用于测序读值的分组和折叠的系统和方法
文献发布时间:2023-06-19 10:18:07
相关申请的引用
本申请要求2018年10月31日提交的美国临时申请62/753,786号的优先权,将其内容通过援引方式整体并入本申请。
版权告知
本专利文件的公开内容的一部分包含受版权保护的材料。虽然版权所有人不反对任何人复制专利文件或专利公开内容,因为它出版在专利和商标局专利文件或记录中,但是版权所有人仍保留所有版权。
背景技术
技术领域
本申请大体涉及处理核苷酸测序数据的领域,更具体地涉及利用局部敏感哈希(locality sensitive hashing)来折叠(collapse)核苷酸测序数据。
读值折叠是一种计算方法,其将从测序系统输出的核苷酸测序读值鉴定为源自相同来源的脱氧核糖核酸(DNA)分子。测序系统可以是二代测序(NGS)系统,例如来自Illumina公司(San Diego,Ca)的NextSseq仪器。读值折叠可包括使用统计方法来减少在这些读值集合中发现的假错误。读值折叠产生的计算机错误减少可用于二代测序(NGS)中的应用,例如检测具有超低等位基因分数的变体,并且能够为临床应用带来提高的变体判断特异性。
发明概述
本申请公开了用于折叠测序读值和鉴定多个不同测序读值中的相似核苷酸序列的系统和方法。在一个实施方案中,系统包括非暂时性存储器,非暂时性存储器被配置成存储可执行指令和将核苷酸测序读值存储在多个箱中的第一哈希数据结构。该系统还可以包括由可执行指令编程以执行包括以下步骤的方法的硬件处理器:接收多个核苷酸测序读值,例如,配对末端测序读值的核苷酸测序读值1;对于每个核苷酸测序读值:从核苷酸测序读值的第一标识符序列生成多个第一标识符子序列;通过对所述多个第一标识符子序列应用哈希处理来生成所述核苷酸测序读值的第一名签;基于所述第一名签,将所述核苷酸测序读值分配到所述第一哈希数据结构的至少一个第一具体箱;以及在一个或多个核苷酸测序读值被分配的情况下,确定所述第一哈希数据结构的每个第一具体箱的核苷酸序列。
本申请的另一个实施方案是一种计算机实施的方法,其包括接收多个核苷酸测序读值,例如核苷酸测序读值;对于每个核苷酸测序读值:从所述核苷酸测序读值的第一标识符序列生成多个第一标识符子序列;通过对所述多个第一标识符子序列应用哈希处理来生成所述核苷酸测序读值的第一名签;基于所述第一名签,将所述核苷酸测序读值分配到第一数据结构的第一具体箱;以及在一个或多个核苷酸测序读值被分配的情况下,确定第一数据结构的每个第一具体箱的核苷酸序列。
另一个实施方案包括用于鉴定相似核苷酸测序读值的系统和方法。在一个实例中,系统包括非暂时性存储器,所述非暂时性存储器被配置以存储:可执行指令,用于存储多个测序读值配对的第一哈希数据结构和第二哈希数据结构;以及硬件处理器,所述硬件处理器由所述可执行指令编程以执行包括以下步骤的方法:接收配对的第一问询核苷酸测序读值和第二问询核苷酸测序读值;分别从所述第一问询核苷酸测序读值和所述第二问询核苷酸测序读值生成多个第一问询标识符子序列和多个第二问询标识符子序列。第一和第二问询核苷酸测序读值可以是一对配对末端测序读值的读值。方法包括通过分别对所述多个第一问询标识符子序列和所述多个第二问询标识符子序列应用哈希处理,生成所述第一核苷酸测序读值和所述第二核苷酸测序读值的第一问询名签和第二问询名签;分别使用所述第一问询名签和所述第二问询名签,从所述第一哈希数据结构和所述第二哈希数据结构中找回一个或多个第一存储配对和一个或多个第二存储配对,其中所述第一对和所述第二对中的每一对包括第一存储核苷酸测序读值和第二存储核苷酸测序读值;以及将第一存储配对和第二存储配对中均存在的每一对第一存储核苷酸测序读值和第二存储核苷酸测序读值分别确定为与第一问询测序读值和第二问询测序读值相似的第一测序读值和第二测序读值。
另一个实施方案是一种计算机实施的方法,其包括接收配对的第一问询核苷酸测序读值和第二问询核苷酸测序读值;分别从第一问询核苷酸测序读值和第二问询核苷酸测序读值生成多个第一问询标识符子序列和多个第二问询标识符子序列;分别对所述多个第一问询标识符子序列和所述多个第二问询标识符子序列应用哈希处理,生成第一核苷酸测序读值和第二核苷酸测序读值的第一问询名签和第二问询名签;以及从第一哈希数据结构中找回一个或多个第一存储配对,存储多个测序读值配对,使用第一问询名签和第二问询名签,其中第一对中的每一个包括相似于第一问询核苷酸测序读值和第二问询核苷酸测序读值的第一存储核苷酸测序读值和第二存储核苷酸测序读值。
本说明书中描述的主题的一个或多个实施方案的细节在附图和以下描述中阐述。根据说明书、附图和权利要求书,其它特征、方面和优点将变得更清晰。发明概述和以下详细描述都不旨在限定或限制本申请主题的范围。
附图简要说明
图1示出了折叠测序读值的示意图。
图2A-2D示出了基于局部敏感哈希的读值分组(grouping)和折叠的示意图,其包括叠板分割(图2A),最小哈希(图2B),局部敏感哈希(LSH)插入(图2C)和LSH问询(图2D)。给定问询测序读值(未示出)的情况下,可以生成问询测序读值的最小哈希或名签232,并且可以将最小哈希232划分成用于对哈希表格1和2问询的两部分232a、232b(224a、224b)。问询测序读值与存储在哈希表格1和2(224a,224b)中的序列ACTGGAC 204相似,但不相同。生成问询测序读值的最小哈希232的方式相似于图2A和2B所示的测序读值ACTGGAC(204)生成最小哈希212的方式。由于哈希表格1(224a)不包括部分232a中的一个作为已有箱228a的密匙212a,因此在哈希表格1(224a)中没有找到与问询测序读值相似的测序读值。由于哈希表格2(224b)包括部分232b中的一个作为已有箱228b的密匙212b,因此问询测序读值相似于与已有箱228b相关联的测序读值204。然后,存储在已有箱228b中的序列ACTGGAC(204)作为与问询测序读值相似的测序读值被返回。
