一种拷贝数变异的检测方法、装置以及计算机可读介质
文献发布时间:2024-04-18 20:01:23
技术领域
本发明属于生物信息学技术领域,具体涉及一种拷贝数变异的检测方法、装置以及计算机可读介质。
背景技术
人类基因组中存在大量的变异,根据碱基数目,其可分为单核苷酸变异(SingleNucleotide Variant,SNV)和结构变异(Structural Variation,SV)。拷贝数变异(CopyNumber Variation,CNV)是结构变异的一种形式,是指与参照基因组相比,大小在50bp到数十Mb DNA片段的缺失、插入、复制和复杂多位点变异。近年来的研究表明,基因组片段的CNV通过改变基因剂量或染色体构象影响基因的表达,在疾病进展、表型多态性和进化研究中占据重要的地位。目前在全基因组范围内寻找CNV主要基于两种技术,分别是基因芯片技术(DNA chip)及新一代测序技术(Next Generation Sequencing,NGS)。
现有技术,如专利公告号为CN108427864B的中国发明专利,公开了一种拷贝数变异的检测方法、装置及计算机可读介质,通过采用T分布拟合获得平均测序深度,进而计算拷贝数基线及分析存在异常的基因片段。该分析方法可有效消除各个检测区域因NGS试验方法导致的测序深度的波动性对检测结果的影响和不同panel间检测结果不一致的问题。但该方法需要大量正常样本构成基线,使得检测效率和难度大大增加。
发明内容
为解决上述问题,本发明提供一种拷贝数变异的检测方法、装置以及计算机可读介质。避免由基线样本引入额外的误差,并节省了建立基线样本的成本,且基于低深度全基因组测序,检测标志物不受到引物设计的影响,更容易形成标准化检测。
为了实现上述目的,本发明所采用的技术方案如下:
一方面,本发明提供一种拷贝数变异的检测方法,包括:对肿瘤样本和正常样本取样,其中正常样本需包括有与所述肿瘤样本严格配对的白细胞,进行全基因组高深度测序,测序深度为20-30×,计算出肿瘤纯度最低检出限,并基于所述肿瘤纯度最低检出限计算相应灵敏性和特异性所对应的理论t值;对待测样本进行低深度测序,测序深度至少为1×,构建CNV阳性状态和CNV阴性状态t分布曲线,针对待确定CNV状态的logR值,分别计算其在上述t分布对应的t统计量,通过对比t统计量与理论t值,判定CNV状态。
进一步的,肿瘤样本的取样例数不少于10例,正常样本的取样例数不小于20例。
进一步的,计算肿瘤纯度最低检出限,包括:通过所述肿瘤样本以及与之配对的正常样本计算出肿瘤纯度;基于所述肿瘤纯度和正常样本按照预设纯度针对每一例肿瘤样本进行reads抽取和混样,并计算t分布曲线的标准差;基于拷贝数构建CNV阳性状态和CNV阴性状态t分布曲线,并计算在一定特异性范围下所对应的特异性t值,并基于所述特异性t值计算灵敏性;将同一预设纯度的混合样本的灵敏性取均值,并与所述特异性范围进行配对组合;通过约登指数将不同预设纯度的特异性与灵敏性进行计算,得到满足预设条件下的肿瘤纯度最低检出限。
进一步的,混样后,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb;采用CBS算法对CNV变异的断点位置对染色体进行分割,将具有相同拷贝数的连续bin区划分为同一个segment;针对基因水平的拷贝数变异分析采用bin区来构建t分布曲线;针对染色体水平的拷贝数变异采用segment来构建t分布曲线。
进一步的,t分布曲线的标准差的计算方法为:
logR=log
其中A为通过矫正后的reads数;
;
其中Var(logR
针对染色体水平,特定长度的segment,t分布的标准差为:
;
其中N代表特定segment的bin个数;
针对基因水平而言,t分布的标准差为:
。
进一步的,基于拷贝数构建CNV阳性状态和CNV阴性状态t分布曲线,包括:确定CNV阴性分布拷贝数C
计算CNV阳性分布均值X
;
;
