一种BRD4和Src双靶点抑制剂及其制备方法与应用
文献发布时间:2024-04-18 19:58:26
技术领域
本发明涉及药物开发技术领域,尤其涉及一种BRD4和Src双靶点抑制剂及其制备方法与应用。
背景技术
过去几十年,药物研究大部分集中于设计或寻找作用于单靶点的高选择性的药物分子。目前,靶向单一靶点的药物已经开发了许多,一系列“单分子-单靶点”的靶向药物的成功上市有效缓解了大批患者的病痛。但是,单靶点的作用机制依然具有一定的局限性。在治疗多因素疾病如肿瘤、心血管系统和内分泌系统疾病时,由于体内复杂的信号通路,单靶点药物通常疗效不足。同时,长期作用一个单一靶点,会诱发代偿机制的激活,进而出现耐药或者毒性不良反应。
针对这个问题,多靶点治疗是更好的选择。目前的多靶点治疗方法包括联合用药、多组分药物和多靶点药物。联合用药,也叫做鸡尾酒疗法,需要同时使用多种不同机制的药物,患者依从性差;而多组分药物则是以固定剂量的不同药物制成单一剂型,但是普适性低。相比于上述两种方法,多靶点药物具有更简单的药物与药物相互作用,更高的患者依从性,更好掌控的药代动力学。
目前已经报道了一些多靶点药物,例如靶向表皮生长因子受体(EGFR)和Src激酶的偶联物、靶向雌激素受体α(ERα)和EGFR的偶联物、靶向组蛋白去乙酰化酶(HDAC)和EGFR的偶联物、靶向含溴结构域蛋白4(BRD4)和HDAC的偶联物、以及靶向Zeste增强子同源物2(EZH2)和BRD4的偶联物。其中,厄洛替尼和伏立司他的偶联物CUDC-101已进入I期临床试验,这体现了多靶点药物的设计具有很大潜力。
乳腺癌(BC)是女性中最常见的恶性肿瘤,占女性癌症的31%,其中三阴性乳腺癌(TNBC),占原发性乳腺癌的9-19%。三阴性乳腺癌具有高侵袭性、易复发性,往往预后较差,生存率低,而目前针对三阴性乳腺癌,只有有限的治疗选择。
BRD4是溴结构域和末端外结构域(BET)家族的成员,通过调节RNA聚合酶II(PolII)活性或读取组蛋白上乙酰化赖氨酸残基促进癌基因转录参与癌症发生和发展。最近,在对三阴性乳腺癌的大规模基因组分析时,发现了BRD4表达异常对三阴性乳腺癌细胞群的存活至关重要。因此,BRD4抑制剂是一种潜在的三阴性乳腺癌治疗药物。Src是一种非受体酪氨酸激酶,与肿瘤的进展和转移密切相关。经多个研究报道,Src抑制剂对乳腺癌治疗有效,特别是侵袭性三阴性乳腺癌亚型。其中,达沙替尼被证明可以抑制乳腺癌细胞生长,调节激素和EGFR信号通路,并改变细胞迁移。然而,达沙替尼在三阴性乳腺癌患者的I/II期研究中显示出的疗效有限。虽然达沙替尼和BRD4抑制剂(如JQ1)联合使用对JQ1耐药的三阴性乳腺癌细胞系SUM159R表现出协同作用,但由于Src和BRD4是独立的蛋白,在联合用药时需要同时使用BRD4抑制剂和Src抑制剂,且联合用药通常伴随着患者依从性差、不可预测的药代动力学特征和药物相互作用等问题。
因此,现有技术还有待于改进和发展。
发明内容
鉴于上述现有技术的不足,本发明的目的在于提供一种新型的BRD4和Src双靶点抑制剂及其制备方法与应用,以解决单靶点抑制剂面临的耐药性及治疗效果不佳、联合用药患者依从性较差等问题。
本发明的技术方案如下:
本发明的第一方面,提供一种BRD4和Src双靶点抑制剂,其中,所述BRD4和Src双靶点抑制剂包括式I所示化合物、式II所示化合物、式I所示化合物的药学上可接受的盐、式II所示化合物的药学上可接受的盐、式I所示化合物的溶剂化物、式II所示化合物的溶剂化物中的至少一种;
式I;
式II;
其中,
本发明中,含酯键的碳链中可以含有环状结构(如六元环)及杂原子(如N等),含聚乙二醇的碳链中也可以含有杂原子(如N等),具体地,
、/>