图3显示了为配对末端测序读值的读值1和读值2产生虚拟通用分子索引(vUMI)的示意图。
图4A和4B示出了从虚拟UMI生成k-mer(图4A)和拼贴k-mer(图4B)的示意图。
图5示出了通过它们的序列和它们的哈希对序列读值进行分箱的流程图。可以从120个可能的物理UMI条形码之一中选择每个物理UMI条形码,使得对于每一对序列读值1和读值2存在120×120=14,400个物理UMI条形码的组合。在流程图中,可以基于配对的物理UMI条形码的组合,将每一对测序读值1和读值2分配到14,400个UMI箱中的一个。本申请的读值折叠方法可以应用于每个UMI箱的测序读值。
图6A-6F是示出利用局部敏感哈希的读值折叠和具有相似性能的基于比对的读值折叠的示例性曲线图。
图7是示出利用局部敏感哈希的读值折叠的示例性方法的流程图。
图8是示出利用局部敏感哈希来鉴定相似读值的示例性方法的流程图。
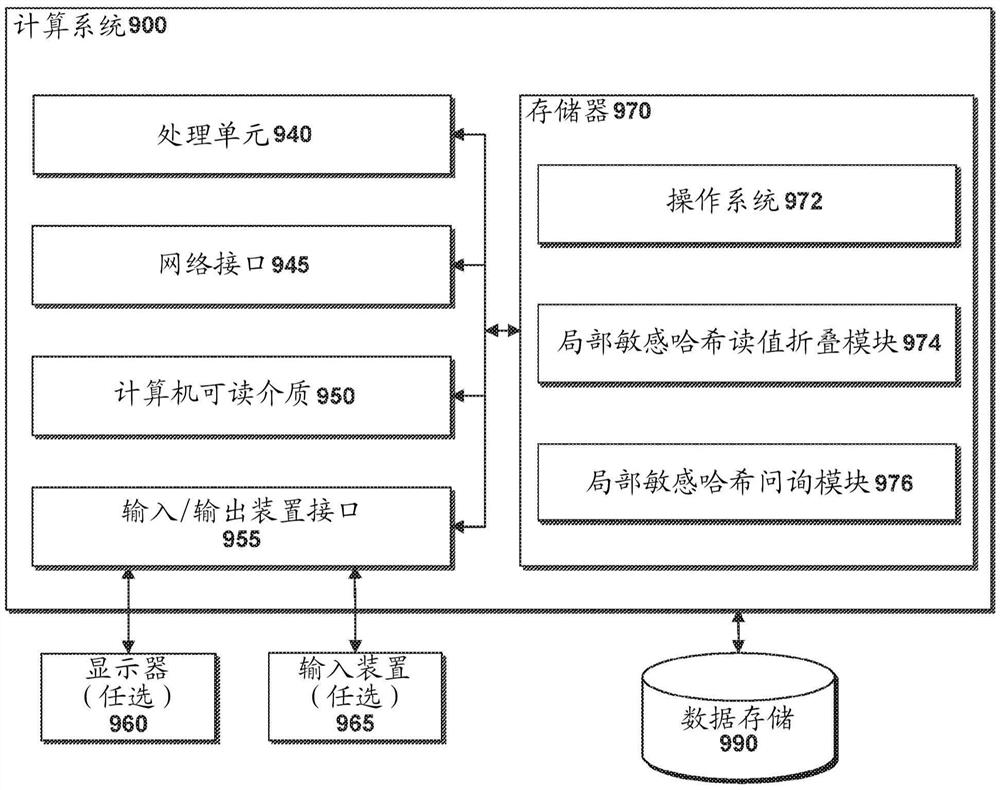
图9是被配置为利用局部敏感哈希来实施读值折叠和问询的示例性计算机系统的框图。
发明详细描述
下面的详细描述参考附图,附图构成详细描述的一部分。在附图中,除非上下文另有规定,相似符号通常标识相似组件。在详细描述、附图和权利要求中描述的示例性实施方案不意味着是限制性的。可以利用其它实施方案,并且可以进行其它改变,这不脱离本申请提出的主题的精神或范围。容易理解的是,本文一般性描述的和附图中示出的本申请的各个方面可以以多种不同的配置来设置、替换、组合、分离和设计,所有这些都本申请中明确地考虑并且构成本申请的一部分。
读值折叠是一种计算方法,其将核苷酸测序读值鉴定为源自相同来源的脱氧核糖核酸(DNA)分子,并且随后使用统计方法来减少在这些读值集合中发现的假错误。参照图1,给定具有正链108a和负链108b的相同DNA分子108的所有双重读值104+r1、104+r2、104-r1、104-r2,读值折叠可以包括将读值104+r1、104+r2、104-r1、104-r2分组在一起。读值折叠可以包括使用统计投票来减少假错误,例如使用单链折叠来确定核苷酸链的核苷酸序列,例如DNA分子108的正链108a的序列。读值折叠可以包括以高置信度推断原始DNA分子108的序列,例如以双链折叠从正链108a的序列和负链108b的序列确定DNA分子108的核苷酸序列。本申请所示的系统和方法可利用局部敏感哈希(LSH)和虚拟标识符序列(vID序列)来进行读值折叠。
读值折叠可以产生高质量的读值。读值折叠可能需要利用标识符序列(ID序列)112a、112b'、112a'、112b对样本进行测序。这种标识符序列在本申请中也称为"物理标识符序列"(pID序列)。这些标识符序列可以是通用分子指数(UMI)条形码。这样的标识符序列112a、112b'、112a'、112b在区分可能看起来非常相似的读值和分子时能够提高分辨率,但是在具体情况下可以在没有这样的标识符序列的情况下实施读值折叠。读值折叠可导致计算机错误减少。这种错误减少对于二代测序(NGS)内的许多应用是有用的。
该方法的一个应用是检测仅存在于超低等位基因分数中的变体,例如循环肿瘤DNA(ctDNA)。另一个应用是在临床应用中提高变体判断特异性。由于读值折叠有效地将DNA片段的所有重复观察(例如DNA片段的PCR重复)组合成单个代表,因此读值折叠具有显著减少需要在下游处理的数据量的益处。去除重复的观察或读值可以导致数据大小的十倍或更多倍的减小。
原始读值折叠方法可以涉及穷举配对序列比较,这需要O(n
常规读值折叠方法可以使用比对位置和UMI条形码信息的组合来鉴定重复读值的组。这些读值折叠方法的一个缺点在于,需要已经比对和分类的输入读值。在读值折叠之前对读值进行比对和分类的预处理是具有挑战性的。首先,常规读值处理需要O(n*log(n))预处理。比对"n"个读值可能需要O(n)的运行时间。对"n"个读值进行分类需要O(n*log(n))的运行时间。比对和分类"n"个读值的预处理需要运行时间O(n*log(n))。
使用常规方法的折叠读值的整个过程具有O(n*log(n))的下限运行时间。这种超线性函数意味着运行时间比输入大小增长得更快。随着生成更多的原始测序数据,常规读值折叠带来的计算成本比正被处理的测序数据的量增长得更快。此外,常规读值折叠受限于良好表征的参考物。基于核苷酸比对的读值折叠将其使用限制在存在高质量参考序列的应用中,因为这些参考序列是实现良好比对以及随后精确折叠所必需的。例如,对于结构变体、重复扩增和重复基因组区域可能不能获得相当好的比对。对比对的依赖性使得在新的或未知的方面应用读值折叠技术(以及UMI技术)变得困难,从而限制了UMI实现的错误减少技术的通用性。由于没有高质量的"参考物"来帮助鉴定测序错误,读值折叠在这些领域中未能具有很大的影响。
本申请公开了使用局部敏感哈希(LSH)进行读值折叠的系统和方法。常规读值折叠方法需要比对信息,因为对于已经共享UMI条形码的子分组读值,没有好的替代方法。本申请所公开的基于LSH的读值折叠方法克服了此缺点,且消除了折叠对需要比对的依赖性,从而实现了算法复杂度的巨大增益,以及读值折叠和相关技术的普遍适用性。
在一个实施方案中,基于LSH的读值折叠方法不具有O(n*log(n))的运行时间,并且不需要良好表征的参考物。例如,读值折叠方法可以根本不需要比对信息,需要更少的分类比对。在一个实施方案中,该方法依赖于物理标识符序列(pID序列),如物理UMI条形码(pUMI条形码),和虚拟标识符序列(vID序列),如虚拟UMI条形码(vUMI条形码),以鉴定存在于各种核苷酸片段上的重复读值组。物理标识符序列在本文也被称为"标识符序列"(ID序列)。物理UMI条形码在本文也被称为"UMI条形码"。虚拟标识符序列可以是用作"虚拟法人"标识符序列的读值的子序列,以鉴定重复读值组。vUMI条形码可以是用作"虚拟的"条形码的读值的子序列,以鉴定重复读值的组。物理标识符序列或物理UMI条形码可以是在测序文库准备期间添加到核苷酸片段的标识符序列或条形码。
