丰度得到。
优选地,S4中,半胰蛋白酶多肽的鉴定原则是:在鉴定序列前一位氨基酸不是R或K的肽为半胰蛋白酶N端肽(不包含蛋白质的N端)。鉴定序列的最后一个氨基酸缺少R或K,则是半胰蛋白酶C端肽(不包含蛋白质的C端)。
蛋白质组学样品制备过程中蛋白质被胰蛋白酶水解后产生的肽段的前面一位氨基酸应该是K或R,而最后一位氨基酸也应该为K或R。如果数据中检测到了半胰蛋白酶,说明有胰蛋白酶以外的其他蛋白酶参与了蛋白质的水解,导致了肽段前面一位氨基酸或最后一位氨基酸不是K或R,因此半胰蛋白酶可作为蛋白质在生物体内被其他蛋白酶水解的标志,而完全胰蛋白酶可以作为蛋白质在生物体内未被其他蛋白酶水解的标志。但是研究蛋白质水解程度不能仅仅依赖于半胰蛋白酶,因为半胰蛋白酶丰度的改变可能仅仅是由于对应的蛋白质总量的改变(合成增加或减少),而蛋白质水解的程度并没有改变。因此需要将半胰蛋白酶多肽的相对丰度归一化到完全胰蛋白酶多肽的相对丰度来比较不同样本间蛋白质水解的程度的变化,这样可以排除蛋白质总量变化这一因素。
优选地,PEAKS DB数据库执行搜索的参数为:母离子(precursor ion)的质量偏差为10ppm,碎片离子(product ion)的质量偏差为0.02Da;半胱氨酸的氨基甲基化被设定为固定修饰;每个肽的最大可变翻译后修饰为3个,包括蛋白质N末端的乙酰化、甲硫氨酸的氧化、天冬酰胺和谷氨酰胺的脱酰胺化以及谷氨酰胺转化为焦谷氨酸;酶为胰蛋白酶,酶切方式为半特异性(semi-specific),未被酶切位点最多为3个;假阳性率(false discoveryrate)设为1%。
优选地,MaxQuant执行搜索的参数为:初次搜索(first search)质量偏差为20ppm,主要搜索(main search)质量偏差为4.5ppm;酶为胰蛋白酶,酶切方式为半特异性(semi-specific),未被酶切位点最多为2个;半胱氨酸的氨基甲基化被设定为固定修饰;每个肽的最大可变翻译后修饰数为5个,包括蛋白质N末端的乙酰化、甲硫氨酸的氧化、天冬酰胺和谷氨酰胺的脱酰胺化以及谷氨酰胺转化为焦谷氨酸;假阳性率(false discoveryrate,FDR)设为1%,保留后验错误概率(posterior error probability,PEP)小于5%的肽段用于后续分析。
优选地,pFind执行搜索的参数为:pFind执行搜索的参数为:母离子的质量偏差为10ppm,碎片离子的质量偏差为20ppm,搜库模式为开放式搜库(open-search),酶为胰蛋白酶,酶切方式为半特异性,未被酶切位点最多为3个;FDR设为1%。
本发明还提供上述方法的应用。
具体地,上述方法用于捕获肠道微生物蛋白质水解的特征。提供了菌群结构和蛋白质丰度之外的不同层次的信息,这项分析是基于这样的假设,即相似的蛋白水解程度应该导致相似的半胰蛋白酶多肽的相对丰度,本发明研究发现447个粪便宏蛋白质组中的微生物半胰蛋白酶多肽在脂肪酸、羧酸、葡萄糖和盐藻糖的代谢过程、支链氨基酸的生物合成过程、蛋白质运输和细菌型鞭毛介导的细胞运动等几个生物学过程中得到了丰富,这表明它们经历了更广泛的蛋白质水解调节。
或者,上述方法用于研究肠道微生物区系和宿主-微生物相互作用。
本发明上述蛋白质组的挖掘方法也适用于捕获植物和环境微生物的蛋白质水解特征,因此,上述方法可用于探索植物和环境微生物的蛋白质水解规律。
本发明上述方法还可以用于研究与细菌蛋白酶有关的疾病(例如细菌感染、炎症性肠病),通过该方法可以研究细菌蛋白水解程度的变化,从而以相应的细菌蛋白酶为靶标,针对性的开发相应的药物进行调控。
与现有技术相比,本发明具有以下有益效果:
本发明提供了一种以半胰蛋白酶多肽为中心的宏蛋白质组挖掘方法,包括两步搜索、从头测序、开放搜索和多种软件结果匹配,以进行大规模的半胰蛋白酶肽为中心的宏蛋白质组挖掘。这些策略可以减少因数据库不完整和多肽修饰而产生的假阳性识别。以往的研究对低分辨率MS/MS生成的宏蛋白质组学数据集进行了半胰蛋白酶多肽搜索,不可避免地增加了搜索空间,降低了鉴定结果的置信度。在他们的研究中,在一个包含6162,582条序列的宏蛋白大数据库中,当搜索Pyrococcus furiosus蛋白质组时,只有80.2%的鉴定肽被注释为P.furiosus序列。相反,本发明是针对的高分辨率MS/MS数据的多引擎搜索。使用本发明的方法在分析大肠杆菌蛋白质组时,从一个明显更大的宏蛋白质数据库(130,975,891个序列)中鉴定出的肽段有93.4%与传统大肠杆菌参考数据库鉴定出的肽段相一致,显示了本方法有更好的准确性。
附图说明
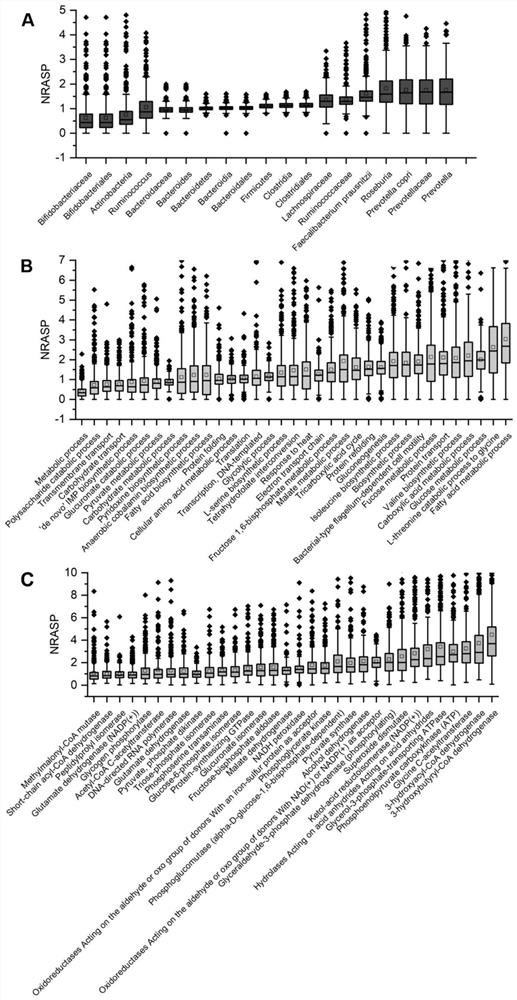
图1为在447份粪便代谢蛋白质组学样本中,来自主要细菌种类和生物过程的半胰蛋白酶多肽的归一化相对丰度(NRASP,半胰蛋白酶多肽丰度/完全胰蛋白酶多肽丰度),不同的细菌种类(A)、生物学过程(B)和酶(C)的功能按升序排列;框图表示中位数(框中间的线)、第25百分位数和第75百分位数;虚线表示四分位范围(IQR)的1.5倍,离群值显示为点;
图2为大肠杆菌蛋白质组在热应激诱导下不同生物过程蛋白水解特征的变化(p<0.05)。
具体实施方式
下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
下述实验例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
数据集:分析了2个公开发表的健康和IBD肠道宏蛋白质组的人群的数据集,数据集1(PXD008675)由来自89名受试者的447个粪便宏蛋白质组组成,这些受试者的年龄在6-58岁,中位数为22.8岁,包括24名非IBD对照组、39名CD患者,26名UC患者;在这些样本中,分别有272个样本具有匹配的宏基因组,184个样本具有匹配的宏蛋白质组;我们还分析了蛋白质组数据集(PXS000498)以研究热应激对大肠杆菌K-12蛋白水解调节的影响。
宏蛋白数据库:一个全面的人体肠道微生物蛋白质数据库由以下部分组成:(1)基于来自1070个人(760个欧洲人、368个中国人和139个美国人样本)的1267个肠道宏基因组的integrated gene catalog(IGC)数据库;(2)从健康成人粪便中培养的215个菌株的序列数据;(3)Culturable Genome Reference(CGR)数据库,包含了6000株从健康人类粪便中分离的肠道菌的1520个非冗余、高质量基因组;(4)UniProtKB(版本2017_06)和NCBI RefSeq(版本90)中的所有古生菌、细菌和真菌序列。上述微生物序列数据库附加了UniProt人类参考蛋白质组,它包括膳食有机物组成的食物数据库,例如生物普通小麦(Triticumaestivum)、水稻(Oryza sativa subsp.japonica)、大豆(Glycine max)、玉米(Zea mays)、花生(Arachis hypogaea)、马铃薯(Solanum tuberosum)、番茄(Solanum lycopersicum)、猪(Sus scrofa)、牛(Bos taurus)、鸡(Gallus gallus)、羊(Ovis aries)、鱼(Salmo salar和Oncorhynchus mykiss)、虾(Artemia sp.和Litopenaeus vannamei),和一个常见污染物数据库(