本发明中,将BRD4抑制剂JQ1和Src抑制剂达沙替尼的分子片段,通过具有特殊结构的Linker偶联,获得了一系列对乳腺癌尤其是三阴性乳腺癌具有抗肿瘤活性的BRD4和Src双靶点抑制剂。具体地,首先分析了JQ1与BRD4、达沙替尼与c-Src的结合模式,以确定这两种化合物上适当的修饰位点,可以通过连接子连接,同时保持它们对每个靶蛋白的效力。如图1所示,JQ1的叔丁基酯从结合袋中突出,而达沙替尼的羟乙基延伸到溶剂区域,在不影响目标活性的情况下提供了足够的结合空间。与此同时,发明人结合连接子对双靶点抑制剂的影响(见下文),选择含酯键的碳链、氨基酸链、含聚乙二醇的碳链中的一种作为连接子,在此基础上,将BRD4抑制剂JQ1和Src抑制剂达沙替尼的分子片段通过上述连接子连接,设计并合成了一系列新型的BRD4和Src双靶点抑制剂,其可以同时作用于BRD4和Src两个靶点,与BRD4和Src两个靶点产生相互作用,从而具有更好的疗效、更低的剂量需求以及更小的组织毒性,且患者依从性好。这一特性使其有效地克服了单靶点抑制剂可能面临的耐药性问题,降低了耐药风险,提高了治疗效果。
连接子对双靶点抑制剂具有如下影响:
活性:连接子影响抑制剂与目标酶的相互作用,从而影响其活性。例如,连接子链太长或太短,可能影响抑制剂与酶的结合能力,从而影响其抑制活性。
稳定性:某些连接子可能会影响抑制剂的稳定性。例如,连接子可能受环境因素的影响,如pH、温度、氧化剂等,从而导致抑制剂分解。
细胞膜通透性:连接子的大小和形状可能会影响抑制剂通过细胞膜的能力,从而影响其在细胞内的分布和作用。
副作用:连接子可能影响抑制剂的副作用。例如,某些连接子可能与其他细胞分子相互作用,从而引起不必要的副作用。
可选地,式I所示化合物中的
、/>
采用上述连接子形成上述式I所示化合物时,式I所示化合物作为整体,一部分化合物中含有没有羟乙基部分的达沙替尼,一部分化合物中含有保留羟乙基部分的达沙替尼,目的在于保持化合物对c-Src的活性。
本发明中,上述各结构中的n各自独立地取正整数,例如
可选地,式II所示化合物中的
、/>
当式II所示化合物采用上述连接子时,化合物中保留了达沙替尼的羟乙基部分,以保持化合物对c-Src的活性。
本发明的第二方面,提供一种BRD4和Src双靶点抑制剂的制备方法,其中,包括如下步骤:
将
;
其中,化合物D选自以下结构中的一种:
、/>
R
、/>
m为正整数。
本发明中,上述结构式中的m各自独立地为正整数。例如,
本发明的第三方面,提供一种BRD4和Src双靶点抑制剂的制备方法,其中,包括如下步骤:
将
;
其中,R
、/>
R
、/>
m取正整数。具体地,上述结构中的m各自独立地为正整数,例如
具体地,上述
、/>
本发明的第四方面,提供一种BRD4和Src双靶点抑制剂的制备方法,其中,包括如下步骤:
将
其中,R
-H、-CH
本发明的第五方面,提供一种本发明如上所述的BRD4和Src双靶点抑制剂在制备用于治疗乳腺癌的药物中的应用,或,提供一种采用本发明如上所述的制备方法制备得到的BRD4和Src双靶点抑制剂在制备用于治疗乳腺癌的药物中的应用。
可选地,所述乳腺癌为三阴性乳腺癌。
本发明的第六方面,提供一种药物组合物,其中,所述药物组合物包括本发明如上所述的BRD4和Src双靶点抑制剂和药学上可接受的载体,或所述药物组合物包括采用本发明如上所述的制备方法制备得到的BRD4和Src双靶点抑制剂和药学上可接受的载体。
在本发明中,药物组合物是指本发明化合物与本领域通常接受的用于将生物活性化合物输送至哺乳动物(例如人)的介质的制剂。该介质包括药学上可接受的载体。药物组合物的目的是促进生物体的给药,利于活性成分的吸收进而发挥生物活性。
可选地,所述药学上可接受的载体包括但不限于赋形剂、助流剂、增甜剂、稀释剂、防腐剂、着色剂、矫味剂、表面活性剂、润湿剂、分散剂、助悬剂、稳定剂、等渗剂、溶剂、乳化剂中的至少一种。