在一个实施方案中,该方法将相似读值分组在一起,并且不需要任何参考序列,或者彻底序列比较。该方法可以包括将读值的第一遍原始分组确定在由UMI条形码(例如物理UMI,虚拟UMI或上述的组合)定义的箱中。该方法将虚拟UMI与局部敏感哈希相结合。由于该方法允许在没有比对信息的情况下将具有相似序列的读值分组在一起,因此该方法将读值折叠的过程脱离于比对限制。该方法可以包括确定相似序列,包括检查该箱中的其它测序读值。
该方法可用于折叠来自任何样品的读值,例如DNA或RNA,与样品所来源的生物体无关。此外,由于哈希处理是O(1)恒定时间操作,且哈希处理需要针对n个读值中的每一者执行固定次数,所以所述方法实现在O(n)运行时间中运行读值折叠。例如,对于n个读值中的每一个,必须执行一次、两次、三次或更多次哈希处理。这种读值折叠运行时间减少了对于用NGS生成的日益增大的数据集所需的处理时间。O(n)运行时间能够显著降低二次分析的运行时间复杂度,并允许对任何样本灵活地应用读值折叠。
在一个实施方案中,DNA测序仪器可以实施本申请公开的基于LSH的读值折叠方法。例如,该方法可以实现为用于减小计算机错误的仪器整合方法,因为该方法不需要用于读值折叠的参考序列。该方法可以通过借助于NGS的重复率来实现错误减少,从而对于所有的序列读值都可以实现大大减小错误率。该方法还可以显著减少用户必须处理的测序数据量,从而增加基因组分析的可及性。该方法可以被用作测序仪机载技术,以便为使用者输出更少的,更高质量的读值,从而降低下游分析的复杂性。
在一个实施方案中,测序读值不与UMI条形码相关或不使用UMI条形码产生。例如,可以在虚拟UMI上进行对局部敏感哈希,以对核苷酸读值进行分组。作为另一个例子,可以使用"分层"虚拟UMI策略来模拟由物理UMI条形码提供的分箱功能。该方法可以生成两种类型的虚拟UMI,一种用作物理UMI的模拟,一种用作虚拟UMI。
重复标记是减少PCR引入的偏倚的生物信息学方法。本申请公开的内容包括用于将相似读值序列分组在一起并且用局部敏感哈希标记重复的系统和方法。
本申请公开的内容包括用于读值折叠的系统和方法。在一个实施方案中,该方法使用虚拟标识符(vID)序列,例如虚拟通用分子索引(vUMI),利用局部敏感哈希,以允许相似读值的无参考分组,而无需实施彻底配对比较。测序读值的虚拟标识符序列,例如虚拟UMI,是指测序读值本身内的任何子串或子序列,包括可能不连续的子串。虚拟标识符序列不同于物理标识符(pID)序列。物理标识符序列,例如物理UMI(pUMI),是指在测序文库制备期间添加的标识符序列或UMI条形码。
局部敏感哈希(LSH)是一种计算方法,其将"相似"数据放置到相同的计算"箱"中,而无需实施彻底配对比较。数据相似性是指读值的序列相似性,其可以用诸如Levenshtein距离,Hamming距离或Jaccard距离之类的方法来计算。LSH函数可以"哈希(hash)"与每一读值相关联的虚拟UMI,且使用该结果将每一读值放置在一个箱中,同在一起的是具有相似虚拟UMI的读值。本申请所公开的应用于测序读值的LSH使得能够基于虚拟UMI将具有包含错误的虚拟UMI的测序读值分组在一起。测序技术时常不会产生无错误的测序读值。因此,能够将可能携带小突变的相似序列分在一箱并快速找到,这对于执行实施读值折叠所必需的读值分组是重要的。因为测序读值中的小突变通常难以预测,所以用于相似测序读值分组的一般方法可能比用于假定特定突变模式的相似测序读值分组的特定方法更有用。在一些实施方案中,本文公开的方法的容错特性在大约20%的测序读值中起作用。如果不进行校正,这些测序读值可能极大地影响折叠的准确性,并且随后在变体判断中表现为大量的假阳性。
在一个实施方案中,可以基于通过(例如词典编排式)分类虚拟UMI而从测序读值生成的虚拟UMI来鉴定相似测序读值。基于分类虚拟UMI的读值折叠方法可以不考虑虚拟UMI中的突变,并且可能具有O(n*log(n))的运行时间复杂度。在另一个实施方案中,相似测序读值可以使用具有虚拟UMI的原始标准哈希处理来鉴定。与基于LSH的读值折叠方法相比,基于原始标准哈希处理的读值折叠方法可以具有相似性能。这种读值折叠方法不具有容错特性。在一个实施方案中,相似的测序读值可以通过与测序读值相关联的UMI聚类来鉴定。基于聚类的读值折叠方法可以处理轻微错配的虚拟UMI条形码,但是将引入O(n
LSH是概率数据处理。利用LSH将相似读值数据放置在同一箱中的概率是高的。存在相似数据不落入同一箱中的小的非零概率。可能存在相似数据不落入同一个箱中或者不同数据落入同一个箱中的小几率。在一个实施方案中,LSH被设计成使利用LSH将相似读值数据放置在同一箱中的概率最小化。在一个实施方案中,基于LSH的读值折叠方法可被配置以用于最大程度地找回和实施对每一箱中的每一项目(通常少于5个项目)基于比对的核查。
局部敏感哈希(LSH)使每条数据,例如数据的虚拟UMI,通过"哈希"函数,该函数的结果被用于将该数据放置到一个箱中。利用LSH,相似数据应该落在相同的(或附近的)箱中,使得能够对相似数据进行非常快速的问询。基于LSH的读值折叠可以包括叠板分割(shingling),最小哈希和局部敏感哈希。叠板分割包括将输入数据分解成长度为k的字符的重叠集合或叠板。最小哈希包括使每个"叠板(shingle)"通过一组哈希函数以生成数据的指纹或名签。局部敏感哈希包括使用指纹将数据放置到"箱"中,其中相似数据可能共享相似分箱方案。例如,测序读值可以被分解成测序读值的长度为k的子序列。每个子序列可以通过一组哈希函数来产生测序读值的名签。名签可用于将测序读值置于一个或多个箱中,其中相似测序读值可能共享相似或相同的名签。
