基于蛋白质工程的集成序列与结构特征的深度学习模型及预测方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于蛋白质突变预测技术领域,具体涉及一种基于蛋白质工程的集成序列与结构特征的深度学习模型及预测方法。
背景技术
蛋白质对生命活动的维持有重要作用。它们具有催化、结合和运输等多种功能,承载着细胞内大部分的代谢活动。自然界提供了大量具有实际应用潜力的蛋白。但这些蛋白往往不具备能满足生物工程需要的最优功能。定向进化是一项通过局部搜索来优化蛋白功能的技术。在这一过程中,通过迭代的过程进行选择和积累突变,也就是在每一代中测试成百上千种突变,以得到具有多个氨基酸点位突变的突变体。这项技术对蛋白序列空间的探索程度是非常有限的。因此定向进化技术需要高效的高通量筛选或者大量的实验测试,以获得所需功能更强大的突变体(特别是具有多个氨基酸突变的深度突变体),这种做法对实验技术和实验成本来说都是巨大的挑战。
由于实验筛选是定向进化的瓶颈,能在计算机上实现对突变(特别是高阶突变)影响的检测便变得十分重要。
而深度学习在蛋白质设计工程中已有广泛的应用。但目前的高精度算法对蛋白质实验数据量要求较大,且在缺乏实验数据的情况下对多点位突变影响的预测精度仍然不足。
发明内容
本发明要解决的是现有深度学习在对蛋白质从序列到功能的预测上对实验数据量需求量大且对于缺乏实验数据的多点位突变预测精度不足的技术问题,从而提供一种基于蛋白质工程的集成序列与结构特征的深度学习模型及预测方法。
为解决上述技术问题,本发明所采用的技术方案为:
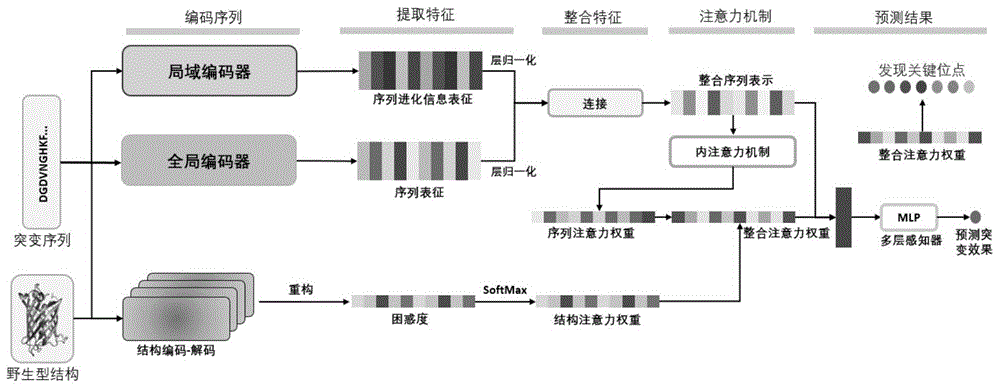
一种基于蛋白质工程的集成序列与结构特征的深度学习模型,包括局域编码器、全局编码器、结构编码器、注意力层、输出层;
所述局域编码器的输入为突变序列,局域编码器使用多重序列对比方法对突变序列编码输出编码了同族蛋白进化信息的张量I,张量I的尺寸为L*256。
在局域编码器中,突变序列通过Bi-LSTM层会转换成一个待训练的张量I
使用多重序列比对方法获得野生型序列的同源约束关系的张量I
将张量I
所述全局编码器的输入为突变序列,全局编码器使用蛋白质语言模型对突变序列进行编码输出编码了包含蛋白质共性生化特征和进化信息的张量II;张量II的尺寸为L*256;
所使用的蛋白质语言模型是已完成大规模训练的蛋白质语言模型,经过大量数据训练后(序列数据量超过千万量级),蛋白质语言模型会具备出色的提取特征能力;而且所使用的蛋白质语言模型是基于无监督学习的蛋白质语言模型;经过大量数据训练的蛋白质语言模型可以充分表征蛋白质的生物学特征和进化多样性,这些信息更多代表整个自然界所有的蛋白质序列的共性,并被编码到该模块输出的张量中,因为该蛋白质语言模型是无监督的,所以在本发明中直接使用即可,输入突变序列后即可得到编码后的张量II。
蛋白质语言模型会先将突变序列编码为向量,且编码过程会将氨基酸彼此之间的联系融入其中。该蛋白质语言模型是在拥有2.5亿条序列的UniRef50数据库上进行预训练,训练时蛋白质语言模型会根据部分氨基酸被遮掩的序列编码和完整序列编码的差异来调整参数。
所述结构编码器的输入为突变序列和野生型结构,结构编码器使用开源的无监督模型对突变序列折叠成野生型结构的概率评估,输出包含蛋白质结构信息的张量III,张量III为长度为L的一维输出向量,该张量III即为无监督模型的打分,该打分旨在评估该序列折叠成给定蛋白质野生型结构的能力,更高的分数意味着该突变序列包含的突变比其他的更有利。
在结构编码器中,对于野生型结构使用开源的esm-if1模型获得饱和单点突变评分矩阵,饱和单点突变评分矩阵的尺寸为L×20;
对突变序列进行one-hot编码,得到编码矩阵,编码矩阵的尺寸为L×20;
计算饱和单点突变评分矩阵和编码矩阵在每一个氨基酸位置的交叉熵,计算结果进行softmax后得到张量III,张量III中元素即为概率表征突变序列中对应位置的氨基酸是否为esm-if1模型认定的最优氨基酸。
所述注意力层的输入为代表蛋白质序列信息的张量IV,张量IV是张量I和张量II层归一化后拼合而成,张量I和张量II具有相同的尺寸,因此可以将直接对齐拼接并通过注意力机制变换为单个向量,张量IV的尺寸为L×512;