有益效果:本发明将BRD4抑制剂JQ1和Src抑制剂达沙替尼的分子片段,通过具有特殊结构的Linker偶联,获得了一系列新型的BRD4和Src双靶点抑制剂。所述BRD4和Src双靶点抑制剂可以同时作用于BRD4和Src两个靶点,与BRD4和Src两个靶点产生相互作用,对乳腺癌尤其是三阴性乳腺癌具有抗肿瘤活性,且患者依从性好,有效地克服了单靶点抑制剂可能面临的耐药性问题,降低了耐药风险,提高了治疗效果。
附图说明
图1为BRD4和Src双靶点抑制剂的设计理念示意图,其中(A)为BRD4晶体结构与JQ1结合的共晶结构示意图,(B)为达沙替尼与c-Src结合的共晶结构示意图,(C)为通过连接JQ1和达沙替尼来设计BRD4和Src双靶点抑制剂的示意图。
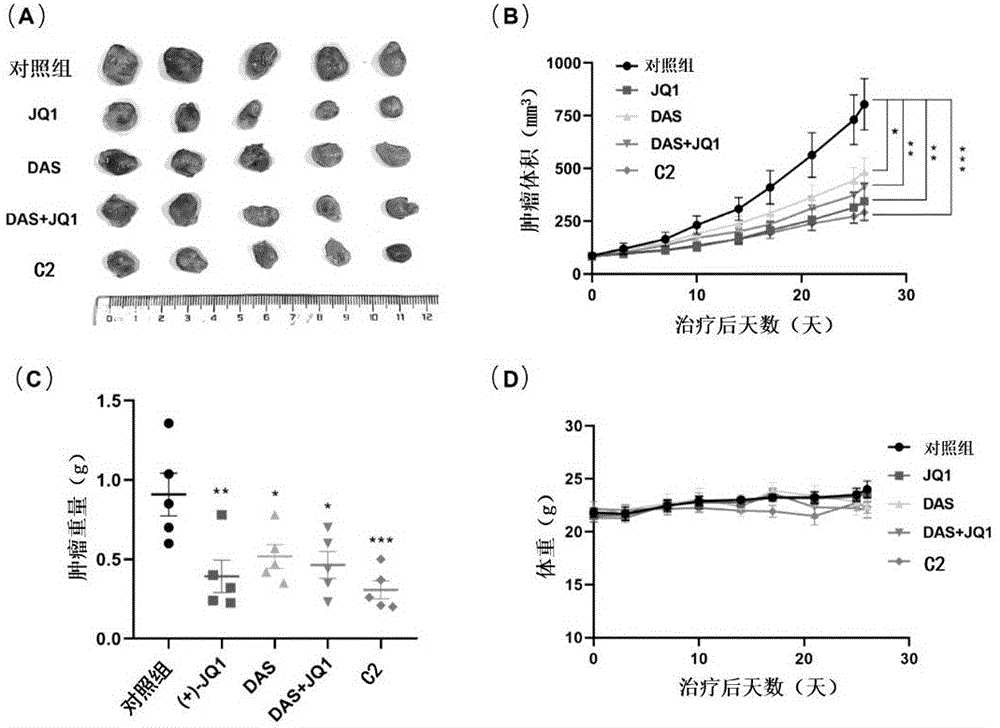
图2为不同组别抗癌效果对比图,其中(A)为不同组别肿瘤大小实物图,(B)为不同组别肿瘤体积随治疗后天数的变化结果图,(C)为不同组别肿瘤重量结果图,(D)为不同组别小鼠体重随治疗天数的变化结果图。
其中,“*”表示p<0.05,“**”表示p<0.01,“***”表示p<0.001。
具体实施方式
本发明提供一种BRD4和Src双靶点抑制剂及其制备方法与应用,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
除非另有定义,本文所使用的所有的技术术语和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。
下面通过具体的实施例进行详细说明。
以下实施例及相关测试中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所有试剂或仪器未注明生产厂商者,均为通过正规渠道商购买得到的常规产品。
以下实施例中化合物A1-A5、B1-B5的结构式为:
,其中的Linker如下表1所示;
以下实施例中化合物C1-C9的结构式为:
表1、化合物A1-A5、B1-B5、C1-C9的结构式中的Linker
以下实施例及相关测试中部分符号或单位的含义:
IC
M:mol/L,例如叔丁醇钾的四氢呋喃溶液(1.0M,0.6mL,0.6mmol)表示摩尔浓度为1.0mol/L的叔丁醇钾的四氢呋喃溶液;
N:当量浓度,例如4N HCl表示4mol/L盐酸溶液;
DAS:达沙替尼。
实施例1化合物6、7、10、11的合成
合成路线如下:
,其中,a-g表示的试剂与反应条件为:
a表示:2-丁酮,硫(S),吗啉,乙醇,70°C,回流;
b表示:Fmoc-Asp(Ot-Bu)-OH(
c表示:哌啶,DMF;
d表示:SiO
e表示:叔丁醇钾,四氢呋喃(THF),PO(OEt)
f表示:4N HCl,乙酸乙酯;
g表示:1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI),对羟基苯甲腈(HOBT),DIPEA,DMF;
h表示:LiOH,甲醇,H
i表示:20% 三氟乙酸(TFA),二氯甲烷(DCM)。
化合物6的合成:
在室温下,将硫(960mg,30mmol)加入到化合物1(也即4-氯苯甲酰乙腈,5.38g,30mmol)、2-丁酮(2.16g,30mmol)和吗啉(2.61g,30mmol)的乙醇(60mL)溶液中,在70℃下回流反应12小时后,冷却至室温,使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产物使用乙酸乙酯/石油醚体系进行硅胶柱层析色谱分离纯化,得到的黄色固体即为化合物2(4.77g,60%)。