图2A-2D示出了基于局部敏感哈希的读值分组和折叠的示意图,其包括叠板分割(图2A),最小哈希(图2B),局部敏感哈希(LSH)插入(图2C)和LSH问询(图2D)。叠板分割包括以m碱基对增量移动k碱基的滑动窗口,从而将虚拟UMI 204分解为k-mer"叠板"208a-208d(图2A)。例如,4个碱基的滑动窗口可以以1个碱基对的增量移动,以将虚拟UMI 204分解为4-mer叠板。图2A示出了4个序列的叠板。第一叠板是GGAC(208a),第二叠板是TGGA(208b),第三叠板是CTGG(208c),第四叠板是ACTG(208d)。
最小哈希包括通过使叠板的k-mer集合的经过几个哈希函数216并取最小哈希(MinHash)来产生叠板的k-mer集合的哈希"名签"212。图2B示出了4个叠板GGAC、TGGA、CTGG和ACTG的集合可以经过8个哈希函数216,以分别为每个叠板生成8个哈希函数的8个元素的输出220a-220d。哈希函数的数目可以是8、16、32、64、128、256、512、1024或更多。可以采用哈希输出220a-220d的对应元素的最小值来计算最小值的最小哈希212。最小哈希的Jaccard距离是真实Jaccard距离的近似值。哈希函数越多,近似越好。
现在参考图2C,基于从测序读值204计算的最小哈希212或最小哈希212的子序列212a、212b,可以将测序读值204插入哈希表格224a、224b。图2C说明基于最小哈希212的子序列212a、212b,可以将测序读值204插入哈希表格224a、224b中。序列读值204的LSH插入消耗哈希"名签"212,然后将名签212分割成小块或子序列212a、212b。然后将这些小块212a、212b用作哈希表格224a、224b中的密匙,特别是哈希表格224a、224b的箱228a、228b中的密匙。这种分块和哈希方案对于箱的"宽度"以及对于更高的召回或更高的特异性是可调的。如图2C所示,相同的测序读值204被放置在两个不同哈希表格224a、224b中的两个不同的箱中。只要两个测序读值共享或存储在一个或多个箱中,则测序读值可以被认为是相似的。
图2D示出了利用LSH确定问询测序读值是否与存储在哈希表格中的测序读值相似或相同。在给定问询测序读值的情况下,系统使问询测序读值通过最小哈希,并针对所有哈希表格问询最小哈希。问询序列的名签232可以被分割成两个小块232a、232b,它们被针对所有的哈希表格224a、224b进行问询。由于哈希表格2(224b)包括其中一个小块232b作为已有箱228b的密匙212b,因此问询序列与已有箱228b所关联(诸如存储在已有箱228b中)的测序读值204相似或相同。
虚拟UMI是测序读值本身的子序列。虚拟UMI可以跨越直到整个核苷酸读值,并且可以是连续的子序列或非连续的子序列。例如,测序读值的虚拟UMI可以是来自测序读值的5'端的25个碱基对(bp)。图3显示了为配对末端测序读值的读值1(R1)和读值2(R2)产生虚拟通用分子索引(vUMI)的示意图。对应于被测序的DNA片段308的正链或负链的配对末端测序读值的读值1304r1和读值2304r2可以被处理以产生虚拟UMI 312r1、312r2。
图4A和4B示出了从虚拟UMI生成k-mer(图4A)和拼贴k-mer(图4B)的示意图。两组k-mer的Jaccard相似性是两组都包含的k-mer的数目。如果为虚拟UMI生成7个k-mer 412,则两个相同的虚拟UMI的Jaccard相似性可以是7/7。如图4A所示,虚拟UMI 404和除了插入408之外的相同虚拟UMI 404i的Jaccard相似性可以是2/12,因为包括插入408或其3'的所有k-mer 412a都受到影响。使用跨越虚拟UMI的重叠k-mer允许更好的插入和缺失容忍度。如图4B所示,在图4A所示的相同插入的情况下,如果为虚拟UMI 404、404i生成16个k-mer412,则虚拟UMI 404和除了插入408之外的相同虚拟UMI 404i的Jaccard相似性很高,在14/18处。Jaccard相似性仍然相当高,因为只有包括插入408的k-mer 412a受到插入408的影响。在叠板分割后更大的相似性导致更相似的最小哈希特征,进而帮助改变的序列落在相同的LSH箱中。
在一个实施方案中,LSH本身不直接存储或索引待折叠的读值组。相反,LSH有助于找到与给定问询读值相似的读值。一旦找到相似读值,就将其存储在常规哈希表格中,其中密匙是第一次开始该组的"质心"读值。换句话说,LSH数据结构包含与前述常规哈希表格相同的"密钥"(并且两者同步更新),其中这些"密钥"是用作"组锚点"的读值,如果其他读值与其相似,则其他读值被分配到这些"组锚点"。LSH使得输入的读值能够快速地找到它可能匹配的密钥/锚点,从而不需要彻底检查或比较。实际的分组或测序读值可以存储在常规哈希表格中。
在检查LSH箱的相似序列之后,可以执行比对得分检查。比对得分检查确保不相同的虚拟UMI不会被等同或认为相似。对于虚拟UMI被认为是等同,虚拟UMI的最小比对得分可能是必需的。当针对LSH匹配执行比对检查时,可以使用最佳匹配来确定比对得分高于阈值(即,具有最高比对得分)。更多的错配,如单核苷酸变体(SNV)和插入和缺失(indel),降低比对得分。"滑动"比对减少匹配的数量,这相应地减少容许的不匹配的数量。在一个实施方案中,可以执行虚拟UMI的全局比对,这会带来昂贵的计算成本。
来自每个读值对的两个虚拟UMI是同一DNA片段的身份的"独立的"条形码或独立度量物。通过为每个虚拟UMI保持两个单独的LSH数据结构,可以减少假阳性。因为在每个箱中放置更少的最小哈希名签,所以存在较低的计划外冲突的几率。通过使两个独立问询的结果相交,消除了大部分剩余的假阳性命中,而不会丢失正确的命中。
