含吡非尼酮缓释组合物用于治疗和逆转人脂肪性肝炎的药物用途
文献发布时间:2023-06-19 19:28:50
本申请是申请日为2017年11月09日、申请号为201780076430.3、发明名称为“包含吡非尼酮的缓释组合物用于治疗和逆转人脂肪性肝炎(NAFLD/NASH)的药物用途”的专利申请的分案申请。
技术领域
本发明涉及包含吡非尼酮(PFD-LP)的缓释片剂型药物组合物用于诱导肝脏脂质积累或脂肪变性的减少/消除的用途,及其在逆转晚期肝纤维化和非酒精性脂肪肝病/非酒精性脂肪性肝炎(NAFLD/NASH)中的治疗应用。
背景技术
肝硬化
肝硬化是晚期肝纤维化(ALF)的最后阶段,其定义为细胞外基质(ECM)或间质性瘢痕的过度积累,其是慢性肝损伤的结果。
由于肝脏的再生能力,肝硬化是一种可以缓慢发展的病理过程。另外,已知它是涉及遗传因子的过程。
针对晚期肝纤维化的医学治疗仍在继续变化,导致抗纤维化和预防性治疗的实施,这些治疗目前似乎为全世界数百万患者带来了希望。
ALF的特征在于ECM合成的增加以及其组成的变化,这导致肝组织的异常重塑和随后的器官结构的扭曲。这造成肝功能降低。
病理生理学
这种疾病的病理生理学是通过炎症过程给出的,其中诸如白细胞介素-6(IL-6)、白细胞介素-1β(IL-1β)和肿瘤坏死因子-α(TNF-α)的促炎细胞因子表达增加。炎症伴随着氧化应激,所述氧化应激由以下引起:诸如超氧化物歧化酶(SOD)和过氧化氢酶(CAT)的抗氧化酶的表达减少,以及超氧阴离子、羟基阴离子和过氧化氢的自由基的产生增加等等。同样,首先由转化生长因子β1(TGF-β1)介导的纤维化过程诱导ECM(细胞外基质)的主要成分1型胶原(COL-1)的表达增加,有利于肝星状细胞增殖的生长因子也增加,并且在金属蛋白酶组织抑制剂1(TIMP-1)中也是如此。除了上述信息外,ECM金属蛋白酶的表达也有所降低。所有这些都导致ECM的合成和降解之间的不平衡,这有利于其积累。
细胞外基质的产生
ECM的细胞成分的定义允许在针对该疾病的药物治疗领域中取得进展。肝星状细胞(HSC)是肝硬化中ECM的主要来源。这些细胞存在于狄氏间隙,是肝血窦和肝细胞之间的部位。HSC是维生素A以类维生素A的形式存储的主要部位,这种维生素是在静止表型中表征HSC的维生素。
然而,HSC可通过化学引诱物细胞因子(趋化因子)迁移。可以作为趋化因子的各种分子中有PDGF、MCP-1和CXCR3。最近,已经描述了PDGF参与该过程的机制。据说它通过在其他细胞的膜中扩散来实现,通过拖拽到其他部位使它们移动。
纤维化
HSC不仅通过ECM产生的增加而且通过ECM产生细胞的增殖形成纤维化。肝脏瘢痕的主要研究成分是1型胶原蛋白(COL-1)。越来越多的刺激和通路调节其转录和转录后的表达。最有效的刺激是由TGF-β1观察到的,其由旁分泌和自分泌剂给出。TGF-β1级联中的下游信号传导包括称为Smads的双功能分子家族。TGF-β1还刺激ECM的其他成分的产生,例如纤连蛋白和蛋白多糖。TGF-β1对HSC的刺激是由氢的过氧化和C/EBPβ的依赖机制介导的。
脂质过氧化是重要的产物,其也会在ECM的产生中形成刺激。HSC的抗氧化能力的丧失会增强效果。
结缔组织生长因子(CTGF/CCN2)也被认为是HSC中用于产生ECM的重要刺激。它也可能由高血糖和高胰岛素血症的时期激活。已经认为CTGF的刺激依赖于TGF-β1。
收缩性
HSC的收缩性可能是肝硬化时门静脉阻力早期或晚期增加的主要决定因素。由于血窦收缩和整个肝硬化的收缩性,胶原带阻止门静脉通路中的正常血液流动。
