一种酚苷类化合物及其制备方法和用途
文献发布时间:2023-06-19 09:23:00
技术领域
本发明属于植物天然化合物领域,具体涉及一种酚苷类化合物及其制备方法和用途。
背景技术
现医疗卫生领域由于抗生素的广泛使用和滥用,导致大肠杆菌对当前使用的主流抗生素产生了不同程度的耐药性,给临床治疗带来了巨大困难,如要从根本上解决由大肠杆菌引发的痢疾和炎症等肠道疾病,需要拓宽思路寻找新的治疗方法,以便克服日益增加的病原菌耐药性难题。大量的研究结果显示,植物来源的天然化合物存在大量的抗菌化合物,同时由于天然化合物高效低毒的特点受到药物开发科研工作者的广泛关注,因此天然化合物在开发抑/抗菌药物方面有巨大潜力。
毛叶山桐子(Idesia polycarpa Maxim.var.vestita Diels)是大风子科山桐子属落叶乔木,该属在全球仅有1种,在我国分布于秦岭淮河以南及台湾地区,国外主要分布在日本、朝鲜半岛及俄罗斯远东地区。研究发现毛叶山桐子果实富含具有广谱生物活性的酚苷类成分,该类成分具有抗炎、抗菌的活血,在化妆品和医药等领域具有潜在的开发和应用价值。然而,由于山桐子果实成分复杂,且含有大量的天然同分异构体化合物,因此,对其中天然异构体成分的分离存在一定难度,也致使针对其特定成分的作用研究受到了限制。
同分异构体是重要的有机化工原料,又是多种农药、医药中间体的起始原料。因同分异构体在化学结构上的相似性,也使得大多数同分异构体的理化性质相近,这一特性导致用常规的分离方法难以对天然植物中的同分异构体达到理想的分离效果。对植物中同分异构体含量的测定及分离鉴定,过去一般沿用填充柱色谱法,但是填充柱柱效较低,所以分离能力受到限制,很多杂质得不到有效分离,且分析时间可能较长。此外,大部分同分异构体易受光照和温度影响而发生结构变化,传统的分离方法无法达到同分异构体在分离过程中所需的严格分离条件,从而导致其活性发生改变,也造成无法对植物中新结构且具有极大潜在开发价值的同分异构体进行研究。因此,开发新的分离方法对植物中同分异构体的分离鉴定及其在天然医药开发利用具有重要的意义。
发明内容
为解决上述问题,本发明提供了一种酚苷类化合物,它是下式I所示结构化合物,或其立体异构体及其混合物形式;
进一步地,它是下式II或式III结构化合物或其混合物形式;
本发明还提供了一种前述酚苷类化合物的制备方法,它是从毛叶山桐子果实中提取,具体包括如下步骤:
1)取毛叶山桐子果实,干燥粉碎过筛,加正己烷脱脂,得脱脂果渣,
2)取步骤1)脱脂果渣,加乙醇提取,提取液过滤,浓缩,得浸膏;
3)取步骤2)浸膏,加水溶解,再加乙酸乙酯萃取,上层萃取液除去溶剂得粗提物;
4)取步骤3)粗提物,加高速逆流色谱流动相和固定相组成的混合溶液溶解,溶解液用高速逆流色谱分离,收集52-70min时间段流分,除去溶剂得酚苷类混合物;
5)取酚苷类混合物,加甲醇溶解,溶解液用制备高效液相色谱分离,收集15.5-32.9min时间段流分,得酚苷类化合物;
所述高速逆流色谱条件为:
以溶剂体系震荡分层后的上层液体为固定相,下层液体为流动相,转速850rpm,流速4~5mL/min,温度28~30℃;所述溶剂体系是由体积比2:5:2:5的正己烷-乙酸乙酯-甲醇-水组成;
所述制备高效液相色谱条件为:
固定相:C18键合硅胶柱;流动相:体积比68~71:29~32的水-甲醇;流速4~5ml/min;柱温40℃。
进一步地,步骤1)所述毛叶山桐子果实与正己烷的质量体积比为1g:20~40ml,优选1g:20ml。
进一步地,步骤1)所述粉碎过60目筛。
进一步地,步骤2)所述果渣与乙醇的质量体积比为50~200g:4000ml,优选100g:4000ml。
更进一步地,所述乙醇为50~75%乙醇,优选60%乙醇。