独立的LSH数据结构可以用于读值1和读值2上的虚拟UMI。LSH可以被配置用于具有次优特异性的非常高的召回率。为了提高特异性,可以采用两个正交问询之间的交互来提高特异性,而不会对灵敏度产生很大的影响。将虚拟UMI与读值1和读值2分开也提高了在更多重复区域中的特异性。
在一个实施方案中,每个k-mer可以有64个哈希。读值1具有64个元素的最小哈希名签,读值2具有64个元素的最小哈希名签。基于读值1的最小哈希名签和读值2的最小哈希名签,读值1和读值2的配对可以分别存储在双箱LSH结构的两个哈希表格中。读值1的最小哈希名签可以被分成子序列,使得读值1和读值2的配对可以被存储在一个哈希表格的多个箱中以及另一个哈希表格的多个箱中。在不同的实施方案中,哈希数字可以是不同的,例如8、64、256、1024及更大。以稍微减缓的运行时间为代价,可以使用更多的哈希来实现更精确的数据结构性能,并且随着运行时间的加快,可以使用更少的哈希来实现精确度稍差的数据结构性能。每个k-mer被哈希相同的次数,无论是64次、8次还是n次,以确保最小哈希名签具有一致的大小。
在一个实施方案中,读值1和读值2被认为是原子性不可分的单元,并且该原子单元由来自读值1的虚拟UMI的最小哈希名签和来自读值2的虚拟UMI的最小哈希名签(vUMI 1和vUMI 2)共同提及。在每个哈希表格内,最小哈希名签可以被分成多个部分,使得原子读值1/读值2配对被存储在多个箱中,对于名签的每个小块存储一次。
下面示出双箱LSH结构的示例性伪码。
下面示出在LSH中使用双箱LSH结构的示例性伪代码。通过插入一对没有存储在双箱LSH结构中的相似虚拟UMI的虚拟UMI,允许将来的问询找到插入的配对。本质上,为家族创建新的"种子",其可能在下一次匹配。
图5示出了通过序列读值的序列和序列读值的哈希对序列读值进行分箱的流程图。图5所示的分箱过程是可并行的,这可能导致具有数千个线程的线性加速。所示的分箱过程需要具有最小存储器使用的中间高速缓存。可以从120个可能的物理UMI条形码之一中选择每个物理UMI条形码,使得存在120×120=14400个物理UMI条形码的组合,并且每一对序列读值1和读值2可以具有14,400个物理UMI条形码组合之一。LSH可以并行地应用于与每一对物理UMI相关联的测序读值。
单链折叠是指以相同的顺序折叠共享相同物理+虚拟UMI对的所有序列。因为这些序列具有相同顺序的条形码,这表明这些序列不仅来自相同的DNA分子,而且来自该DNA分子的相同链。在单链折叠中,组/家族中的每个读值具有相同的第一UMI,相同的第二UMI,相同的第一虚拟UMI和相同的第二虚拟UMI。多个读值可以满足这种条件,在这种情况下,它们都被认为是来自同一分子的同一链的读值。
在执行单链折叠之后,可以执行双链折叠。在双链折叠中,在给定被折叠的读值对的情况下,尝试寻找相反的顺序的具有相同物理和虚拟UMI配对的另一单链分子,这相似于寻找相同DNA分子的相反链。如果发现这种双链匹配,则执行双链折叠。
因为双链折叠是在单链折叠之后执行的,所以所有的链特异性副本都已经通过单链折叠而被去除。在双链折叠中,发现来自同一分子的相反的链的已经折叠的读。例如,给定具有第一UMI x,第二UMI y,第一虚拟UMI a和第二虚拟UMI b的折叠读值对,双链折叠查找将具有其第一UMI y,其第二UMI x,其第一虚拟UMI b和其第二虚拟UMI a的相反的链的读值对。由于在输出文件(例如"fastq"文件)中如何报告读值的语义,可能不需要相反的链的读值对的反向互补。这种两级单链、然后交叉链折叠使得能够在下游分析中使用一些先进的变体判断技术。
发现常规的基于比对的方法和基于LSH/虚拟UMI的方法之间的读值折叠结果是相当的,无论在比对总结度量的水平,还是当用于变体判断时,诸如结构变体判断和小变体判断。为了发现相似的核苷酸序列,可以检查该箱中的其它项目。因此,本文公开的基于虚拟UMI的方法可以用于折叠用于变体判断的测序读值。
图6A-6F是示出利用局部敏感哈希的读值折叠和基于比对的读值折叠在用
表2显示对融合判断特异性的改善不会对NovaSeq
表1.融合判断假阳性
表2.融合判断召回。
总之,这些数据表明,与基于比对的折叠相比,基于LSH的读值折叠是有利的,并且在总体级别度量以及变体判断方面匹配或超越现有的性能。
图7是示出使用局部敏感哈希的读值折叠的示例性方法700的流程图。方法700可以包含在存储于计算系统的计算机可读介质(例如一个或多个磁盘驱动器)上的一组可执行程序指令中。例如,图9中所示和下面更详细描述的计算系统900可以执行一组可执行程序指令,以实现方法700。当方法700被启动时,可执行程序指令可以被加载到诸如RAM的存储器中,并且由计算系统900的一个或多个处理器执行。虽然结合图9所示的计算系统900描述了方法700,但是该描述仅仅是说明性的,而不是限制性的。在一些实施方案中,方法700或其各个部分可以由多个计算系统串行地或并行地执行。计算系统900可以包括第一哈希数据结构,例如哈希表格,用于将核苷酸测序读值存储在多个箱中。
在方法700开始于框704之后,方法700进行到框708,其中计算系统接收多个第一核苷酸测序读值。多个第一核苷酸测序读值可以与相同的物理标识符序列相关联。多个第一核苷酸测序读值可以不与任何物理标识符序列相关。
方法700从框708前进到框712,其中计算系统从每个第一核苷酸测序读值的第一标识符序列生成多个第一标识符子序列。生成多个第一标识符子序列可以包括从测序读值的第一标识符序列生成多个k-mer。子序列可包含核苷酸插入、核苷酸缺失、核苷酸取代或上述的组合。两个连续的第一标识符子序列可以重叠。例如,两个连续的第一标识符子序列重叠k-1个核苷酸。例如,多个第一标识符子序列包含多个4-mer,并且其中所述第一标识符序列包含约25个核苷酸。第一标识符序列可以是测序读值1的子序列。子序列可以是测序读值1的连续子序列。子序列可以是测序读值1的非连续子序列。