其中
进一步的,所述灵敏性的计算方法为:
通过特异性t值t
;
基于所述灵敏性t值t
进一步的,针对染色体水平,确定约登指数等于98%的肿瘤纯度作为该染色体的肿瘤纯度最低检出限;针对基因水平,采用约登指数增长的饱和点确定肿瘤纯度最低检出限,规定肿瘤纯度上升5%,约登指数上升首次小于2%的点作为约登指数增长的饱和点。
进一步的,计算待测样本CNV阳性状态分布和CNV阴性状态分布的t统计量,
当
当
其中,t
本发明还提供了一种拷贝数变异的检测装置,包括:肿瘤纯度最低检出限计算模块,用于对肿瘤样本和正常样本进行全基因组高深度测序,测序深度为20-30×,计算出肿瘤纯度最低检出限,并基于所述肿瘤纯度最低检出限计算相应灵敏性和特异性所对应的t值;CNV判定模块,对待测样本进行低深度测序,测序深度至少为1×,构建CNV阳性状态和CNV阴性状态t分布曲线,针对待确定CNV状态的logR值,分别计算其在上述t分布对应的t统计量,通过对比t统计量与理论t值,判定CNV状态。
进一步的,所述肿瘤纯度最低检出限计算模块包括:样品高深度测序单元,用于对肿瘤样本和正常样本进行全基因组高深度测序,测序深度为20-30×,其中正常样本中有与所述肿瘤样本相同数量的样本进行配对;测序数据预处理单元,用于对测序数据比对、排序、去重、去除低质量序列以及建立索引;肿瘤纯度估计单元,基于所述肿瘤样本和与之配对的相同数量的正常样本进行分析,估计肿瘤样本的肿瘤纯度;模拟样本构建单元,针对每例肿瘤样本和正常样本,按照预设纯度进行reads抽取和混样;覆盖度信息统计单元,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb,对bin区内reads数目进行矫正,并对矫正之后的reads数进行log
进一步的,模型方差计算单元中,
;
其中Var(logR
针对染色体水平,特定长度的segment,t分布的标准差为:
;
其中N代表特定segment的bin个数;
针对基因水平而言,t分布的标准差为:
。
进一步的,所述CNV判定模块包括:待测样本的低深度测序单元,用于对待测样品进行低深度全基因组测序,测序深度至少为1×;测序数据预处理单元,用于对测序数据比对、排序、去重、去除低质量序列以及建立索引;覆盖度信息统计单元,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb,对bin区内reads数目进行矫正,并对矫正之后的reads数进行log
本发明还提供了计算机可读介质,记载有可以运行上述拷贝数变异的检测方法的程序。
本发明实施例提供的技术方案带来的有益效果包括:
1. 本发明不需要大量正常样本构成的基线,避免由基线样本引入额外的误差,并节省了建立基线样本的成本;2. 本发明不受样本类型的限制,可以应用于组织和液态活检的CNV检测;3. 由于本发明基于低深度全基因组测序,检测标志物不受到引物设计的影响,更容易形成标准化检测。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例所提供的肿瘤纯度最低检出限推断的原理图;
图2为本发明实施例所提供的CNV判定原理示意图;
图3为本发明实施例1中对脑胶质瘤1p/19q共缺失的检测结果,向下箭头表示染色体发生拷贝数缺失,向右箭头表示染色体没有发生拷贝数变异;
图4为本发明实施例1中对脑胶质瘤7号染色体扩增和10号染色体缺失的检测结果,向上箭头表示染色体发生拷贝数扩增,向下箭头表示染色体发生拷贝数缺失,向右箭头表示染色体没有发生拷贝数变异;
图5为本发明实施例1中对脑胶质瘤EGFR基因高倍扩增的检测结果,三角形代表EGFR发生高倍扩增,正方形代表EGFR未发生高倍扩增;
图6为本发明实施例1中对脑胶质瘤CDKN2A/B纯合缺失的检测结果,三角形代表基因CDKN2A/B纯合缺失,正方形代表CDKN2A/B未发生纯合缺失;