统计分析方法:利用主成分分析(PCA)和偏最小二乘判别分析(PLS-DA)对裂解位点附近的氨基酸频率进行多元分析,并采用Bayesian PCA(BPCA)估算缺失值。在R(vesion3.5.3)和RStudio(version 1.1.383)中使用Kruskal-Wallis检验和Dunn-Bonferroni检验,P值小于0.05来检测各组之间显著不同的变量(存在于至少75%的样本中)。使用Bray-Curtis距离的主坐标分析(PcoA)来确定多组学数据的β多样性。
实施例1不同软件执行搜索的表现
使用MLI数据集和大型宏蛋白质数据库,我们比较了不同的商业软件(ProteomeDiscoverer、PEAK、ProteinPilot和Byonic)和开源软件(MaxQuant、MSFragger和pFind)在几个36核服务器(安装有192G内存)上搜索半胰蛋白酶肽的性能。Proteome Discoverer、Byonic、MaxQuant、pFind和ProteinPilot在一个月内没有完成搜索,而MSFragger因内存不足错误而崩溃。只有PEAK在一个月内完成了分析,因此使用一个156核的高性能计算集群进行进一步的高通量分析,该集群在2周内完成了数据库搜索。
实施例2数据库搜索
数据库搜索流程一般包括两个主要步骤:(1)从头测序(de-novo sequencing),并使用大的宏蛋白数据库(large database)以及PEAKS软件执行第一次搜索,得到至少一个肽被鉴定出来的蛋白质并生成一个相应的小的蛋白质数据库(reduced database);(2)使用reduced database和多种软件进行第二次搜索,提高鉴定半胰蛋白酶多肽的准确性。
为了应对在宏蛋白质组半胰蛋白酶多肽鉴定中增加的搜索空间和时间,首先在配置有Intel(R)Xeon(R)156核处理器和1.5TB 2666MHz内存的集群上使用PEAKS DB进行搜索,软件首先进行从头测序,接下来使用下列参数进行数据库搜索:母离子的质量偏差为10ppm,碎片离子的质量偏差为0.02Da;半胱氨酸的氨基甲基化被设定为固定修饰;每个肽的最大可变翻译后修饰为3个,包括蛋白质N末端的乙酰化、甲硫氨酸的氧化、天冬酰胺和谷氨酰胺的脱酰胺化以及谷氨酰胺转化为焦谷氨酸;酶为胰蛋白酶,酶切方式为半特异性,未被酶切位点最多为3个;假阳性率设为1%。
这里使用两步法搜索策略,是为了增加搜库的灵敏度,在第一步搜索中由至少一个肽被鉴定的蛋白质被保留用于第二轮多引擎搜索,第二步搜索使用PEAKS DB、MaxQuant(版本1.6.2)和pFind(版本3.1.5)。
使用Andromeda引擎执行MaxQuant(版本1.6.2.10)搜索。设置参数如下:初次搜索质量偏差为20ppm,主要搜索质量偏差为4.5ppm;酶为胰蛋白酶,酶切方式为半特异性,未被酶切位点最多为2个;半胱氨酸的氨基甲基化被设定为固定修饰;每个肽的最大可变翻译后修饰数为5个,包括蛋白质N末端的乙酰化、甲硫氨酸的氧化、天冬酰胺和谷氨酰胺的脱酰胺化以及谷氨酰胺转化为焦谷氨酸;假阳性率设为1%,保留后验错误概率小于5%的肽段用于后续分析;“Second peptides”选项在MS/MS谱中搜索共碎片肽。启用“match betweenruns”选项,设置使用0.7分钟的匹配时间窗口和20分钟的校准时间段。蛋白质和肽的定量使用无标记定量(LFQ)算法,最小比计数为1,最小邻域数和平均邻域数分别为3和6。
使用pFind进行数据库搜索,母离子的质量偏差为10ppm,碎片离子的质量偏差为20ppm,搜库模式为开放式搜库(open-search),酶为胰蛋白酶,酶切方式为半特异性,未被酶切位点最多为3个。