在框712产生第一标识符子序列之后,方法700进行到框716,在框716,计算系统通过对多个第一标识符子序列应用哈希处理来为第一核苷酸测序读值产生第一名签。第一名签可以匹配第一哈希数据结构的第一具体箱的密钥。第一名签和第一哈希数据结构的第一具体箱的密钥可以是相同的。
生成第一名签可以包括为每个第一标识符子序列确定多个哈希。生成第一名签可以包括从多个第一标识符子序列的相应哈希确定第一名签的每个第一元素。第一名签的每个第一元素可以是多个第一标识符子序列的相应哈希的最小值。第一名签的每个第一元素是多个第一标识符子序列的相应哈希的最小值、平均值、中位值或最大值。
该方法从框716前进到框720,其中基于第一名签,计算系统将第一核苷酸测序读值分配到第一哈希数据结构的至少一个第一具体箱。在一个实施方案中,分配第一核苷酸测序读值包括从第一核苷酸测序读值的第一名签确定第一名签的多个子序列;以及基于第一名签的子序列,将第一核苷酸测序读值分配到多个第一哈希数据结构中的每个第一哈希数据结构的第一具体箱。在另一个实施方案中,分配第一核苷酸测序读值包括:从第一核苷酸测序读值的第一名签确定第一名签的多个子序列;以及基于第一名签的多个子序列,将所述第一核苷酸测序读值分配到第一哈希数据结构的多个第一具体箱。方法700在框728结束。
在一个实例中,第一具体箱是第一哈希数据结构的现有箱,并且其中第一核苷酸测序读值和分配到第一哈希数据结构的第一具体箱的另一第一核苷酸测序读值的名签的比对得分高于比对得分阈值。在另一个实例中,第一具体箱是第一哈希数据结构的现有箱,并且其中第一核苷酸测序读值和分配到第一哈希数据结构的第一具体箱的任何第一核苷酸测序读值的名签的最高比对得分高于比对得分阈值。在另一个实例中,第一具体箱是第一哈希数据结构的新箱,并且其中第一核苷酸测序读值和分配到第一哈希数据结构的任何现有箱的任何第一核苷酸测序读值的名签的比对得分低于比对得分阈值。
在框720处将第一核苷酸测序读值分配到第一具体箱后,方法700进行到框724,其中在一个或多个第一核苷酸测序读值被分配后,计算系统确定第一哈希数据结构的每个第一具体箱的核苷酸序列。确定核苷酸序列可以包括确定分配到第一具体箱的一个或多个第一核苷酸测序读值的共有序列。确定共有序列可以包括将分配到第一具体箱的最频繁的第一核苷酸测序读值确定为第一具体箱的共有序列。共有序列可以包含对于分配到第一具体箱的第一核苷酸测序读值的每个相应位置最频繁的核苷酸碱基。确定共有序列可以包括将分配到第一具体箱的具有最高质量分数的第一核苷酸测序读值确定为第一具体箱的共有序列。最高质量分数可以基于具有最高质量分数第一核苷酸测序读值上的每个碱基的质量分数来确定。确定核苷酸序列可以包括选择分配到第一具体箱的一个或多个第一核苷酸测序读值的序列作为第一具体箱的代表性序列。确定核苷酸序列可以包括确定分配到第一具体箱的一个或多个第一核苷酸测序读值中的两个的比对得分高于比对得分阈值。
每个第一核苷酸测序读值可以与第二核苷酸测序读值相关联。第一核苷酸测序读值和第二核苷酸测序读值可以形成配对末端核苷酸测序读值。计算系统可以从第二核苷酸测序读值的第二标识符序列生成多个第二标识符子序列;以及通过对所述多个第二标识符子序列应用哈希处理来产生第二核苷酸测序读值的第二名签。
在不同的实施方式中,分配第一核苷酸测序读值可以是不同的。例如,分配第一核苷酸测序读值包括基于第一名签将包括第一核苷酸测序读值和第二核苷酸测序读值的一对测序读值分配到第一哈希数据结构的第一具体箱。作为另一个实例,分配第一核苷酸测序读值包括基于第二名签将第二核苷酸测序读值分配到第一哈希数据结构的第二具体箱。作为另一个实例,分配第一核苷酸测序读值包括基于第二名签将包括第一核苷酸测序读值和第二核苷酸测序读值的一对测序读值分配到第二哈希数据结构的第二具体箱。
作为实例,分配第一核苷酸测序读值包括基于第一核苷酸测序读值的第一名签的多个子序列和第二核苷酸测序读值的第二名签的多个子序列分别为将包括第一核苷酸测序读值和核苷酸测序读值的一对测序读值分配到第一哈希数据结构的第一具体箱和第二数据结构的第二具体箱。计算系统可以存储第一数据结构和第二数据结构,分别用于存储第一哈希数据结构的箱的密匙和第二哈希数据结构的箱的密匙。分配一对测序读值可以包括确定第一名签和第二名签被存储在第一数据结构和第二数据结构中;以及分别使用第一存储密钥和第二存储密钥将一对测序读值分配到第一哈希数据结构的第一具体箱和第二哈希数据结构的第二具体箱。该对测序读值和包括与第一存储密钥相关联的第一测序读值和与第二存储密钥相关联的第二测序读值的一对测序读值的比对得分高于比对得分阈值。
分配一对测序读值可以包括确定存储在第一数据结构中并与第一名签相关联的第一哈希数据结构的一个或多个第一密钥;确定存储在第二数据结构中并与第二名签相关联的第二哈希数据结构的一个或多个第二密钥;确定包括与第一存储密匙相关联的第一测序读值和与第二存储密匙相关联的第二测序读值的一对测序读值,该对测序读值与包括与任何第一存储密匙相关联的第一测序读值和与任何第二存储密匙相关联的第二测序读值的任何对测序读值具有最高比对得分;以及分别使用与具有最高比对得分的该对第一测序读值和第二测序读值相关联的第一存储密匙和第二存储密匙,将该对测序读值分配到第一哈希数据结构的第一具体箱和第二哈希数据结构的第二具体箱。与第一存储密匙相关联的第一测序读值可以具有与任何第一存储密匙相关联的第一测序读值与第一名签的最高比对得分。与第二存储密钥相关联的第二测序读值可以具有与任何第二存储密匙相关联的第二测序读值与第二名签的最高比对得分。