进一步地,步骤2)所述提取为先浸渍提取再超声提取,浸渍提取温度50~80℃,时间30min~8h,超声提取温度50℃,时间20min,所述提取次数为4~8次,优选5次。
进一步地,步骤3)所述浸膏与水的质量体积比为20g:200ml;所述加乙酸乙酯萃取10~15次,每次乙酸乙酯与水等体积,合并上层萃取液。
进一步地,步骤4)所述高速逆流色谱流动相和固定相组成的混合溶液中流动相和固定相的体积比为2:1;所述粗提物与高速逆流色谱流动相和固定相组成的混合溶液的质量体积比为15~20mg:20ml,优选20mg:20ml。
进一步地,步骤5)所述酚苷类混合物与甲醇的质量体积比为1g:10ml;所述高速逆流色谱条件中波长312nm;所述制备高效液相色谱条件中波长为312nm。
进一步地,所述甲醇为60~80%甲醇,优选80%甲醇。
进一步地,所述高速逆流色谱条件中流速5mL/min,温度30℃,收集52-59min时间段流分,除去溶剂得酚苷类混合物;所述制备高效液相色谱条件中流动相为体积比70:30的水-甲醇,流速5ml/min,收集18.7-21.5min时间段流分,得式II化合物,收集28.6-31min时间段流分,得式III化合物。
进一步地,所述高速逆流色谱条件中流速4mL/min,温度30℃,收集57-61min时间段流分,除去溶剂得酚苷类混合物;所述制备高效液相色谱条件中流动相为体积比71:29的水-甲醇,流速4ml/min,收集19.7-23.5min时间段流分,得式II化合物,收集29.6-32.9min时间段流分,得式III化合物;
进一步地,所述高速逆流色谱条件中流速5mL/min,温度28℃,收集62-70min时间段流分,除去溶剂得酚苷类混合物;所述制备高效液相色谱条件中流动相为体积比68:32的水-甲醇,流速4ml/min,收集15.5-17.2min时间段流分,得式II化合物,收集21.8-23.1min时间段流分,得式III化合物。
本发明最后提供了一种前述化合物在制备具有抗菌作用的药物中的用途。
本发明酚苷类化合物是从毛叶山桐子果实中分离得到的两种新型单体化合物,其丰富了天然抗菌化合物库,经试验证明,本发明酚苷类化合物在低浓度下对大肠杆菌的增值产生了显著的抑制效果,这也为山桐子天然化合物在抗菌药物开发方面奠定了基础。本发明从毛叶山桐子果实提取分离酚苷类化合物的方法,克服了传统分离方法无法实现同分异构体的有效分离,以及容易导致化合物活性发生改变的技术难题,具备工业生产推广应用价值。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
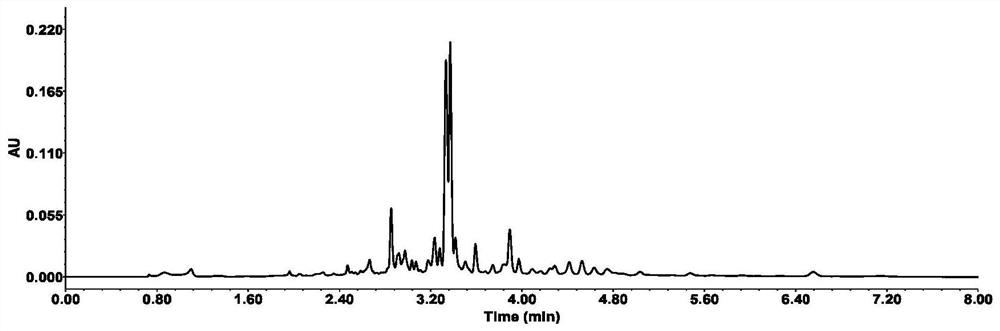
图1毛叶山桐子果实乙酸乙酯相成分分析UPLC色谱图,纵坐标为AU(吸光值),横坐标为min(保留时间);
图2毛叶山桐子果实乙酸乙酯相高速逆流色谱分离色谱谱图,纵坐标为紫外吸收AU,横坐标为min(保留时间);
图3.高速逆流色谱流分UPLC图;
图4.制备色谱分离图;
图5.制备色流分分析UPLC色谱图;
图6.单体化合物质谱图;
图7单体化合物化学结构。
图8不同浓度的Idepolycoside A和Idepolycoside B(10,20,40μM)对大肠杆菌的抑制效果。
具体实施方式
实施例1本发明酚苷类化合物的制备
1.毛叶山桐子果实500g,干燥粉碎,过60目筛,加10L正己烷脱脂,得脱脂果渣;
2.称取100g脱脂果渣,加入60%乙醇溶剂4000ml,50℃浸渍30min后,超声(180w、50℃、59kHz)提取20min,过滤,重复以上步骤5次,弃去滤渣,合并滤液,用旋转蒸发仪浓缩滤液至粘稠浸膏状,得提取物浸膏41.5g;
