一种美托洛尔类原料药或制剂中N-亚硝基美托洛尔的检测方法
文献发布时间:2024-04-18 19:58:30
技术领域
本发明属于药物分析检测技术领域,具体涉及一种美托洛尔类原料药或制剂中N-亚硝基美托洛尔的检测方法。
背景技术
有机酸美托洛尔,如酒石酸美托洛尔、琥珀酸美托洛尔等,为选择性肾上腺素B
亚硝胺类杂质属于ICH M7(R1)(《评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险》)指南中提及的“关注队列”物质。以药物为基质的亚硝胺物质(NDSRI,nitrosamine drug substance related impurities)指的是那些药物分子本身参与亚硝胺杂质形成的类型,这些药物中通常都含有二级或三级胺结构,与药物中微量的亚硝酸生成亚硝胺物质。N-亚硝基美托洛尔为美托洛尔药物中特有的NDSRI遗传毒性杂质,结构见下式。欧洲药品管理局(EMA)及美国食品药品监督管理局(FDA)均要求对NDSRI遗传毒性杂质严格控制。因此,能够准确检测N-亚硝基美托洛尔对于该类药物的临床用药的安全性具有非常重要的意义。
经检索,目前未见N-亚硝基美托洛尔检测方法的有关报道。亚硝胺常见亚硝胺的检测方法主要有液质联用法,气质联用法等。亚硝胺杂质限度低,要求供试品溶液浓度高,导致基质干扰大,不容易检测,因此亟需开发一种灵敏度高、专属性良好、准确度高的针对N-亚硝基美托洛尔的检测方法。
发明内容
本发明提供了一种能够准确测定美托洛尔类原料药或制剂中N-亚硝基美托洛尔的检测方法,克服了由于限度低,供试品溶液浓度高,导致基质干扰带来难检测的问题。
本发明的技术方案为:一种美托洛尔类原料药或原料药尔中N-亚硝基美托洛尔的检测方法,包括以下步骤:
(1)用甲醇水溶液对待测样品进行溶解,得到供试品溶液;
(2)以甲醇水溶液为溶剂,对N-亚硝基美托洛尔对照品进行溶解和定容,配制得到对照品溶液;
(3)采用LC-MS/MS法检测供试品溶液和对照品溶液,使用外标法计算得到供试品中N-亚硝基美托洛尔的含量。
LC-MS/MS法中的液相色谱条件为:
色谱柱:十八烷基硅烷键合硅胶,HPH填料色谱柱,
流动相A:0.05~0.2%甲酸水溶液,优选为0.1%甲酸水溶液,
流动相B:0.05~0.2%甲酸水溶液,优选为0.1%甲酸甲醇溶液,
进样量:1~7μl,
流速:0.3~1.0ml/min,
梯度梯度洗脱条件如下:
梯度洗脱条件优选为
质谱分析条件为:
进一步的,
所述美托洛尔类原料药指美托洛尔在药学上可接受的有机酸盐。
所述的步骤(1)中,待测样品为美托洛尔制剂时,剂型优选为片剂。
所述的步骤(1)中,待测样品为美托洛尔制剂时,供试品溶液的配制方法为:将制剂研成粉末,称取适量,用甲醇水溶液溶解,10000rpm离心10min,取上清液为供试品溶液。
所述甲醇水溶液为体积比30-70%的甲醇水溶液;优选体积比50%的甲醇水溶液。
所述的步骤(1)中,供试品溶液中含有机酸美托洛尔的浓度为10~75mg/mL,优选为50mg/ml;
所述的步骤(2)中,对照品溶液中N-亚硝基美托洛尔的浓度为0.4~3ng/mL,优选为2ng/ml。
所述步骤(3)中,优选的,色谱柱为Agilent InfinityLab Poroshell HPH-C18色谱柱。在一具体实施方案中,色谱柱为Agilent InfinityLab Poroshell HPH-C18 4.6*150mm 2.7μm,流速为1.0ml/min。在另一具体实施方案中,色谱柱为Agilent InfinityLabPoroshell HPH-C18 3.0*100mm
2.7μm,流速为0.4ml/min。
同现有技术相比,本发明的有益效果体现在:
本发明提供了一种能够准确测定美托洛尔类原料药或制剂中N-亚硝基美托洛尔的检测方法,克服了由于限度低,供试品浓度高,导致基质干扰带来难检测的问题。通过筛选色谱条件使得N-亚硝基美托洛尔与美托洛尔及其他杂质进行分离,避免共流出;优化质谱条件使检测限达0.121ng/mL,定量限达0.404ng/ml,相当于供试品溶液浓度的0.008ppm,且定量限浓度经过准确度的验证。本方法专属性、灵敏度、准确度、线性、重复性均符合验证要求,对美托洛尔类原料药及制剂的质量控制及临床用药安全性具有十分重要的意义。
附图说明
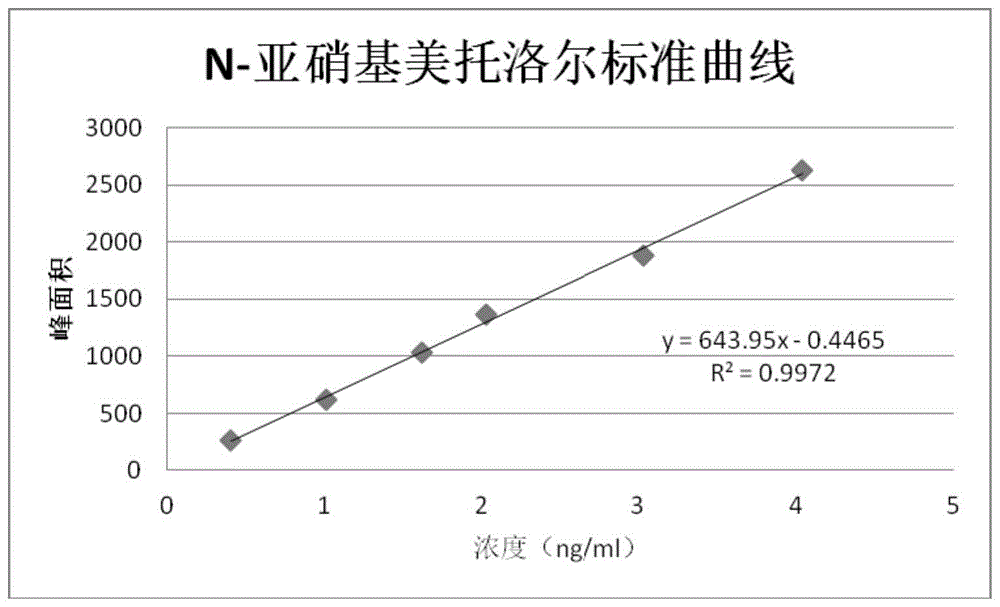
图1为N-亚硝基美托洛尔的标准曲线。
图2为实施例1中空白溶剂谱图。
图3为实施例1中N-亚硝基美托洛尔对照品溶液谱图。
图4为实施例1中酒石酸美托洛尔原料药溶液谱图。
图5为实施例1中100%加标溶液谱图。
图6为实施例2中空白溶液谱图。
图7为实施例2中琥珀酸美托洛尔片供试品溶液的紫外色谱图。
图8为实施例2中N-亚硝基美托洛尔对照品溶液谱图。
图9为实施例2中琥珀酸美托洛尔片供试品溶液的谱图。
图10为实施例2中100%加标溶液的谱图。
具体实施方式
下面结合实施例对本发明进行详细的说明,以帮助理解本发明的精神实质,但该实施例仅是代表性的,不以任何方式限制本发明的范围。
本发明中,如无特别说明,溶剂的比例为体积比。
1.试剂
分析天平:XPR105
甲醇:LC-MS
甲酸:LC-MS
水:UP水
2.对照品
N-亚硝基美托洛尔对照品及供试品酒石酸美托洛尔原料药由浙江普洛家园药业有限公司提供,琥珀酸美托洛尔片由浙江普洛康裕制药有限公司提供。
实施例1
色谱条件:
液相色谱三重四级杆质谱联用仪:Agilent1290II+6470B,
色谱柱:Agilent InfinityLab Poroshell HPH-C18,4.6*150mm 2.7μm,
流动相A:0.1%甲酸水溶液,
流动相B:0.1%甲酸甲醇溶液,
进样量:5μl,
流速:1.0ml/min,
梯度梯度洗脱条件为:
质谱分析条件为:
供试品处理:
溶剂:50%的甲醇水溶液。
对照储备溶液:取N-亚硝基美托洛尔对照品20mg,精密称定,至50mL容量瓶中,用甲醇溶解稀释后并定容至刻度,摇匀。精密移取0.5ml上述溶液至100ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。再移取溶液1.0ml上述溶液至10ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
对照品溶液:精密移取0.5ml对照储备溶液至50ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
供试品溶液:称取酒石酸美托洛尔原料药供试品250mg,精密称定,于10ml离心管中,加5.0ml溶剂溶解,摇匀。
100%加标溶液:称取酒石酸美托洛尔供试品250mg,精密称定,于10ml离心管中,加5.0ml对照品溶液溶解,摇匀。
分别取空白溶剂、对照品溶液、供试品溶液和100%加标溶液,按上述LC-MS条件进行检测,记录谱图;结果见图2~图5。
图2表明,溶剂不干扰测定。
图3表明,杂质N-亚硝基美托洛尔响应较高,峰形较好。
图4表明,酒石酸美托洛尔原料药中N-亚硝基美托洛尔有少量检出。
图5表明,实施例1的检测条件专属性良好,可以有效分离检测酒石酸美托洛尔中N-亚硝基美托洛尔,可用于酒石酸美托洛尔中N-亚硝基美托洛尔的分离测定。
实施例2
色谱条件:
液相色谱三重四级杆质谱联用仪:LC40+8060NX,
色谱柱:Agilent InfinityLab Poroshell HPH-C18,3.0*100mm 2.7μm,
流动相A:0.1%甲酸水溶液,
流动相B:0.1%甲酸甲醇溶液,
进样量:7μl,
流速:0.4ml/min,
梯度梯度洗脱条件为:
质谱分析条件为:
供试品处理