图7为本发明实施例2中在肠癌组织和血液cfDNA的CNV检测结果,向上箭头表示染色体发生拷贝数扩增,向下箭头表示染色体发生拷贝数缺失,向右箭头表示染色体没有发生拷贝数变异;
图8为本发明实施例3中症状没有好转的动态监测样本的CNV检测结果, 向上箭头表示染色体发生拷贝数扩增,向下箭头表示染色体发生拷贝数缺失,向右箭头表示染色体没有发生拷贝数变异,三角形代表基因发生高倍扩增,正方形代表基因未发生高倍扩增,基因从左到右分别为:TERT、IL7R、EGFR、MYC;
图9为本发明实施例3中症状明显好转的动态监测样本的CNV检测结果,向上箭头表示染色体发生拷贝数扩增,向下箭头表示染色体发生拷贝数缺失,向右箭头表示染色体没有发生拷贝数变异,三角形代表基因发生高倍扩增,正方形代表基因未发生高倍扩增。
具体实施方式
下面通过具体实施方式对本发明作进一步详细说明。但本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
本文使用的词语“包括”、“包含”、“具有”或其任何其他变体意欲涵盖非排它性的包括。例如,包括列出要素的工艺、方法、物品或设备不必受限于那些要素,而是可以包括其他没有明确列出或属于这种工艺、方法、物品或设备固有的要素。除非上下文明确规定,否则单数形式“一个/种”和“所述(该)”包括复数个讨论对象。
本发明提供的检测方法仅仅是用于通过测序结果判定是否存在着拷贝数变异现象,并非是用于疾病的诊断和治疗目的。
本发明中“待测样本”是指需要进行检测,并判定该样本上的一个或者多个区域或基因是否存在有拷贝数变异,可以通过组织或液态活检方式获取。“肿瘤样本”是指通过活检,检测出具有肿瘤细胞的样本,“正常样本”是指血液样本中离心得到的白细胞。
如本文所用的,术语“比对”是指将测序序列与参考基因组序列进行比较并且由此确定该测序序列在参考基因组中具体位置的过程。如果该参考序列含有该测序序列,则可以将其映射至参考序列中的某个特定位置。作为结果,比对可得到测序序列对应的基因组位置,并可进而判别其是否属于某一特定区域。术语“参考基因组”或“参考序列”是指生物体或病毒的已知基因组序列(无论是部分的或完整的),它可以用于对来自受试者的测序序列进行参比。例如,用于人类受试者以及许多其他生物体的参考基因组可见于美国国家生物技术信息中心(ncbi.nlm.nih.gov),对于人类样品来说,参照序列可以是基因组hg18或hg19版本。目前hg19的相关数据库相对较多且hg19对应的碱基量比hg18要多,所以优先选择hg19。
术语“测序序列(read)”是指来自核酸样品经测序后所获得的短片段序列。典型地,测序序列代表样品中的相邻碱基对的短序列。测序序列通过碱基对序列(ATCG符号)代表。它可以存储在存储设备中,且经过适当处理后可确定其是否与参考基因组序列匹配。测序序列可以直接地从测序装置中或者间接地从涉及样品的存储序列信息中获得。一般而言,测序序列是有一定长度(例如,至少约30bp)的DNA序列,可以通过比对来确定其在染色体对应的具体位置。
每个目标区域上的测序深度信息是比对结果中位于区域内的测序片段数目;位点的测序深度信息是比对结果中包含该位点的测序片段数目。
本发明提供的拷贝数变异的检测方法,包括:
S1.对肿瘤样本和正常样本取样,其中正常样本中有与所述肿瘤样本相同数量的样本进行配对,进行全基因组高深度测序,测序深度为20-30×,计算出肿瘤纯度最低检出限,并基于所述肿瘤纯度最低检出限计算相应灵敏性和特异性所对应的t值。
1.样本高深度测序。为了得到具有足够的样本,肿瘤样本的取样例数不少于10例,正常样本的取样例数不小于20例。本实施例中,选取10例肿瘤组织样本和20例白细胞样本,进行全基因组测序,得到样本的测序数据。在20例白细胞样本中,其中10例白细胞样本是与肿瘤组织配对的,可用于估计对应样本肿瘤组织的纯度。
2.测序数据预处理。对所述10例肿瘤组织样本和20例白细胞样本的测序数据进行预处理,包括了比对、排序、去重、去除低质量序列、建立索引五个步骤。