3.称取提取物浸膏20g,用200ml蒸馏水溶解,然后等体积加入乙酸乙酯萃取10次,合并乙酸乙酯萃取相,减压蒸发脱去乙酸乙酯溶剂,得乙酸乙酯相提取物12.8g;
4.选取高速逆流色谱溶剂系统:正己烷—乙酸乙酯—甲醇—水(v/v/v/v2:5:2:5);高速逆流色谱分离条件:转速为850rpm,流速为5mL/min,波长为312nm,温度30℃;
5.步骤3中所述乙酸乙酯相提取物10g加入到高速逆流色谱分离体系上下相混合溶液中(上下相溶剂体积比为2:1),质量和体积比为20mg:20ml,超声混匀待用,将溶解好的乙酸乙酯提取物用高速逆流色谱进行分离,收集在52-59min处出现的色谱峰,除去溶剂后即为同分酚苷同分异构体的混合物,共得到两个酚苷异构体的混合物。经UPLC检测,两个异构在此流分中的含量达到84%,故需要进一步纯化分离。
6.取步骤5所得混合物,溶于10倍量(v/w,ml/g)的80%甲醇溶剂中,使用制备高效液相色谱进行精制。
色谱柱填料:C18键合硅胶柱(250mm*30mm)
流动相A相为:水
流动相B相为:甲醇
梯度程序为:0~30min,A:70%,B:30%
流速:5ml/min
柱温:40℃
检测波长:312nm
7.根据计算机实时采集的色谱峰图收集在18.7-21.5min和28.6-31min出现的两段流分,即分别为Idepolycoside A和Idepolycoside B,
8.取步骤7两段流份,按照步骤6所述制备高效液相色谱条件再次精制,减压浓缩脱去溶剂得Idepolycoside A和Idepolycoside B。
实施例2本发明酚苷类化合物的制备
1.毛叶山桐子果实500g,干燥粉碎,60目筛,加10L正己烷脱脂,得脱脂果渣;
2.称取100g脱脂果渣,加入60%乙醇溶剂4000ml,80℃浸渍5h后,超声(180w、50℃、59kHz)提取20min,过滤,重复以上步骤5次,弃去滤渣,合并滤液,用旋转蒸发仪浓缩滤液至粘稠浸膏状,得提取物浸膏49.25;
3.称取提物浸膏20g,用200ml蒸馏水溶解,然后等体积加入乙酸乙酯萃取12次,合并乙酸乙酯萃取相,减压蒸发脱去乙酸乙酯溶剂,得粗制品13.1g;
4.选取高速逆流色谱溶剂系统:正己烷—乙酸乙酯—甲醇—水(v/v/v/v2:5:2:5);高速逆流色谱分离条件:转速为850rpm,流速为4mL/min,波长为312nm,温度30℃
5.步骤3中所述乙酸乙酯相提取物10g加入到高速逆流色谱分离体系上下相混合溶液中(上下相溶剂体积比为2:1),质量和体积比为15mg:20ml,超声混匀待用。将溶解好的乙酸乙酯提取物用高速逆流色谱进行分离,收集在57-61min处出现的色谱峰,除去溶剂后即为同分酚苷同分异构体的混合物,共得到4.2g含有两个酚苷异构体的混合物。经UPLC检测,两个异构在此流分中的含量达到84%,故需要进一步纯化分离;
6.将步骤5中所述的混合物4g溶于40ml的80%甲醇溶剂中,使用制备高效液相色谱进行精制;
色谱柱填料:C18键合硅胶柱(250mm*30mm)
流动相A相为:水
流动相B相为:甲醇
梯度程序为:0~30min,A:71%,B:29%
流速:4ml/min
柱温:40℃
检测波长:312nm
7.根据计算机实时采集的色谱峰图收集在19.7-23.5min和29.6-32.9min出现的两段流分,即分别为Idepolycoside A和Idepolycoside B,
8.取步骤7两段流份,按照步骤6所述制备高效液相色谱条件再次精制,减压浓缩脱去溶剂得Idepolycoside A和Idepolycoside B,其分别为1.57g和0.8g(纯度经UPLC分析纯度分别为98.3%和98.1%)。