内皮素-1和一氧化氮是HSC收缩性的主要调节剂。该列表还包括血管紧张素原II、类二十烷酸、心房利钠肽、生长抑素、一氧化碳等。细胞骨架α-SMA蛋白表达增加;这赋予了HSC潜在的收缩性。
直接背景技术
发达国家的主要公共卫生问题是超重和肥胖[1]。因此,与代谢综合征相关的诸如非酒精性脂肪肝病/非酒精性脂肪性肝炎(以下称为NAFLD/NASH)的病理学;继续增加他们在全世界的患病率。预计非酒精性脂肪肝将成为未来几十年肝脏相关发病率和死亡率的主要原因。
酒精引起的脂肪肝的发病机制以及由诸如非酒精性脂肪性肝炎的非酒精消耗引起的发病机制是基于导致肝脏脂肪变性的甘油三酯的积累。在储存异常的慢性脂质后,炎症形成第二种损伤,由炎症反应和细胞和细胞因子的产生和浸润协调[2]。最严重的非酒精性脂肪肝的组织学形式是非酒精性脂肪性肝炎(NASH),其特征在于存在或不存在肝硬化时炎性细胞的浸润和肝损伤。
据估计,西方国家20-30%的人患有非酒精性脂肪肝;发展这种状况的倾向似乎与种族-民族起源有关,由45%的西班牙裔、33%的高加索人和24%的非洲裔美国人代表。已经观察到,在存在正常血糖和正常或中等体重的情况下,NAFLD的特征在于实验室水平和临床数据类似于在糖尿病和肥胖中观察到的[1,3]。
最近的研究表明,增加膳食胆固醇的量会导致更严重的炎症、肝细胞受损和纤维化[4]。另一方面,高碳水化合物饮食与胰岛素水平的增加有关,这有助于提高甘油三酯水平[5]。果糖和蔗糖在代谢紊乱的发展中具有特别重要的作用。此外,研究表明,高果糖饮食与肝脏炎症、NASH和既定纤维化的发展有关[6]。响应于慢性过量的卡路里,已经观察到促炎症轴的主要参与者IL-17的上调。这种事件与肝损伤有关,后者由Kupffer细胞和肝星状细胞(HSC)的介质释放导致,而与肝细胞、内皮细胞或中性粒细胞无关[7]。
关于肥胖,脂肪生成以复杂的方式受激素、脂质、糖代谢物和脂肪因子以及其他炎性细胞因子的调节。总之,这些因素加剧了NASH的发作,NASH可促进几种促炎细胞因子诸如TGF-β1、IL-1β、IL-16的分泌,从而导致肝脏炎症[8]。此外,已观察到IL-17A刺激肝细胞产生C反应蛋白(CRP)[9]。最近的研究表明,诸如TGF-β1和IL-6的免疫调节细胞因子的组合抑制了导致免疫耐受性的丧失的调节性T细胞(Treg)的形成,引起Th17细胞产生增加,从而导致更高水平的形成促炎性反馈响应的IL-17A[10,11,12]。
调节脂肪酸氧化和合成的转录因子与非酒精性脂肪肝的发展以及抗炎过程有关[13,14]。在这个意义上,核受体PPARalfa、PPARgamma和LXR代表了旨在改善肝脏脂肪变性和炎症的治疗策略的主要目标。PPARalpha通过增加诸如CPT1A的基因的表达调节脂肪酸的氧化,CPT1A又将脂质内化到线粒体中以从中获取能量[15]。另一方面,长期以来,LXR因其通过促进诸如SREBP1的另一种主转录因子的表达来提高脂肪生成速率的能力而闻名[16]。
最后,已经表明LXRα的活化抑制了编码炎症介质的基因的表达[14,16]。
吡非尼酮
吡非尼酮是一种由小分子组成的药物,其化学名称为5-甲基-1-苯基-2-(1H)吡啶酮。它是一种非肽合成分子,分子量为185.23道尔顿。其化学式为C
已知吡非尼酮发挥其治疗作用的机制之一是通过调节几种细胞因子的作用。吡非尼酮是纤维化细胞因子和TNF-α的有效抑制剂。
已发现缓释型吡非尼酮(PFD-LP)的新的分子作用机制,表明该化合物具有降低酒精性和非酒精性脂肪性肝炎的有害作用的活性,这是本发明的目的。
发明目的
本发明的目的是缓释片剂形式的药物组合物用于逆转和治疗酒精性和非酒精性脂肪性肝炎(NAFLD/NASH)的用途,所述药物组合物包含100mg、200mg、300mg、400mg或600mg吡非尼酮。