图8是示出使用局部敏感哈希来鉴定相似读值的示例性方法800的流程图。方法800可以包含在存储在计算系统的计算机可读介质(例如一个或多个磁盘驱动器)上的一组可执行程序指令中。例如,图9中所示和下面更详细描述的计算系统900可以执行一组可执行程序指令以实现方法800。当方法800被启动时,可执行程序指令可以被加载到诸如RAM的存储器中,并且由计算系统900的一个或多个处理器执行。虽然参考图9所示的计算系统900描述了方法800,但是该描述仅仅是说明性的,而不是限制性的。在一些实施方案中,方法800或其各个部分可以由多个计算系统串行地或并行地执行。计算系统可以存储用于存储多个序列读值对的第一哈希数据结构和第二哈希数据结构。
每一对测序读值可以包括第一核苷酸测序读值和第二核苷酸测序读值,其中每一对测序读值被分配到第一哈希数据结构的多个第一箱中的一个使基于通过对第一核苷酸测序读值的第一标识符序列的第一标识符子序列进行哈希处理而产生的该对测序读值的第一核苷酸测序读值的第一名签。每一对测序读值被分配到第二哈希数据结构的多个第二箱中的一个可以基于通过对第二核苷酸测序读值的第二标识符序列进行哈希处理而产生的该对测序读值的第二核苷酸测序读值的第二名签。
在方法800开始于框804之后,方法800进行到框808,其中计算系统接收配对的第一问询核苷酸测序读值和第二问询核苷酸测序读值。方法800从框808前进到框812,其中计算系统分别从第一问询核苷酸测序读值和第二问询核苷酸测序读值生成多个第一问询标识符子序列和多个第二问询标识符子序列。在框812生成问询标识符子序列之后,方法800进行到框816,在框816,计算系统通过分别对多个第一问询标识符子序列和多个第二问询标识符子序列应用哈希处理而生成第一核苷酸测序读值和第二核苷酸测序读值的第一问询名签和第二问询名签。计算系统在框808-816处执行的步骤可参考图7所描述的框708-716所描述。
在框816之后,该方法可以包括正交问询。例如,该方法从框816前进到框820,其中计算系统分别使用第一问询名签和第二问询名签从第一哈希数据结构和第二哈希数据结构中找回一个或多个第一存储配对和一个或多个第二存储配对,其中第一对和第二对中的每一个都包括第一存储核苷酸测序读值和第二存储核苷酸测序读值。在框820处找回测序读值配对之后,方法800进行到框824,在框824处,计算系统将第一存储配对和第二存储配对中均存在的第一存储核苷酸测序读值和第二存储核苷酸测序读值中确定为相似于问询测序读值1和问询测序读值2的测序读值1和测序读值2。方法800在框828结束。
每一对测序读值可以与第一标识符序列和第二标识符序列相关联。计算系统可以确定第一对测序读值的第一标识符序列和第二标识符序列与第二对测序读值的第一标识符序列和第二标识符序列是相同的;确定第一对测序读值和第二对测序读值的核苷酸序列。
在一个实施方案中,方法800可以包括在框808处接收第一问询核苷酸测序读值。接收第一问询核苷酸测序读值可以包括接收配对的第一问询核苷酸测序读值和第二问询核苷酸测序读值。方法800可以包括在框812处从第一问询核苷酸测读值的生成多个第一问询标识符子序列。产生多个第一问询标识符子序列可以包括从第二核苷酸测序读值产生多个第二问询标识符子序列。在框816,方法800可以包括通过对多个第一问询标识符子序列应用哈希处理来产生第一核苷酸测序读值的第一问询名签。产生第一问询名签可以包括通过对多个第二问询标识符子序列应用哈希处理来产生第二核苷酸测序读值的第二问询名签。方法800可以包括在框820处使用第一问询名签从第一哈希数据结构找回一个或多个第一存储核苷酸测序读值。第一存储核苷酸测序读值中的每一个可以相似于第一问询核苷酸测序读值。找回一个或多个第一存储核苷酸测序读值可以包括从第一哈希数据结构找回一个或多个第一存储配对,存储多个测序读值对,使用第一问询名签和第二问询名签。第一对中的每一对可分别包括与第一问询核苷酸测序读值和第二问询核苷酸测序读值相似的第一存储核苷酸测序读值和第二存储核苷酸测序读值。
图9示出了被配置为实现本申请所公开的代谢物、注释和基因整合系统的示例性计算设备900的一般结构。图9所示的计算设备900的一般体系结构包括计算机硬件和软件组件的布置。计算设备900可以包括比图9所示的元件更多(或更少)的元件。然而,为了提供有效的公开内容,不必示出所有这些常规元件。如图所示,计算设备900包括处理单元940,网络接口945,计算机可读介质驱动器950,输入/输出设备接口955,显示器960和输入设备965,它们都可以通过通信总线彼此通信。网络接口945可以提供到一个或多个网络或计算系统的连接。因此,处理单元940可以经由网络从其他计算系统或服务器接收信息和指令。处理单元940还可以与存储器970互相通信,并且还通过输入/输出设备接口955为任选的显示器960提供输出信息。输入/输出设备接口955还可以接受来自任选的输入设备965的输入,例如键盘,鼠标,数字笔,麦克风,触摸屏,手势识别系统,语音识别系统,游戏手柄,加速度计,陀螺仪或其它输入设备。
存储器970可以包含供处理单元940执行的计算机程序指令(在一些实施方案中被分组为模块或组件),以实现一个或多个实施方案。存储器970一般包括RAM、ROM和/或其它永久性、辅助性或非暂时性的计算机可读介质。存储器970可以存储操作系统972,操作系统972提供由处理单元940在计算设备900的一般管理和操作中使用的计算机程序指令。存储器970还可以包括用于实现本申请的方面的计算机程序指令和其它信息。
例如,在一个实施方案中,存储器970包括基于局部敏感哈希的读值折叠模块974,用于使用局部敏感哈希来折叠测序读值,例如参照图7所述的读值折叠方法700。额外地或作为替代地,存储器970可以包括局部敏感哈希问询模块976,用于鉴定问询测序读值的相似核苷酸测序读值,例如参考图8描述的鉴定方法800。此外,存储器970可以包括用于存储读值折叠和/或相似核苷酸测序读值鉴定的数据和读值的结果的数据存储器990和/或一个或多个其它数据存储器,或与数据存储器990和/或一个或多个其它数据存储器通信,