溶剂:同实施例1。
对照储备溶液:同实例1。
对照品溶液:精密移取0.5ml对照储备溶液至100ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
供试品溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml溶剂溶解,10000rpm离心10min,取上清液为供试品溶液。
100%加标溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml对照品溶液溶解,10000rpm离心10min,取上清液为100%加标溶液。
分别取空白溶剂、对照品溶液、供试品溶液和100%加标溶液,按上述LC-MS条件进行检测,记录谱图;结果见图6~图10。
图6表明,空白溶剂无干扰。
图7,图8表明,供试品溶液中主峰在2.5min之前出峰,且在N-亚硝基美托洛尔出峰处无明显干扰,证明专属性良好,可用于琥珀酸美托洛尔片中N-亚硝基美托洛尔的分离测定。
图9表明琥珀酸美托洛尔片中N-亚硝基美托洛尔有少量检出。
图10表明实施例2的检测条件专属性良好,可以有效分离检测琥珀酸美托洛尔片剂中N-亚硝基美托洛尔。
实施例3
灵敏度
液相及质谱条件同实施例1。
对照储备溶液:同实施例1。
定量限溶液:精密移取0.1ml对照储备溶液,于50ml容量瓶中,用溶剂稀释并定容至刻度,摇匀。
检测限溶液:精密移取3.0ml定量限溶液,于10ml容量瓶中,用溶剂稀释并定容至刻度,摇匀。
取定量限溶液及检测限溶液各5μl注入液相质谱仪,记录并计算目标杂质检测限、定量限溶液的浓度及其信噪比。
灵敏度结果:检测限溶液的浓度为0.121ng/ml,信噪比为23,相当于供试品溶液浓度的0.002ppm;定量限溶液的浓度为0.404ng/ml,信噪比为52,相当于供试品溶液浓度的0.008ppm。
实施例4
线性范围
液相及质谱条件同实施例1。
对照储备溶液:同实例1。
精密量取对照储备溶液适量,用溶剂分别配制成含杂质0.4ng/mL、1ng/mL、1.6ng/mL、2ng/mL、3ng/mL、4ng/mL的线性溶液,取上述溶液各5μL,注入液相质谱仪,记录谱图,以N-亚硝基美托洛尔浓度和相对应的峰面积做线性回归。
表1线性测定结果
图1为线性实验的标准曲线图,N-亚硝基美托洛尔仪器响应值在0.404ng/mL~4.04ng/mL内表现出良好的线性关系,线性相关系数为0.9986,满足检测要求。
实施例5
酒石酸美托洛尔原料药中N-亚硝基美托洛尔准确度
液相及质谱条件同实施例1。
对照储备溶液同实施例1。
定量限加标储备液:精密移取0.1ml对照储备溶液至50ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
100%加标储备液:精密移取0.5ml对照储备溶液,于50ml容量瓶中,用溶剂稀释并定容至刻度,摇匀。
150%加标储备液:精密移取0.75ml对照储备溶液至50ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
供试品溶液:称取酒石酸美托洛尔供试品约250mg,精密称定,于10ml离心管中,加5.0ml溶剂溶解,摇匀。同法配制3份。
定量限加标溶液:称取酒石酸美托洛尔供试品250mg,精密称定,于10ml离心管中,加5.0ml定量限加标储备液溶解,摇匀。同法配制3份。
100%加标溶液:称取酒石酸美托洛尔供试品250mg,精密称定,于10ml离心管中,加5.0ml 100%加标储备液溶解,摇匀。同法配制3份。
150%加标溶液:称取酒石酸美托洛尔供试品250mg,精密称定,于10ml离心管中,加5.0ml的150%加标溶液储备液,摇匀。同法配制3份。
精密量取上述各溶液5μL,依次注入液相质谱仪,记录谱图。
表2准确度测定结果