向Fmoc-Asp(Ot-Bu)-OH(12.1g,29.4mmol)的DMF(15mL)溶液中加入HBTU(10.61g,28mmol)和DIPEA(10mL),在室温下搅拌5分钟后加入化合物2(3.71g,14mmol),室温下反应20小时。然后使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产物使用乙酸乙酯/石油醚体系进行硅胶柱层析色谱分离纯化,得到棕色油状化合物3(5.62g,61%)。
向化合物3(5.62g,8.54mmol)的DMF溶液(10mL)中加入哌啶(145mg,1.7mmol),室温反应30分钟后,用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产品使用乙酸乙酯/石油醚体系进行硅胶柱层析色谱分离纯化,得到黄色固体即为化合物4(2.46g,66%)。
在室温下,将化合物4(2.46g,5.64mmol)溶解在甲苯(70mL)中,然后加入SiO
在-78℃下,将叔丁醇钾的THF溶液(1.0M,0.6mL,0.6mmol)加入到化合物5(228mg,0.54mmol,1.0当量)的THF(3.6mL)溶液中,得到混合物。将混合物升温至-10℃,并在室温下搅拌30分钟。然后,将混合物冷却至-78℃,并向反应混合物中加入PO(OEt)
化合物7的合成:
向化合物6(551.2mg,1.21mmol)的乙酸乙酯(6.0mL)溶液中加入4N HCl(7.0mL),在室温下搅拌3小时。然后使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产物使用乙酸乙酯/石油醚体系进行硅胶柱层析色谱分离纯化,得到化合物7(339.5mg,70%)。
化合物10的合成:
向化合物7(120mg,0.3mmol)的DMF(3mL)溶液中加入HOBT(127.2mg,0.6mmol)、EDCI(86.25mg)和DIPEA(116.4mg),在室温下搅拌5分钟,然后加入化合物8(也即6-氨基己酸甲酯,60mg,0.33mmol),室温反应6小时。将纯化的产物(150.5mg,95%)溶解在甲醇(1mL)和H
化合物11的合成:
向化合物7(120mg,0.3mmol)的DMF(3mL)溶液中加入HOBT(127.2mg,0.6mmol)、EDCI(86.25mg)和DIPEA(116.4mg),在室温下搅拌5分钟,然后加入化合物9(也即3-(2-(2-(2-氨基乙氧基)乙氧基)乙氧基)丙酸叔丁酯,83.2mg,0.3mmol),室温反应6小时。将纯化的产物(176mg,89%)溶解在DCM(2mL)中,并加入TFA(0.4mL),在室温下搅拌2小时,使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产品使用乙酸乙酯/石油醚体系进行硅胶柱层析色谱分离纯化,得到化合物11(153.2mg,95%)。
实施例2 BRD4和Src双靶点抑制剂(化合物A1、A3、B1、B3和B4)的合成
合成路线如下:
,
其中,a表示的试剂与反应条件为:EDCI,HOBT,DIPEA,DMF。
对上述合成路线进行如下解释:
当化合物12中的R=H时,其为化合物12a;当化合物12中的R=
对于产物化合物,当Linker为
化合物A1的合成:
向化合物7(40mg,0.1mmol)的DMF(2mL)溶液中加入HOBT(42.4mg,0.2mmol)、EDCI(28.75mg)和DIPEA(38.8mg),在室温下搅拌5分钟,然后加入化合物12a(48.8mg,0.11mmol),室温下反应6小时。使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产品使用甲醇/DCM体系进行硅胶柱层析色谱分离纯化,得到的白色粉末即为化合物A1(49.6mg,60%)。其核磁共振氢谱数据为