在一些实施方案中,本文所公开的用于测序读值的分组和折叠的方法在专用硬件中实现,专用硬件被设计或编程为以比通用计算机处理器更高的效率来计算本文所公开的方法。例如,处理单元940可以是现场可编程门阵列(FPGA)或专用集成电路(ASIC)。
在一个实例中,局部敏感哈希(LSH)操作可由FPGA加速。在一些实施方案中,FPGA对LSH操作的加速可以取决于构建和问询每个UMI的哈希表格所需的存储,并且还取决于存储带宽是否变成软件中的瓶颈。如果在FPGA中实现与测序读值相关联的UMI的聚集(clustering),则也可以有益地在相同硬件内加速读值折叠方法。
在一些实施方案中,可以对一个或多个专用集成电路(ASIC)进行编程以执行本申请所述的一个或多个相应基因组分析模块或其它计算机的功能。ASIC包括集成电路,集成电路包括一个或多个可编程逻辑电路,可编程逻辑电路相似于本申请所述的FPGA之处在于ASIC的数字逻辑门可使用硬件描述语言(例如VHDL)来编程。然而,ASIC与FPGA的不同之处在于,ASIC仅可编程一次且一旦被编程就不能动态地重新配置。此外,本申请的各个方面不限于使用FPGA或ASIC来实现测序读值的分组和折叠。相反,处理单元940的任何基因组分析模块或其它计算机可以使用一个或多个中央处理单元(CPU)、图形处理单元(GPU)或能通过软件指令的执行实现测序读值的分组和折叠的任何组合来实现。
在一些实施方案中,使用诸如FPGA、ASIC、CPU、GPU或上述的组合的集成电路来实现测序读值的分组和折叠可包括单个FPGA,单个ASIC,单个CPU,单个GPU或其任何组合。额外地或作为替代地,使用诸如FPGA、ASIC、CPU、GPU或上述的组合的集成电路来实现测序读值的分组和折叠可包括多个FPGA,多个ASIC,多个CPU或多个GPU或其任何组合。使用诸如多个FPGA的额外集成电路来实现测序读值的分组和折叠可以减少执行诸如映射、比对、P-HMM概率计算和变体判断的二级分析操作所花费的时间量。在一些实施方案中,使用FPGA来实现这些二级分析操作可以将完成这些二级分析操作所花费的时间从24小时或更长减少到少至30分钟或更短。在一些实施方案中,使用多个FPGA来执行这些二级分析操作可以导致在少至5分钟内完成这些二级分析操作。
关于本申请中基本上任何复数和/或单数术语的使用,本领域技术人员可以根据上下文和/或应用从复数转换成单数和/或从单数转换成复数。为了清楚起见,本申请可以清楚地阐述各种单数/复数排列。
本领域技术人员能理解,通常,本申请使用的术语,特别是在权利要求(例如,权利要求的主体)中,通常意图为“开放”术语(例如,术语“包括”应被解释为“包括但不限于”,术语“具有”应被解释为“至少具有”,术语“包括”应被解释为“包括但不限于”等)。本领域的技术人员还能理解,如果希望在权利要求中进入特定数量,则在权利要求中会明确地叙述这种意图,并且在没有这种叙述的情况下,不存在这种意图。例如,为了帮助理解,以下权利要求可以包含使用短语"至少一个"和"一个或多个"来引入权利要求表述。然而,这种短语的使用不应被解释为暗示通过不定冠词"a"或"an"引入的权利要求表述将包含这种引入的权利要求表述的任何具体权利要求限制为仅包含一个此表述的实施方案,即使当同一权利要求包括短语"一个或多个"或"至少一个"和不定冠词如"a"或"an"时(例如,“a”和/或“an”应被解释为意指“至少一种”或“一种或多种”);对于用于引入权利要求表述的定冠词的使用也是如此。此外,即使明确地陈述了所引入的权利要求表述的具体数目,本领域技术人员能认识到,这种陈述应被解释为表示至少为所陈述的数目(例如,没有其它修饰词的仅“两个…”这样的陈述意指至少两个或两个或更多个)。此外,在类似于"A、B和C等中的至少一个"的那些情况下,通常使用这种造句是指本领域技术人员能理解的惯例(例如,“具有A、B和C中的至少一个的系统”将包括但不限于系统具有单独的A、单独的B、单独的C、A和B一起、A和C一起、B和C一起、和/或A、B、C一起等)。在相似于“A、B或C等中的至少一个”的那些情况下,通常使用这种造句是指本领域技术人员能理解的惯例(例如,“具有A、B或C中的至少一个的系统”将包括但不限于系统具有单独的A、单独的B、单独的C、A和B一起、A和C一起、B和C一起、和/或A、B、C一起等)。本领域的技术人员能进一步理解,无论在说明书、权利要求书或附图中,实际上任何表示两个或多个替换术语的分离的单词和/或短语都应该被理解为考虑包括这些术语中的一个、这些术语中的任一个、或这两个术语的可能性。例如,短语“A或B”将被理解为包括“A”或“B”或“A和B”的可能性。
此外,在本申请的各个特征或各个方面是根据马库什分组来描述的情况下,本领域技术人员将认识到,本申请也由此根据马库什分组的任何单个成员或成员亚组来描述。
如本领域技术人员将理解的,出于任何和所有目的,例如就提供书面描述而言,本申请公开的所有范围还涵盖其任何和所有可能的子范围及其子范围的组合。任何列出的范围可以容易地被认为充分地描述并且使得相同的范围能够被分解为至少相等的两份、三份、四份,五份,十份等。作为非限制性实例,本申请讨论的每个范围可以容易地分解成下三分之一,中三分之一和上三分之一等。本领域技术人员还能理解,所有如“至多”、“至少”、“大于”、“小于”等的语言包括所列举的数字,并且是指随后可被分解成如上所述的子范围的范围。最后,本领域技术人员能理解的,范围包括每个单独的成员。因此,例如,具有1-3个制品的一组是指具有1、2或3个制品的一组。相似地,具有1-5个制品的一组是指具有1、2、3、4或5个制品的一组,以此类推。
虽然本文已经公开了各个方面和各种实施方案,但是其它方面和实施方案对于本领域技术人员来说也是清楚的。本文公开的各个方面和实施方案是为了说明的目的,而不是为了限制,真正的范围和精神由权利要求书指示。
- 用于测序读值的分组和折叠的系统和方法
- 用于在高性能计算环境中允许多个全局标识符(GID)子网前缀值同时用于传入分组处理的系统和方法