已知可接受标准为回收率在75-120%之间,RSD值小于10.0%。表3显示,在3个加标水平上,利用此方法检测美托洛尔的回收率为87.5%~109.3%,RSD为8.8%,符合标准,说明本法的回收率较高。
实施例6
琥珀酸美托洛尔片中N-亚硝基美托洛尔准确度
液相及质谱条件同实施例2。
对照储备溶液同实施例1。
定量限加标储备液:精密移取0.1ml对照储备溶液至100ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
100%加标储备液:精密移取0.5ml对照储备溶液,于100ml容量瓶中,用溶剂稀释并定容至刻度,摇匀。
150%加标储备液:精密移取0.75ml对照储备溶液至100ml容量瓶中,用稀释剂稀释并定容至刻度,摇匀。
供试品溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml溶剂溶解,10000rpm离心10min,取上清液为供试品溶液。同法配制3份。
定量限加标溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml定量限加标储备液溶解,10000rpm离心10min,取上清液为供试品溶液。同法配制3份。
100%加标溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml100%加标储备液溶解,10000rpm离心10min,取上清液为供试品溶液。同法配制3份。
150%加标溶液:将琥珀酸美托洛尔片(标示量为25%)研成粉末,称取500mg,于10ml离心管中,加5.0ml150%加标储备液溶解,10000rpm离心10min,取上清液为供试品溶液。同法配制3份。
精密量取上述各溶液5μL,依次注入液相质谱仪,记录谱图。
表2准确度测定结果
已知可接受标准为回收率在75-120%之间,RSD值小于10.0%。表3显示,在3个加标水平上,利用此方法检测美托洛尔的回收率为98.5%~107.8%,RSD为4.2%,符合标准,说明本法的回收率较高。
实施例7
重复性
液相及质谱条件同实施例1。
对照品溶液:同实施例1。
供试品溶液:同实施例1。
100%加标溶液:同实施例1,同法配制6份。
精密量取上述各溶液5μL,依次注入液相质谱仪,记录谱图。
表3重复性测定结果
已知可接受标准RSD值小于8%,供试品测试结果的RSD小于5%,重复性较好。
对比例1
与实施例1不同之处在于,对比例1选用ESI作为离子源,不同填料的色谱柱(Agilent Eclipse C18 RRHD,1.8μm,2.1×50mm),具体分析参数见色谱及质谱条件。
1.色谱条件
仪器:LC40D+8060NX,
色谱柱:Agilent Eclipse C18 RRHD,1.8μm,2.1×50mm,
流动相A:0.1%甲酸水溶液,
流动相B:0.1%甲酸(甲醇:乙腈=1:1),
进样量:3μl,
流速:0.3ml/min,
梯度洗脱条件:
2.质谱条件
3.供试品处理
稀释剂:水:乙腈:甲酸=800:200:4。
对照品溶液:配制成2ng/ml的溶液。
供试品溶液:称取适量酒石酸美托洛尔原料药,用稀释剂配制成浓度为50mg/ml溶液。
100%加标溶液:称取适量酒石酸美托洛尔原料药,用对照品溶液配制成浓度为50mg/ml溶液。同法配制2份。
4.进样验证初步回收率:
取上述溶液各3μL,注入液相质谱仪,记录谱图。
表4对比例1的初步回收率
由表4可知,在该条件下,初步回收率低于50%,回收率低。该条件下未能完全分离目标杂质与供试品,且在ESI离子源的模式下受基质干扰更大,无法进行准确检测。
- 一种阿加曲班原料药或制剂中N-亚硝基二甲胺、N-亚硝基二乙胺的分析方法
- 一种恩他卡朋原料或其制剂中N-亚硝基二甲胺与N-亚硝基二乙胺的检测方法