本发明的另一个目的是缓释片剂形式的药物组合物用于消退晚期肝纤维化的用途。
本发明的另一个目的是缓释片剂形式的药物组合物用于消退NAFLD/NASH的用途,其通过降低血清胆固醇和甘油三酯而发生。
本发明的另一个目的是缓释片剂形式的药物组合物用于NAFLD/NASH的消退和治疗的用途,其通过降低肝脏脂肪积累(大泡性脂肪变性(macrosteatosis)和小泡性脂肪变性(microsteatosis)的形式)的含量而发生。
此外,本发明的另一个目的是缓释药物组合物中的吡非尼酮用于逆转和治疗晚期肝纤维化和NAFLD/NASH的用途,其治疗效果归因于吡非尼酮,其作为PPARγ(过氧化物酶体增殖物激活受体γ)、PPARα(过氧化物酶体增殖物激活受体α)、LXR和CPT-1的激动剂。伴随这些作用,吡非尼酮-LP诱导NFkB主基因表达的降低,所述NFkB主基因为肝脏炎症过程的诱导物。
本发明通过上述目的进行说明,但不限于此;用于治疗或消退晚期肝纤维化、酒精性和非酒精性脂肪性肝炎(NAFLD/NASH),证明包含吡非尼酮的缓释片剂形式的药物组合物有利于减少吡非尼酮的肝毒性作用,这已在现有技术中公开。
附图说明
从以下对所附权利要求和附图的目的和优选实施方式的详细描述中,本发明的其他特征和优点将变得清楚,其中:
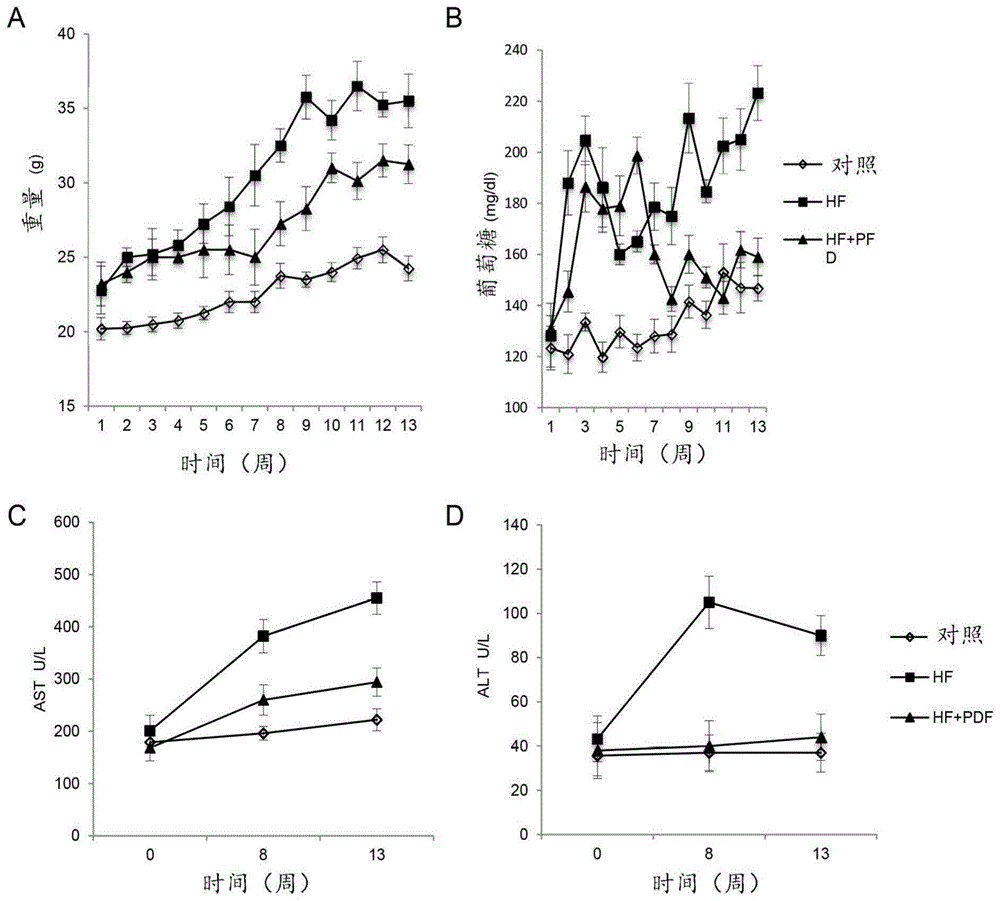
图1(A-D):示出了小鼠的体重、葡萄糖水平和ALT和AST肝酶的水平。
图2:包括在对照、HF和用PFD-LP治疗的小鼠肝脏组织的组织学检查。
图3(A-F):示出了包含在每个所示组中的小鼠血清中的促炎细胞因子的分析。
图4:示出了促纤维化和促炎标记基因的表达。
图5:示出了NAFLD/NASH中脂肪代谢的介质的表达的蛋白免疫印迹,评估了肝脏组织中关键代谢转录因子LXR和PPARα。
图6:示出了在肝脏中炎症过程调节中关键转录因子的表达的蛋白免疫印迹,诸如PPARγ和NFkB。
具体实施方式
在实验中使用的动物
将6至8周龄的小鼠,雄性C57BL/6NHsd(Harlan,墨西哥城),在12小时光照/黑暗循环中在22±2℃的气氛中饲养。将5-7只小鼠随机分配至标准饲料(对照)或高脂肪/高碳水化合物(HF)饮食16周。对照组接受饮食Harlan TM-2018(18%卡路里来自脂肪)并且可自由获取纯净水,而HF组接受Harlan饮食TD-06414(60%卡路里来自脂质)并可以自由获得富含浓度为42g/L的高果糖(55%果糖和45%蔗糖的比例)的水。PFD小鼠组(HF+PFD)接受HF饮食8周,然后HF饮食8周和100mg/kg/天PFD缓释制剂。所有方案均接受0.1ml载体。禁食一夜后,分析血液样本。从开始到处死每周记录小鼠和葡萄糖的重量。
体重、血糖、胆固醇、甘油三酯、VLDL胆固醇和氨基转移酶。
患有由高脂肪饮食(FH)诱导的脂肪性肝炎(NASH/NAFLD)小鼠的特征显示在图1A-1D中。两组之间存在显著差异,HF组小鼠体重最高(相比对照高达37%),HF+PFD-LP组体重比HF组低11%(图1A)。关于血糖,与对照组相比,在HF组中观察到更高的血清葡萄糖水平,在第9周至第13周有显著差异。然而,在治疗的最后五周,HF+PFD-LP与对照组之间没有显著差异(图1B)。我们还发现HF组的AST血清水平(图1C)、ALT(图1D)和胆固醇、甘油三酯和VLDL-胆固醇(表1)与HF+PFD或对照组相比更高。
表1.血清中胆固醇、甘油三酯和VLDL-胆固醇的测定
该值是平均值±SD。小鼠的数量在括号中表示。
a
b
组织学