实施例3本发明酚苷类化合物的制备
1.毛叶山桐子果实500g,干燥粉碎,60目筛,加20L正己烷脱脂,得脱脂果渣。
2.称取100g脱脂果渣,加入60%乙醇溶剂4000ml,80℃浸渍8h后,超声(180w、50℃、59kHz)提取20min,过滤,重复以上步骤5次,弃去滤渣,合并滤液,用旋转蒸发仪浓缩滤液至粘稠浸膏状,得提取物浸膏51.7g。
3.提取物浸膏20g用200ml蒸馏水溶解,然后等体积加入乙酸乙酯萃取15次,合并乙酸乙酯萃取相,减压蒸发脱去乙酸乙酯溶剂,得粗制品13.5g。
4.选取高速逆流色谱溶剂系统:正己烷—乙酸乙酯—甲醇—水(v/v/v/v2:5:2:5);高速逆流色谱分离条件:转速为850rpm,流速为5mL/min,波长为312nm,温度28℃
5.步骤3中所述乙酸乙酯相提取物10g加入到高速逆流色谱分离体系上下相混合溶液中(上下相溶剂体积比为2:1),质量和体积比为15mg:20ml,超声混匀待用,将溶解好的乙酸乙酯提取物用高速逆流色谱进行分离,收集在62-70min处出现的色谱峰,除去溶剂后即为同分酚苷同分异构体的混合物,共得到3.4g含有两个酚苷异构体的混合物。经UPLC检测,两个异构在此流分中的含量达到80%,故需要进一步纯化分离。
6.将步骤5中所述的混合物3.4g溶于34ml的80%甲醇溶剂中,使用制备高效液相色谱进行精制。
色谱柱填料:C18键合硅胶柱(250mm*30mm)
流动相A相为:水
流动相B相为:甲醇
梯度程序为:0~30min,A:68%,B:32%
流速:4ml/min
柱温:40℃
检测波长:312nm
7.根据计算机实时采集的色谱峰图收集在15.5-17.2min和21.8-23.1min出现的两段流分,即分别为Idepolycoside A和Idepolycoside B。
8.取步骤7两段流份,按照步骤6所述制备高效液相色谱条件再次精制,减压浓缩脱去溶剂得Idepolycoside A和Idepolycoside B,分别为1.2g和0.5g(纯度经UPLC分析纯度分别为98.8%和97.3%。)。
以下通过试验例来说明本发明的有益效果。
试验例1
1.毛叶山桐子果实500g,干燥粉碎,60目筛,加10L正己烷脱脂,得脱脂果渣。
2.称取100g脱脂果渣,加入60%乙醇溶剂4000ml,50℃浸渍30min后,超声(180w、50℃、59kHz)提取20min,过滤,重复以上步骤5次,弃去滤渣,合并滤液,用旋转蒸发仪浓缩滤液至粘稠浸膏状,得提取物浸膏。
3.提取物浸膏20g用200ml蒸馏水溶解,然后等体积加入乙酸乙酯萃取10次,合并乙酸乙酯萃取相,减压蒸发脱去乙酸乙酯溶剂,得粗制品12.8g。液相检测结果如图1所示。
4.选取高速逆流色谱溶剂系统:正己烷—乙酸乙酯—甲醇—水
a)高速逆流色谱溶剂体系的获得:
分配系数K在0.5到2之间时则可使样品实现较好的分离。分配系数测定方法:配备一定比例的溶剂体系(见下表1),待分层后,得到上下相溶剂备用。准确称取步骤3中的乙酸乙酯相浸膏状浓缩物2g。先用5mL下相溶解,然后加入5mL上相,充分震荡,静置10min分层后,分别取0.5mL上下相,用氮吹仪除去溶剂,再向各样品中分别加入1mL色谱甲醇溶解,离心后过膜,最后用UPLC测定上下相目标峰面积A
按照K值测定方法,得出目标物质分离的最佳HSCCC溶剂体系为正己烷—乙酸乙酯—甲醇—水(v/v/v/v2:5:2:5),结果如表1所示。由表可知,两个酚苷异构体的极性较小,而当正己烷—乙酸乙酯—甲醇—水组成的溶剂系统的上相溶剂极性小于下相极性时,两个化合物的K值均大于2,如溶剂系统正己烷—乙酸乙酯—甲醇—水为3:2:1:5,4:1:1:5,1.5:5:1.5:5和3:5:3:5v/v/v/v。而当溶剂系统正己烷—乙酸乙酯—甲醇—水的组成比例为2:5:2:5v/v/v/v时,两个酚苷异构体的K值均达到最优值,且以此为溶剂系统上相为固定相,下相为流动相时,固定相的保留率约为70%。因此,选择正己烷—乙酸乙酯—甲醇—水(v/v/v/v2:5:2:5)为两个酚苷异构体最佳的分离体系。