化合物A3的合成:
参照化合物A1的合成方法,由化合物10和化合物12a反应得到化合物A3(35.7mg,38%),其为白色粉末。其核磁共振氢谱数据为
化合物B1的合成:
参照化合物A1的合成方法,由化合物7和化合物12b反应得到化合物B1(61mg,70%),其为白色粉末。其核磁共振氢谱数据为
化合物B3的合成:
参照化合物A1的合成方法,由化合物10和化合物12b反应得到化合物B3(44.3mg,45%),其为白色粉末。其核磁共振氢谱数据为
化合物B4的合成:
参照化合物A1的合成方法,由化合物11和化合物12b反应得到化合物B4(26.8mg,25%),其为白色粉末。其核磁共振氢谱数据为
实施例3 BRD4和Src双靶点抑制剂(化合物A2、A4、A5、B2、B5和化合物C1-C9)的合成
化合物A2、A4、A5、B5和C1的合成路线如下:
;
化合物B2和C2-C9的合成路线如下:
,
其中,a表示的试剂与反应条件为:EDCI,HOBT,DIPEA,DMF;b表示的试剂与反应条件为:i)对于-Fmoc时:哌啶,DMF,30-85%;ii)对于-Boc时:4 N HCl,乙酸乙酯。
化合物14(14a-14e)、16的合成:
向化合物13a(97.6mg,0.3mmol)的DMF(3mL)溶液中加入HOBT(127.2mg,0.6mmol)、EDCI(86.25mg,0.45mmol)和DIPEA(116.4mg,0.9mmol),在室温下搅拌5分钟后,加入化合物12a(133.2mg,0.3mmol),室温反应6小时。将纯化产物(112.6mg,0.15mmol,50%)与哌啶(2.6mg,0.03mmol)一起加入到DMF溶液(1mL)中,室温搅拌30分钟,然后加入乙酸乙酯(6mL)和饱和浓盐水(6mL)。使用乙酸乙酯/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压蒸馏除去溶剂,浓缩后的产品使用甲醇/DCM体系进行硅胶柱层析色谱分离纯化,得到化合物14a(55.5mg,70%)。
向化合物13d(68.8mg,0.3mmol)的DMF(3mL)溶液中加入HOBT(127.2mg,0.6mmol)、EDCI(86.25mg,0.45mmol)和DIPEA(116.4mg,0.9mmol),在室温下搅拌5分钟后,加入化合物12b(133.2mg,0.3mmol),室温反应6小时。使用DCM /水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压浓缩。将残留物溶解在乙酸乙酯(4mL)中,并加入4N HCl(3.0mL)。3小时后,减压下浓缩混合物得到化合物14d。
按照化合物14d的合成步骤:
化合物14b由化合物13b和化合物12a反应获得;
化合物14c由化合物13d即(S)-1-(叔丁氧羰基)哌啶-2-羧酸和化合物12a反应获得;
化合物14e由化合物13c即(叔丁氧羰基)-L-脯氨酸和化合物12b反应获得;
化合物16a由化合物15a即(叔丁氧羰基)-甘氨酸和化合物12b反应获得;
化合物16b由化合物15b即(叔丁氧羰基)-DL-丙氨酸和化合物12b反应获得;化合物16c由化合物15c即(叔丁氧羰基)-L-缬氨酸和化合物12b反应获得;
化合物16d由化合物15d即(叔丁氧羰基)-L-亮氨酸和化合物12b反应获得;化合物16e由化合物15e即(叔丁氧羰基)-L-异亮氨酸和化合物12b反应获得;
化合物16f由化合物15f即(叔丁氧羰基)-L-甲硫氨酸和化合物12b反应获得;
化合物16g由化合物15g即(叔丁氧羰基)-L-色氨酸和化合物12b反应获得;
化合物16h由化合物15h即(叔丁氧羰基)-L-苯丙氨酸和化合物12b反应获得;
按照化合物14a的合成步骤:
化合物16i由化合物15i即(S)-2-((((9H-芴-9-基)甲氧基)羰基)氨基)-4-(叔丁氧基)-4-氧代丁酸和化合物12b反应获得。
化合物A2的合成:
参照化合物A1的合成方法,由化合物7和化合物14a反应获得化合物A2(41mg,45%),其为白色粉末。其核磁共振氢谱数据为
化合物A4的合成:
参照化合物A1的合成方法,由化合物7和化合物14b反应获得化合物A4(46.5mg,48%),为白色粉末。其核磁共振氢谱数据为
化合物A5的合成:
参照化合物A1的合成方法,由化合物7和化合物14c反应获得化合物A5(38.5mg,41%),其为白色粉末。其核磁共振氢谱数据为
化合物B2的合成:
参照化合物A1的合成方法,由化合物7和化合物16a反应获得化合物B2(65mg,70%),其为白色粉末。其核磁共振氢谱数据为
化合物B5的合成:
参照化合物A1的合成方法,由化合物7和化合物14d反应获得化合物B5(52mg,53%),其为白色粉末。其核磁共振氢谱数据为
化合物C1的合成:
参照化合物A1的合成方法,由化合物7和化合物14e反应获得化合物C1(48.4mg,50%),其为白色粉末。其核磁共振氢谱数据为
化合物C2的合成:
参照化合物A1的合成方法,由化合物7和化合物16b反应获得化合物C2(58.4mg,62%),其为白色粉末。其核磁共振氢谱数据为
化合物C3的合成:
参照化合物A1的合成方法,由化合物7和化合物16c反应获得化合物C3(68.9mg,71%),其为白色粉末。其核磁共振氢谱数据为
化合物C4的合成:
参照化合物A1的合成方法,由化合物7和化合物16d反应获得化合物C4(34.4mg,30%),其为白色粉末。其核磁共振氢谱数据为
化合物C5的合成:
参照化合物A1的合成方法,由化合物7和化合物16e反应获得化合物C5(83.6mg,85%),其为白色粉末。其核磁共振氢谱数据为
化合物C6的合成:
参照化合物A1的合成方法,由化合物7和化合物16f反应获得化合物C6(52.1mg,52%),其为白色粉末。其核磁共振氢谱数据为
化合物C7的合成:
参照化合物A1的合成方法,由化合物7和化合物16g反应获得化合物C7(79.3mg,75%),其为白色粉末。其核磁共振氢谱数据为
化合物C8的合成:
参照化合物A1的合成方法,由化合物7和化合物16h反应获得化合物C8(58mg,57%),其为白色粉末。其核磁共振氢谱数据为
化合物C9的合成:
参照化合物A1的合成方法,由化合物7和化合物16g反应获得化合物C9(36.7mg,35%),其为白色粉末。其核磁共振氢谱数据为
实施例4化合物20(即带有荧光素标记的JQ1)的合成
其合成路线如下:
,
其中,a表示的试剂与反应条件为:EDCI,HOBT,DIPEA,DMF;b表示的试剂与反应条件为:10% TFA,DCM;c表示的试剂与反应条件为:THF,氩气。
向化合物7(80mg,0.2mmol)的DCM(4mL)溶液中加入HOBT(84.8mg,0.4mmol)、EDCI(57.5mg)和DIPEA(77.6mg),室温搅拌5分钟,然后加入(4-氨基丁基)氨基甲酸叔丁酯(即化合物17,37.7mg,0.2mmol),室温反应6小时。将纯化的产物(93mg,0.16mmol,81%)溶解在DCM(2mL)中,并加入TFA(0.2mL),室温搅拌2小时。使用DCM/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压浓缩,浓缩后的产品使用甲醇/DCM体系进行硅胶柱层析色谱分离纯化,得到化合物18(58.1mg,0.12mmol ,75%)。在氩气保护下,将化合物18(58.1mg,0.12mmol)和化合物19即异硫氰酸荧光素(46.7mg 0.12mmol,1.0当量)溶解在THF(4mL)中,在室温下反应12小时,使用DCM/水体系进行萃取,收集有机相,并用无水硫酸钠干燥,减压浓缩,浓缩后的产物使用甲醇/DCM体系进行硅胶柱层析色谱分离纯化,获得化合物20(51.6mg,50%)。