HF组的肝组织的组织学检查显示出大量的小泡性脂肪变性和具有炎性变化的大泡性脂肪变性(图2)。HF组脂肪变性在腺泡区1主要为大泡性的且严重,腺泡区2出现严重的小泡性脂肪变性和大泡性脂肪变性。腺泡区3发现严重的小泡性脂肪变性和肝细胞中度球囊变性;炎症主要是中性粒细胞和单核细胞的门静脉周围,这也示出了评分为1(0-4)的门静脉纤维化,这种组织学变化与肝纤维化的1C期和3级的NAFLD相容。PFD-LP诱导急剧的肝脏脂肪含量下降。这些结果与大致的肝脏观察结果相关。关于中心和外周脂肪,与HF组相比,HF+PFD-LP示出了较低水平的脂肪。
血清中细胞因子的分析
为了将组织学结果与系统标记相关联,分析了血清IL-17A、IL-6、IL-1β、IFN-γ和TNF-α的促炎细胞因子。与HF组(211±30ng/ml)相比,HF+PFD组(145.8±15.0ng/ml)的血清IL-6水平显著降低(图3A)。与HF组(177.4±20.6ng/ml)相比,HF+PFD组(69.3±13ng/ml)的IL-1β的血清水平显著降低(图3B)。图3C中所示的IFN-γ在PFD-LP组中(18.2±8ng/ml)比在HF组(221.3±50ng/ml)显著减少。类似地,与HF组(254.6±70ng/ml)相比,HF+PFD组(32.9±21.1ng/ml)的TNF-α的血清水平显著降低(图3D)。值得一提的是,与HF组相比(其显示明显增加的IL-17A血清水平为1983.5±400ng/ml),用PFD-LP治疗的小鼠的IL-17A水平(271.1±149.6)显著减少。与对照组相比,HF组中分析的所有细胞因子显示出高度显著的增加。最后,观察到HF+PFD组(37.8±10.4ng/ml)的抗炎细胞因子IL-10的血清水平显著高于HF组(15.4±9.1ng/ml)(图3E)。
促纤维化和促炎标记基因的表达
肝脏信使RNA水平显示在PFD-LP+HF组中TGF-β1表达下降、COL1A1和TNF-α显著下调。此外,与HF组相比,观察到基因CD11b和MCP1表达的显著降低(图4)。
PFD-LP调控肝脏脂肪代谢的调节剂。
为了分析PFD对NAFLD/NASH中脂肪代谢介质调控的影响,评估了在肝组织中关键的代谢转录因子LXR和PPARα。如图5所示,与对照(分别为2,634±1,042和3,890±1,130)和HF组(分别为3375±898和7308±1117)相比,PFD+HF组的LXR(8082±847)和PPARα(11506±1167)的蛋白水平显著增加。为了更好地理解PFD-LP对诸如分别由LXR和PPAR靶向的SREBP1和CPT1A关键蛋白的影响,还对它们进行了评估。如图5所示,与对照(785±396)和HF组(2,622±1,161)相比,PFD-LP诱导前体蛋白SREBP1(6057±847)的增加。然而,其切割形式(活性形式)的SREBP1示出了降低的趋势,但在三组(4567±1620)、HF(1776±893)和PFD+HF(1638±303)之间未发现显著差异。此外,与对照组(4303±820)和HF组(6.172±1.356)相比,在PFD+HF组(9303±809)中观察到CPT1A表达(负责内化肝脏脂肪到线粒体“燃烧”的酶)的显著增加。
最后,图6示出了肝脏中炎症过程调节中的关键转录因子诸如PPARγ和NFkB被特异性调控以导致肝脏炎症减少。
参考文献
1.Marra F,Lotersztajn S.Pathophysiology of NASH:perspectives for atargeted treatment.Curr Pharm Des 2013;19(29):5250–5269.