只有被三个搜索引擎(PEAKS DB、MaxQuant和pFind)识别的肽才会被保留下来,用于进一步分析。
实施例3半胰蛋白酶多肽鉴定以及分类和功能分析
1、半胰蛋白酶多肽的鉴定原则
在鉴定序列前一位氨基酸不是R或K的肽为半胰蛋白酶N端肽(不包含蛋白质的N端)。鉴定序列的最后一个氨基酸缺少R或K,则是半胰蛋白酶C端肽(不包含蛋白质的C端)。根据洗脱时间将源内片段(In-source CID fragment)与蛋白水解衍生的半胰蛋白酶多肽区分开来。与它们的理论保留时间(使用SSRCalc预测)相比,大多数源内片段显示出不同的保留时间。根据FASTA序列条目中相应的检索号,将微生物半胰蛋白酶多肽与人来源的肽和食物来源的肽区分开来。
2、结合半胰蛋白酶和完全胰蛋白酶的数据来定量蛋白质的水解程度
我们通过将半胰蛋白酶肽的相对丰度归一化到完全胰蛋白酶肽的相对丰度,根据半胰蛋白酶肽的归一化相对丰度(normalized relative abundance of semi-trypticpeptides,简称NRASP)来确定蛋白水解程度的变化。这一归一化步骤很重要,因为如果半胰蛋白酶多肽和完全胰蛋白酶多肽的丰度成比例变化,通常表明蛋白水解程度没有变化。然而,在这种情况下,如果只比较半胰蛋白酶多肽,就会出现组间差异。
3、结果
为了提高基于大序列空间的宏蛋白质组分析的灵敏度,我们采用了两步数据库搜索策略。这有效地将宏蛋白质数据库的规模缩小到了传统蛋白质组学分析的数据库规模,从而方便了基于半胰蛋白酶的宏蛋白质组学搜索。此外,通过结合三个常用的软件来提高肽鉴定的可信度。这些软件使用不同的算法进行峰匹配、共流出肽段识别和FDR计算(MaxQuant和pFind使用目标-诱饵策略,PEASK DB使用诱饵-融合方法),从而显著增加了肽识别的置信度。只保留三个软件共同鉴定的肽用于进一步分析。
共检索了12,828,005个MS/MS图谱,从粪便宏蛋白质组中鉴定出3,804,903(29.66%)个二级图谱(PSMs),125,494个肽,其中108,784(86.68%)肽为微生物特有肽(未被人或食物序列共享)。在粪便宏蛋白质组中鉴定出7,969(6.35%)人特异性多肽,其中5,104(64.05%)肽为半胰蛋白酶。基因本体(Gene ontology,GO)分析表明,84.13%的人半胰蛋白酶肽来自潜在的胞外蛋白,只有1.16%的微生物半胰蛋白酶肽来源于潜在的胞外蛋白。
实施例4通过分析大肠杆菌热休克反应中的蛋白水解特征来验证上述方法
我们通过使用已发表的大肠杆菌K12的蛋白质组数据集分析热休克诱导的蛋白水解特征,验证了我们的方法。结合三个搜索引擎,用上述大的宏蛋白数据库鉴定了9937个肽段,而用UniProt E.coli K12参考数据库中鉴定了14111个肽段。两种方法中鉴定肽的数量减少了29.6%,反映了正常的灵敏度损失,因为大数据库产生的序列比常规的参考序列多10,000倍。
在UniProt E.coli K12参考数据库鉴定的全部14111个肽段中,有83.7%的肽段PEP值低于0.01,61.6%的肽段PEP值低于0.001。而在仅被UniProt E.coli K12参考数据库鉴定出(未被宏蛋白质数据库鉴定)的4783个肽段中,PEP值低于0.01的占60.3%,低于0.001的占39.5%。在仅被UniProt E.coli K12参考数据库鉴定出的肽段具有较高的PEP值,这说明低质量的肽谱(PSMs)在大数据库搜索时更容易受到灵敏度降低的影响。同时值得注意的是,单一微生物蛋白质组与肠道蛋白质组有显著差异。最近的研究表明,大型公共数据库组装的宏蛋白数据库和样本匹配参考数据库(sample-matched)对肠道宏蛋白质组学研究产生了相当的结果。因此,我们的方法在肠道宏蛋白质组分析中不会出现明显的灵敏度损失。用巨大的宏蛋白数据库鉴定出的肽段有93.4%与大肠杆菌参考数据库鉴定出的肽段相一致,表明我们的方法具有较高的肽段鉴定准确性。