测试:
(1)在人乳腺癌细胞(MDA-MB-231细胞)上BRD4和Src双靶点抑制剂(实施例2-3中的化合物)的抗增殖活性测试:利用细胞计数试剂盒-8(CCK-8)检测细胞活力。MDA-MB-231细胞接种于96孔板,密度为每孔3000个细胞。过夜孵育后,以梯度浓度(起始浓度为50μM,设置9个梯度浓度,每个梯度浓度用DMEM+10%FBS+1%P/S培养基进行5倍稀释)给药3天。然后用100μL含10% CCK8试剂的新鲜培养基(Bimake;Selleck Chemicals;cat. no. B34304),在37℃、5%CO
表2、在MDA-MB-231细胞上实施例2-3中化合物的抗增殖活性结果
a在MDA-MB-231细胞上化合物的IC
(2)实施例2-3中化合物对BRD4的IC
a、竞争荧光偏振结合试验
通过竞争荧光极化(FP)结合实验确定化合物对BRD4-BD1结构域的抑制作用和结合亲和力。384孔板(Nunc 262260)在BioTek Synergy H1上读取,激发波长为485nm,发射波长为535nm。首先根据实施例4合成FITC标记的JQ1即化合物20。通过监测荧光探针在固定浓度和梯度浓度下的荧光偏振值(毫单位/偏振,mP),确定FITC标记的JQ1到BRD4-BD1的平衡解离常数(K
b、时间分辨荧光共振能量转移(TR-FRET)试验
根据HTRF KinEASE-TK试剂盒(62TK0PEB,Cisbio)的说明,测量不同化合物对c-Src激酶的抑制作用。以达沙替尼(Selleck)为阳性对照。简单地说,384孔分析板(784075,Greiner)每孔加入5μL具有浓度梯度的化合物(设置10个浓度梯度,起始浓度为300nM,每个浓度梯度用激酶缓冲液进行3倍稀释)和5μL 2X SRC(Signalchem)。然后加入5μL的2X TK -底物-生物素和ATP的混合物启动反应。所有成分用1X激酶缓冲液稀释。密封,室温孵育30分钟。之后,每孔加入5μL含有4X Sa-XL 665的HTRF检测缓冲液和5μL TK-antibody-Cryptate。将检测板密封,室温孵育1小时。在Envision 2104多标签读取器(PerkinElmer,USA)上读取615nm(Cryptate)和665nm(XL 665)的荧光信号。计算每孔的A比值(665/615nm)。%抑制率=(1-(化合物比值-对照阳性比值)/(溶媒对照组比值-对照阳性比值))×100%。IC
表3、化合物对BRD4的IC
a化合物对BRD4的IC
(3)动物实验
NOD-SCID小鼠购自广东耀康生物科技有限公司,6-7周龄时在每只小鼠右背皮下注射500万个MDA-MB-231细胞。半个月后,肿瘤大小达到60-70mm
由以上测试结果表明,本发明提供的一系列新型BRD4和Src双靶点抑制剂(化合物A1-A4、B1-B4、C1-C4、C6-C9)均对BRD4和c-Src保持较强的抑制活性,能够用于治疗三阴性乳腺癌。其中,化合物C2对BRD4和c-Src保持最强的抑制活性,具有最优的性能,其抗肿瘤效果优于JQ1、达沙替尼单独使用,也优于两者联合使用。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 一种二氢蝶啶酮类BRD4蛋白抑制剂、制备方法及其应用
- 一类HDAC/ALK双靶点抑制剂及其制备方法与应用
- 一种双靶点化合物及其制备方法和在制备sEH抑制剂与PPARs激动剂中的应用
- 一种APN和AKT双靶点抑制剂及其制备方法和应用