在注意力层中,张量IV在注意力机制下会得到序列注意力权重;
序列注意力权重的获得是深度学习中的注意力机制,编码序列信息的张量IV中每个氨基酸都会得到一个评估其与序列中其他氨基酸相关度大小的权重。该权重是由编码了蛋白质序列信息的张量IV和待学习参数矩阵计算得到的;
计算通式为:
其中,r
张量III在注意力机制下会得到结构注意力权重;
而结构注意力权重,则是编码了蛋白质结构信息的张量III和待学习参数矩阵一起构成结构注意力权重,被用于评估结构编码器的输出中每个氨基酸和其余氨基酸的关联程度;
结构注意力权重的计算通式为:
其中p’
将序列注意力权重和结构注意力权重的平均值作为联合注意力权重;
注意力层根据联合注意力权重和张量IV进行加权求和输出聚合向量,聚合向量的尺寸为1x512;
所述输出层的输入为聚合向量和无监督模型的打分,该无监督模型的打分即为张量III;
在输出层中,先对聚合向量采用ReLU函数处理,得到隐含向量;
根据隐含向量和无监督模型的打分使用Sigmoid函数计算动态权重,该动态权重表示在多大程度上信任无监督模型的打分;
使用线性层计算隐含向量的突变效果打分;
最后,将动态权重*突变效果打分与(1-动态权重)*无监督模型的打分之和作为输出层的输出。
本发明还提供了一种基于蛋白质工程的集成序列与结构特征的深度学习模型的预测方法,步骤为:
训练深度学习模型:
获取训练数据:
训练数据包括蛋白质的野生型序列、野生型结构、突变内容以及突变后评估蛋白质特质的数值打分;
突变内容是指野生型序列中哪个位置的氨基酸突变为哪一种氨基酸,一条突变数据并非;
划分训练集和测试集:
将训练数据中突变内容按8:2的比例划分为训练集和验证集;
训练模型:
野生型序列根据训练集中各突变内容生成突变序列集;
将突变序列集中的每个突变序列依次输入到深度学习模型中进行迭代训练;
每次迭代时:突变序列被局域编码器编码成具有同族蛋白进化信息的张量I;
突变序列被全局编码器编码成包含蛋白质共性生化特征和进化信息的张量II;
突变序列被结构编码器编码成包含蛋白质结构信息的张量III,张量III即为无监督模型的打分;
张量I和张量II分别层归一化后拼接成表征蛋白质序列信息的张量IV;
在注意力层中,张量IV在注意力机制下得到序列注意力权重;
张量III在注意力机制下得到结构注意力权重;
序列注意力权重和结构注意力权重的平均值作为联合注意力权重;
联合注意力权重和张量IV的加权求和得到的聚合向量就是注意力层的输出;
聚合向量输入到输出层,在输出层中,聚合向量先进行ReLU函数处理,得到隐含向量;
隐含向量和无监督模型的打分使用Sigmoid函数计算动态权重;
隐含向量在线性层中得到突变效果打分;
突变效果打分和无监督模型的打分在动态权重的分配下得到该突变序列的突变预测分值;
突变预测分值与对应的突变后评估蛋白质特质的数值打分计算损失函数,损失函数所采用的形式为均方根误差(MSE),并更新深度学习模型的参数,得到更新后的深度学习模型;
将突变序列集下一个突变序列输入到更新后的深度学习模型进行再训练,直至循环结束,得到训练后的深度学习模型;
使用验证集对训练后的深度学习模型进行验证得到验证后的深度学习模型;
目标突变预测:
给定目标突变内容,将目标突变序列输入到验证后的深度学习模型中得到突变预测分值。
作为本发明的一种优选方案,所述训练数据的获取方式跟蛋白质有关,对于具有充足实验数据的蛋白质,直接使用实验数据作为训练数据;
对于实验数据不足甚至缺失的蛋白质,则使用数据增强策略获得训练数据。
作为本发明的一种优选方案,所述的数据增强策略是选择无监督模型;
通过给定突变内容获得该蛋白质大量的低点位突变数据;
将各低点位突变数据使用无监督模型进行打分,得到突变后评估蛋白质特质的数值打分;
低点位突变数据和对应的突变后评估蛋白质特质的数值打分作为训练数据,
训练后获得的深度学习模型对高维度突变进行预测。
作为本发明的一种优选方案,对于实验数据缺失的蛋白质,无监督模型的选择根据蛋白质以及蛋白质特质凭经验选择;对于实验数据不足的蛋白质,先使用各无监督模型分别对已有实验数据中的所有突变进行预测,选择预测结果与实验结果的排序相关性最高的无监督模型。