比对:使用BWA-MAM (v.0.7.12-r1039)将待测样本的fastq文件比对至hg19参考基因组,比对参数为BWA-MAM默认参数,生成待测样本原始bam文件。
排序:使用samtools (v.1.2)软件sort命令对原始bam文件进行排序。
去重:使用Picard (v.1.124)软件MarkDuplicates命令对排序后的bam文件进行重复序列标记和过滤。
去除低质量序列:使用samtools去除MAPQ<20的低质量序列。
建立索引:使用samtools软件的index命令对去除低质量序列后的bam文件构建索引。
3.肿瘤纯度估计。通过所述肿瘤样本以及与之配对的正常样本计算出肿瘤纯度。使用Sclust (v.1.0)软件默认参数对10组肿瘤组织样本和与之配对的白细胞样本的bam文件进行分析,估计肿瘤组织的肿瘤纯度值T。
4.构建模拟样本。基于所述肿瘤纯度和正常样本按照预设纯度针对每一例肿瘤样本进行reads抽取和混样,并计算t分布曲线的标准差。具体的,使用samtools 软件的命令"samtools view –s 12.Y"和"samtools view –s 12.Z"分别对每一例肿瘤组织和20例白细胞样本的bam文件按照预设纯度T' ∈{0.025,0.05, 0.1,0.15,0.2,0.25,0.3,0.35,0.4,0.45,0.5,0.55,0.6}进行reads抽取。因此,得到每一个预设纯度下具有200个混样。
肿瘤细胞抽取的reads比例Y按照式1进行计算:
1
T'为上述预设纯度梯度对应的任意值;T为肿瘤纯度值;R
白细胞样本抽取reads比例Z按照式2进行计算:
2
式2中的R
使用samtools merge将抽取后的每一例肿瘤样本分别和抽取后的20例白细胞样本逐一混合。由此在某一特定预设纯度T'条件下10例肿瘤样本可以产生200个模拟样本,共计200*13个模拟样本。
以肿瘤纯度为0.6的样本(样本1)和正常白细胞样本(样本2)混合为预设纯度为0.025的样本(样本3)为例:假设样本1的reads总数为430000000(测序深度为20×),样本2的reads数目为645000000 (测序深度为30×)。首先,使用步骤4的抽样方法,抽取样本1的reads比例为:
;
即为0.21%,抽取样本2的reads数目的比例为:
;
即为3.19%。使用samtools软件的"samtools view -s 12.0021 样本1.bam"和"samtools view -s 12.0319 样本2.bam",分别对肿瘤样本和白细胞样本进行抽取,使用步骤4的混合方法将抽取的样本1和样本2的reads混合,得到预设纯度为0.025,reads数目为21500000的样本3。
5.覆盖度信息统计。混样后,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb;采用CBS算法对CNV变异的断点位置对染色体进行分割,将具有相同拷贝数的连续bin区划分为同一个segment;针对基因水平的拷贝数变异分析采用bin区覆盖度表征值来构建t分布曲线;针对染色体水平的拷贝数变异采用segment覆盖度表征值来构建t分布曲线。
具体的,使用HMMcopy (v0.99.0)软件对样本的bam文件进行计数,间隔窗口为50-500kb均可,本实施例优选中间隔窗口选择190kb,每个窗口称为一个bin区。
使用R语言的loess函数的默认参数分别对全基因组bin区内reads数目进行GC含量和比对率两步矫正,并对矫正后的reads数进行log2转化,记为logR。
3
其中,A为矫正后的reads数。
利用DNAcopy (v1.64.0)软件包的CBS算法的默认参数,按照发生CNV变异的断点位置对染色体进行分割,将具有相同拷贝数的连续bin区划分成同一区间,记为segment。