表1果渣中四种化合物在不同两相溶剂体系中的K值
b)高速逆流色谱分离条件:转速为850rpm,流速为5mL/min,波长为312nm,温度30℃。乙酸乙酯相高速逆流色谱分离结果,如图2所示。
5.步骤3中所述乙酸乙酯相提取物10g加入到高速逆流色谱分离体系上下相混合溶液中(上下相溶剂体积比为2:1),质量和体积比为20mg:20ml,超声混匀待用。将溶解好的乙酸乙酯提取物用高速逆流色谱进行分离,收集在52-59min处出现的色谱峰,即为同分酚苷同分异构体的混合物。经UPLC检测,两个异构在此流分中的含量达到86%(图3),故需要进一步纯化分离。
6.将步骤5中所述的混合物4g溶于40ml的80%甲醇溶剂中,使用制备高效液相色谱进行精制。
色谱柱填料:C18键合硅胶柱(250mm*30mm)
流动相A相为:水
流动相B相为:甲醇
梯度程序为:0~30min,A:70%,B:30%
流速:5ml/min
柱温:40℃
检测波长:312nm
7.根据计算机实时采集的色谱峰图收集在18.7-21.5min和28.6-31min出现的两段流分(图4),即分别为Idepolycoside A和Idepolycoside B。
8.取步骤7两段流份,按照步骤6所述制备高效液相色谱条件再次精制,减压浓缩脱去溶剂得Idepolycoside A和Idepolycoside B分别为1.56g和0.9g(纯度经UPLC分析纯度分别为98.6%和97.9%,结果如图5所示);
UPLC分析方法:色谱柱为HSS T3(1.8μm,2.1×100mm),柱温箱温度设置为40℃,进样体积为1μL,检测波长设置为275nm和312nm。流动相A为乙腈,B为缓冲盐(含0.1%甲酸和25mM甲酸铵)水溶液,流动相设为梯度,流速为0.5mL/min。流动相梯度设置为:
酚苷类化合物Idepolycoside A和Idepolycoside B质谱图和化学结构见图6和图7,其核磁共振数据如下:
化合物Idepolycoside A:白色粉末,UVλmax(乙腈)=195,223,313nm;ESI-MS m/z:563.17[M-H]
化合物Idepolycoside B:白色粉末,UVλmax(乙腈)=195,223,312nm;ESI-MS m/z:563.19[M-H]
9.称取上述已分离的Idepolycoside A和Idepolycoside B单体化合物,分别配置成3个浓度梯度的工作液(10,20,40μM)。用孔径为4mm打孔器在LB固体培养基上均匀打3个孔,然后向孔中分别加入10μL上述已配置好的不同浓度梯度的Idepolycoside A和Idepolycoside B单体化合物。随后将上述培养板至于37℃培养箱中培养24h后观察抑菌圈的变化。从图8可看出,不同浓度化合物均对大肠杆菌的产生了抑制效果。
综上,本发明从毛叶山桐子果实中分离得到的两种新型酚苷类化合物,丰富了天然抗菌化合物库,经试验证明,本发明酚苷类化合物在低浓度下对大肠杆菌的增值产生了显著的抑制效果,为山桐子天然化合物在抗菌药物开发方面奠定了基础。本发明从毛叶山桐子果实提取分离酚苷类化合物的方法,克服了传统分离方法无法实现同分异构体的有效分离,以及容易导致化合物活性发生改变的技术难题,具备工业生产推广应用价值。
- 一种酚苷类化合物及其制备方法和用途
- 酚苷类化合物及其在制备抗补体药物中的用途