为了验证我们的方法,我们比较了所有样本中发现的185个生物过程的NRASP(作为蛋白质水解监管指标),发现20(约占10.8%)个生物过程的NRASP在对照组和热应激组之间明显不同(P值<0.05,图2)。
热应激会扰乱蛋白质的折叠,导致错误折叠蛋白质的积累,这些错误折叠的蛋白质需要重新折叠成正确的构象。相应的,使用我们的方法发现,热应激下与蛋白质折叠的NRASP减少,而与蛋白质重新折叠相关的NRASP增加。同时我们观察到与甲基化相关的NRASP在热应激下增加,这与最近发现是一致的。综上所述,使用我们的方法得到与蛋白质水解调控的生物学发现具有较高的可信度。
实施例5肽的分类和功能分析
分析使用Unipept(版本4.3.5),使用UniProt 2020.01,基于最低共同祖先(LCA)算法进行分析,所有肽用以下参数进行分析:使I和L相等,过滤重复肽,高级缺失切割处理(Advanced missing cleavage handling)。分类信息是使用UniPept提供的Sunburst视图可视化的。、
研究结果
(1)半胰蛋白酶多肽的相对丰度和分布
图1给出了来自CD(n=204)、UC(n=123)和对照(n=120)组的447个粪便宏蛋白质组中至少在75%样品中鉴定到20个主要细菌种属、35个主要生物学过程和32个酶亚类的NRASP。厚壁菌门(phyla Firmicutes)和拟杆菌门(Bacteroidetes),拟杆菌(Bacteroidia)和梭状芽孢杆菌(Clostridia),拟杆菌目(Bacteroidales)和梭菌目(Clostridiales),拟杆菌科(fBacteroidaceae)和拟杆菌属(Bacteroides)的NRASP的中位数在1左右,表明相应的半胰蛋白酶肽与完全胰蛋白酶肽的相对丰度相当(图1A)。然而,NRASP的中位数在毛螺菌科(Lachnospiraceae)和反刍球菌科(Ruminococcaceae)分别增加到约1.25,罗斯拜瑞氏菌属(genera Roseburia)和普氏菌属(Prevotella)以及普拉梭菌(Faecalibacteriumprausnitzii)和普氏菌(Prevotella copri)的NRASP中位数分别增加到1.5,而放线杆菌门(Actinobacteria)和双歧杆菌目(Bifidobacteriales)的NRASP中位数则下降到约0.5。以上数据表明了不同肠道菌具有不同的蛋白酶水解程度。
大多数生物过程的NRASP的中位数也在1左右波动(图1B)。而异亮氨酸生物合成过程、缬氨酸生物合成过程、细菌型鞭毛依赖细胞运动、蛋白质转运、羧酸代谢过程、岩藻糖代谢过程和葡萄糖代谢过程的NRASP值均增加到1.75-2,脂肪酸代谢过程和L-苏氨酸分解代谢过程的NRASP进一步增加到2.5,多糖分解代谢过程、碳水化合物运输和跨膜运输的NRASP降低到0.75左右,代谢过程的NRASP进一步降低到0.3。
在酶水平上,参与丁酸代谢的3-羟丁酰辅酶A脱氢酶的NRASP最高(中位数>3),其次是参与脂肪酸β氧化的3-羟基丁酰辅酶A脱氢酶,参与L-苏氨酸降解的甘氨酸C-乙酰基转移酶,参与糖异生的磷酸烯醇式丙酮酸羧激酶(ATP),参与支链氨基酸(BCAA)生物合成的酮酸还原异构酶,以及参与抗氧化剂胁迫的超氧化物歧化酶(NRASP中位数2-3,图1C)。
- 一种宏蛋白质组挖掘方法及其在获取肠道微生物蛋白水解特征中的应用
- 一种自然发酵豆酱中微生物胞外宏蛋白质组的提取方法