后续计算过程中,所有基因层面的拷贝数变异分析均根据bin区来计算,染色体层面的拷贝数变异均根据segment来计算。
6.模型方差计算:t分布曲线的标准差的计算方法为:用待计算样本所有segment的logR方差中位数代表样本全基因组范围的噪声(每个样本独立计算一个噪音)。
4
其中Var(logR
t分布的标准差估计:对于染色体水平而言,特定长度的segment,采用抽样分布原理对其标准差进行估计,计算公式如式5:
5
其中M代表样本全基因组范围的噪声,N代表特定segment的bin个数。例如:对某个样本一号染色体长臂(1p)的标准差计算,N代表特定长度染色体的bin数目。例如:1p染色体长度为125000000 bp,以190kb的bin区划分,1p的bin数目为658个。
对于基因水平而言,bin长度设定为1。因此,其分布的标准差即是该样本全基因组范围的噪声的平方根,即为:
7.肿瘤纯度最低检出限的计算,肿瘤纯度最低检出限被定义为在1×测序深度条件下时仍稳定检出CNV的肿瘤纯度最低值。本发明使用每个肿瘤纯度条件下的200个模拟样本进行肿瘤纯度最低检出限的计算。
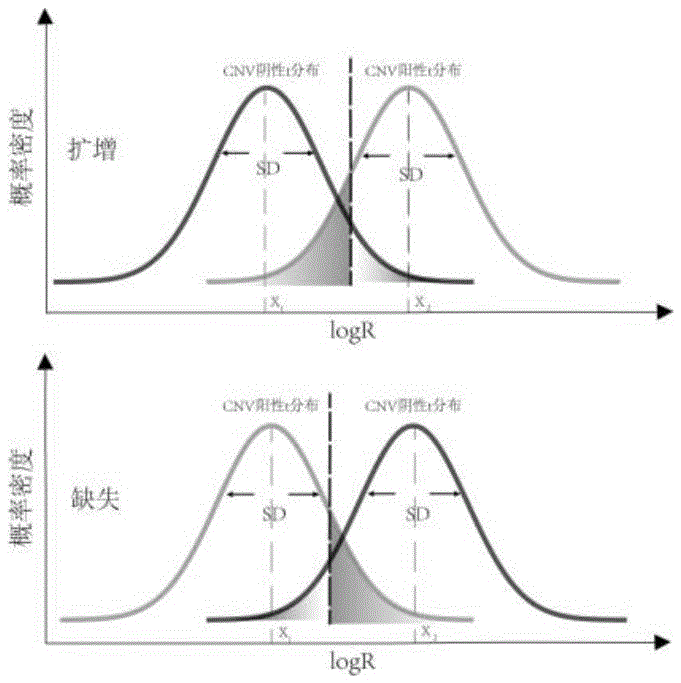
(1) 构建肿瘤纯度最低检出限的计算模型:如图1所示,本发明是基于两个t分布来进行CNV检出的,分别被定义为CNV阳性状态和CNV阴性状态分布,分别对应特定CNV事件发生的有无。其中长度为N bin的segment 其t分布自由度设置为N-1,基因水平的自由度设置为2.1。当模型达到一定的特异性和灵敏性时,CNV阳性分布和CNV阴性分布会处于如图1所示的临界状态。临界状态下的特异性和灵敏性通过后续步骤进行计算。
(2) 模型阴性分布和阳性分布拷贝数的确定:阴性状态分布拷贝数是指没有发生指定CNV事件的拷贝数,阳性状态分布拷贝数是指发生指定CNV事件的拷贝数。本发明旨在判定染色体的扩增/缺失,基因的高倍扩增/纯合缺失。因此,需预先确定每个标志物CNV阴性分布拷贝数(C
(3) t分布均值计算:如图1所示,阳性状态分布均值(X
6
7
公式中C
(4) 计算灵敏性:首先,在特异性∈ {0.900, 0.905,0.910, ..., 0.99}条件下,使用R语言的“qt”函数计算特异性对应的特异性t值t
8
公式中的X
通过上述方法计算每个CNV标志物的特定预设纯度和特异性条件下200样本的灵敏性,并将200个样本的灵敏性取均值,此时,每个预设纯度条件下,均存在19对灵敏性和特异性的组合。
(5) 计算约登指数:通过计算每个预设纯度条件下19对灵敏性和特异性组合的约登指数,确定每个预设纯度条件下最优特异性和灵敏性组合。每个预设纯度条件下的约登指数如式9所示:
YI=max (Spec+Sens-1) 9
YI代表约登指数,Spec代表特异性,Sens代表灵敏性;
染色体水平扩增和缺失的约登指数计算结果如表1所示,基因高倍扩增和纯合缺失的约登指数计算结果如表2所示。
表1. 染色体水平的约登指数统计
表2基因水平的约登指数统计
/>
(6) 肿瘤纯度最低检出限的确定:对于染色体水平而言,确定约登指数等于98%的预设纯度作为该染色体的肿瘤纯度最低检出限,通过计算所有染色体均可稳定检出的预设纯度的最小值作为染色体CNV的肿瘤纯度最低检出限。对于基因水平而言,采用约登指数增长的饱和点确定肿瘤纯度最低检出限,规定预设纯度上升5%,约登指数上升首次小于2%的点作为约登指数增长的饱和点。所有CNV的肿瘤纯度最低检出限如表3所示。
表3. 不同类型CNV的肿瘤纯度最低检出限
S2.对待测样本进行低深度测序,测序深度至少为1×,构建CNV阳性状态和CNV阴性状态t分布曲线,针对待确定CNV状态的logR值,分别计算其在上述t分布对应的t统计量,通过对比t统计量与理论t值,判定CNV状态。
计算待测样本CNV阳性状态分布和CNV阴性状态分布的t统计量,
当
当
其中,t
具体的,包括:
1. 待测样本的低深度测序。对待检验的样本进行低深度全基因组测序,测序深度至少为1×。
2. 测序数据预处理:用于对测序数据比对、排序、去重、去除低质量序列以及建立索引,与S1中的预处理相同。
3. 覆盖度信息统计:与S1中的计算方式相同。
4. 构建待检测CNV标志物的假设检验模型:如图2所示:在进行扩增或者缺失判断时,分别构建两组t分布,即CNV阳性状态分布和CNV阴性状态t分布。两个分布平均数和标准差计算方法如下:
(1) 计算CNV标志物的CNV阴性状态分布和CNV阳性状态分布的logR平均值(X
(2) 待检测样本的标准差计算的计算方法与S1中的步骤6一致,这里采用待检测样本实际segment划分来进行计算;
(3) 构建待检测CNV标志物的假设检验模型:通过计算(1) (2)两步的参数,构建如图2所示的假设检验模型。
5. t检验判断待检测CNV状态:根据待检测CNV标志物构建的假设检验模型,通过两次t检验,判定某个segment/bin是否发生了拷贝数变异。
(1).计算t统计量:分别计算CNV阳性状态分布和CNV阴性状态分布两个分布的t统计量,计算公式如式10和式11所示:
10
11
其中R代表待检测segment/bin对应的logR值,X
(2). 判断CNV状态:CNV判断通过两步t检验完成,当判定扩增CNV时,要求:
并且/>
当判定缺失CNV时,要求:
并且/>
t
6. 输出样本的CNV状态:通过上述计算,确定样本所有待检测CNV的状态。
本发明还保护了一种拷贝数变异的检测装置,包括:肿瘤纯度最低检出限计算模块,用于对肿瘤样本和正常样本进行全基因组高深度测序,测序深度为20-30×,计算出CNV稳定检出所对应的肿瘤纯度最低检出限,并基于所述肿瘤纯度最低检出限计算相应灵敏性和特异性所对应的理论t值;CNV判定模块,对待测样本进行低深度测序,测序深度至少为1×,构建CNV阳性状态和CNV阴性状态t分布曲线,针对待确定CNV状态的logR值,分别计算其在上述t分布对应的t统计量,通过对比t统计量与理论t值,判定CNV状态。
所述肿瘤纯度最低检出限计算模块包括:
样品高深度测序单元,用于对肿瘤样本和正常样本进行全基因组高深度测序,测序深度为20-30×,其中正常样本中有与所述肿瘤样本相同数量的样本进行配对;
测序数据预处理单元,用于对测序数据比对、排序、去重、去除低质量序列以及建立索引;
肿瘤纯度估计单元,基于所述肿瘤样本和与之配对的相同数量的正常样本进行分析,估计肿瘤样本的肿瘤纯度;
模拟样本构建单元,针对每例肿瘤样本和正常样本,按照预设纯度进行reads抽取和混样;
覆盖度信息统计单元,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb,对bin区内reads数目进行矫正,并对矫正之后的reads数进行log
模型方差计算单元,计算全基因组范围内的噪声,基于所述噪声计算染色体水平及基因水平的t分布的标准差;
肿瘤纯度最低检出限计算单元,基于拷贝数构建CNV阳性状态和CNV阴性状态t分布曲线,并计算在一定特异性范围下所对应的特异性t值,并基于所述特异性t值计算灵敏性,将同一预设纯度的混合样本的灵敏性取均值,并与所述特异性范围进行配对组合,通过约登指数将不同预设纯度的特异性与灵敏性进行计算,得到满足预设条件下的肿瘤纯度最低检出限。
模型方差计算单元中,
;
其中Var(logR
针对染色体水平,特定长度的segment,t分布的标准差为:
;
其中N代表特定segment的bin个数;
针对基因水平而言,t分布的标准差为:
。
所述CNV判定模块包括:
待测样本的低深度测序单元,用于对待测样品进行低深度全基因组测序,测序深度至少为1×;
测序数据预处理单元,用于对测序数据比对、排序、去重、去除低质量序列以及建立索引;
覆盖度信息统计单元,计算固定间隔窗口内的reads数,所述固定间隔窗口称为一个bin区,间隔窗口的长度为50-500kb,对bin区内reads数目进行矫正,并对矫正之后的reads数进行log