2.Ferramosca A,Vincenzo Zara.Modulation of hepatic steatosis bydietary fatty acids.World J Gastroenterol 2014;20(7):1746-1755
3.Paschos P,Paletas K.Non alcoholic fatty liver disease and metabolicsyndrome.Hippokratia 2009;13(1):9–19.
4.Van Rooyen DM,Larter CZ,Haigh WG,Yeh MM,Ioannou G,Kuver R,et.al.Hepatic free cholesterol accumulates in obese,diabetic mice and causesnonalcoholic steatohepatitis.Gastroenterology 2011;141:1393–403.
5.Kang H,Greenson JK,Omo JT,Guillot C,Peterman D,Anderson L,et.al.Metabolic syndrome is associated with greater histologic severity,higher carbohydrate,and lower fat diet in patients with NAFLD.Am JGastroenterol 2006;101:2247–53.
6.Lim JS,Mietus-Snyder M,Valente A,Schwarz JM,Lustig RH.The role offructose in the pathogenesis of NAFLD and the metabolic syndrome.Nat RevGastroenterol Hepatol 2010;7:251–64.
7.Harley IT,Stankiewicz TE,Giles DA,Softic S,Flick LM,Cappelletti M,et al.IL-17 signaling accelerates the progression of nonalcoholic fatty liverdisease in mice.Hepatology.2014;59(5):1830–9.
8.
9.Weaver CT,Hatton RD.Interplay between the TH17 and Treg celllineages:a co-evolutionary perspective.Nat Rev Immunol 2009;9:883–9.
10.Korn T,Bettelli E,Oukka M,Kuchroo VK.IL-17 and Th17 Cells.Annu RevImmunol 2009;27:485–517.
11.Tang Y,Bian Z,Zhao L,Liu Y,Liang S,Wang Q,et.al.Interleukin-17exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liverdisease.Clin Exp Immunol 2011;166(2):281-90.
12.Wang Y-X.PPARs:diverse regulators in energy metabolism andmetabolic diseases.Cell Res.2010;20(2):124–37.
13.Yan Xing,Tingting Zhao,Xiaoyan Gao,Yuzhang Wu.Liver X receptorαisessential for the capillarization of liver sinusoidal endothelial cells inliver injury.
14.Yoshikawa T,Ide T,Shimano H,Yahagi N,Amemiya-Kudo M,Matsuzaka T,etal.Cross-Talk between Peroxisome Proliferator-Activated Receptor(PPAR)αandLiver X Receptor(LXR)in Nutritional Regulation of Fatty AcidMetabolism.I.PPARs Suppress Sterol Regulatory Element Binding Protein-1cPromoter through Inhibition of LXR Signaling.Mol Endocrinol.2003;17(7):1240–54.
15.Cui G,Qin X,Wu L,Zhang Y,Sheng X,Yu Q,Sheng H,Xi B,Zhang JZ,ZangYQ.Liver X receptor(LXR)mediates negative regulation of mouse and human Th17differentiation.J Clin Invest.2011;121(2):658–670.
16.Ducheix S,Montagner A,Polizzi A,Lasserre F,Marmugi A,Bertrand-Michel J,et al.Essential fatty acids deficiency promotes lipogenic geneexpression and hepatic steatosis through the liver X receptor.J Hepatol.2013;58(5):984–92.