构建待检测CNV标志物的假设检验单元,基于拷贝数计算CNV阳性状态和CNV阴性状态t分布曲线的logR平均值,计算全基因组范围内的噪声,基于所述噪声计算染色体水平及基因水平的t分布的标准差,构建检测CNV标志物的假设验证模型;
t检测判断待检测CNV状态单元,用于计算CNV阳性状态和CNV阴性状态t分布曲线的t统计量,基于所述肿瘤纯度最低检出限计算相应灵敏性和特异性所对应的t值;通过对比所述t统计量与所述t值判定待检测样本CNV状态;
输出样本待检测CNV的状态单元,确定样本所有待检测CNV状态,并输出。
本发明还保护了一种计算机可读介质,记载有可以运行上述方法的拷贝数变异的检测方法的程序。
显然,本领域的技术人员应该明白,上述的本发明的各模块或各步骤可以用通用的计算装置来实现,它们可以集中在单个的计算装置上,或者分布在多个计算装置所组成的网络上,可选地,它们可以用计算装置可执行的程序代码来实现,从而,可以将它们存储在存储装置中由计算装置来执行,或者将它们分别制作成各个集成电路模块,或者将它们中的多个模块或步骤制作成单个集成电路模块来实现。这样,本发明不限制于任何特定的硬件和软件结合。
实施例1
胶质瘤组织样本的CNV判定。选取1p/19q共缺失、7号染色体扩增和10号染色体缺失(+7/-10)、EGFR基因高倍扩增、CDKN2A/B纯合缺失的阴性样本和阳性样本各1例,并对这些样本进行FISH检测,FISH检测严格按照试剂盒说明书执行,最终确定样本的CNV状态如表4所示。
表4. 脑胶质瘤FISH检测的样本信息表。
对8例脑胶质瘤患者组织样本进行低深度测序,并参考实施例1中CNV判定模块的步骤,对脑胶质瘤用于分级分型的CNV标志物进行检测,其中EGFR高倍扩增C
实施例2
肠癌组织和cfDNA样本的CNV判定。选取一例结直肠癌组织和血液cfDNA配对样本进行低深度测序。使用本发明所提供的拷贝数变异的检测方法对组织和血液cfDNA中已被报道的结直肠癌高频出现的CNV进行判定,包括扩增:7p、7q、8q、13q、20p、20q;缺失:8p、17p、18p、18q。
如图7所示,上述高频出现的CNV在组织(病例9-组织)和血液cfDNA(病例9-血液)中均发生,且组织和cfDNA判定的CNV状态完全一致,说明本申请方法可以应用于血液cfDNA的CNV判定。
实施例3
肺腺癌脑转移cfDNA样本的CNV判定。取两例肺腺癌脑转移病人动态监测的脑脊液样本进行低深度测序,其中病例10为两次随访病情稳定且无进展的样本,其两次随访样本脑脊液异型细胞占比差距较小,分别为:7%和12%。病例11为第二次随访症状好转的样本,两次随访脑脊液异型细胞占比差距较大,分别为:37%和0%。
选取在肺腺癌中高频出现的CNV区域进行判断,包括扩增:1q、5p、7p、8q;缺失:6q、9p、13q、15q;基因高倍扩增:TERT、IL7R、EGFR、MYC。其中TERT、IL7R、EGFR、MYC基因高倍扩增使用参数C
使用本发明所提供的拷贝数变异的检测方法对两次随访病情稳定且无进展样本的低深度数据进行CNV判定,结果如图8所示:两次随访中的所有高频CNV表现一致。
本发明所提供的拷贝数变异的检测方法对两次随访症状有好转样本的低深度数据进行CNV判定,结果如图9所示:随访1表现出多个CNV,包括:1q、5p、7p、8p的扩增,6q、15q的缺失和TERT、EGFR的高倍扩增,而对于随访2而言,本申请方法并未鉴定出CNV事件发生。
综合以上四个实施例,本申请方法可以应用于脑胶质瘤、肠癌、肺腺癌脑转移等多个癌症类型包括组织和cfDNA在内的CNV检测。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 一种无CO2非金属矿物粉体材料的连续烧结装备系统
- 一种可回收再生碳网结合粉体非金属矿物多孔净水材料制备方法