作为本发明的一种优选方案,对于实验数据缺失的蛋白质,直接使用低点位突变数据和对应的突变后评估蛋白质特质的数值打分作为训练数据对深度学习模型进行训练,训练完成后对高维突变进行预测;
对于实验数据不足的蛋白质,使用低点位突变数据和对应的突变后评估蛋白质特质的数值打分作为训练数据对深度学习模型进行预训练;
然后使用具有的实验数据对预训练后的深度学习模型进行二次训练;
训练完成后对高维突变进行预测。
本发明先建立了整合序列和结构信息来预测蛋白质突变效果的深度学习模型。然后结合特定的数据增强策略,以减少深度学习模型对实验样本量的依赖程度。具体是大量来自无监督模型的低质量预测结果会首先被用来对深度学习模型进行预训练,之后对于有实验结果的则用有限数量的高品质实验结果会被用来对模型进行微调。实验表明当后续微调的实验数据量小于40或没有任何实验数据时,仅经过预训练得到的深度学习模型可以在预测高阶突变效果的任务上取得非常高的精度(特别是突变个数大于4的深度突变)。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明深度学习模型的架构图。
图2为本发明局域编码器的架构图。
图3为本发明结构编码器的架构图。
图4为本发明预测流程图。
图5为本发明GFP蛋白在不同体量实验数据下训练的模型的预测结果与实验结果的排序相关性对比图;其中,A-D图分别表示使用40,100,400,1084条单点位实验数据进行微调的模型对2-8点位突变效果的预测结果和实验结果的排序相关性。
图6为本发明无实验数据时使用无监督模型预测结果进行预训练的模型结果,其中,图A是无实验数据时预训练模型和ESM-IF1无监督模型对2-8点位突变效果的预测结果和实验结果的排序相关性;图B是无实验数据时预训练模型和ProGen2无监督模型对2-8点位突变效果的预测结果和实验结果的排序相关性。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例:
一种基于蛋白质工程的集成序列与结构特征的深度学习模型,如图1所示,包括局域编码器、全局编码器、结构编码器、注意力层、输出层;
所述局域编码器的输入为突变序列,局域编码器使用多重序列对比方法对突变序列编码输出编码了同族蛋白进化信息的张量I,张量I的尺寸为L*256。
在局域编码器中,
突变序列通过Bi-LSTM层会转换成一个待训练的张量I
使用多重序列比对方法获得野生型序列的同源约束关系的张量I
将张量I
因为目前许多已有模型会利用多重序列比对方法来提取进化过程中残基之间的约束关系,并将这类信息编码为指导突变预测的重要特征。本实施例中具体使用的原理架构图如图2所示;
该方法会先搜索给定蛋白的同源序列,然后利用HHsuite构建序列比对信息。在这之后,该方法使用一个统计模型来识别进化耦合,该模型通过使用马尔可夫随机场学习同源序列比对生成模型。在这个模型中,每个序列的概率取决于一个能量函数。
这个能量函数被定义为单点约束e
E(x)=∑
其中i和j是序列上残基的位置编号。
因此,序列中第i个氨基酸x
而这些耦合参数e
最终同源序列中每个氨基酸都会被表示成长度为L+1(L即序列长度)的向量,那么整个输入的同源序列就可以通过拼接每个氨基酸的向量被表示成L×(L+1)的矩阵。
由于每个氨基酸局部进化信息的表征长度和序列长度相近,其长为(L+1)的向量会通过全连接层被转化成固定长度为128的向量以避免过拟合问题,该向量就是张量I
突变序列会通过Bi-LSTM层(双向长短记忆网络层)并被转化成大小为L×128的矩阵来进行初始随机化,该矩阵就是张量I
将上述两个矩阵拼接得到的张量I(大小为L×256)即为该模块的输出结果。
所述全局编码器的输入为突变序列,全局编码器使用蛋白质语言模型对突变序列进行编码输出编码了包含蛋白质共性生化特征和进化信息的张量II;张量II的尺寸为L*256;
所使用的蛋白质语言模型是已完成大规模训练的蛋白质语言模型,经过大量数据训练后(序列数据量超过千万量级),蛋白质语言模型会具备出色的提取特征能力;而且所使用的蛋白质语言模型是基于无监督学习的蛋白质语言模型;经过大量数据训练的蛋白质语言模型可以充分表征蛋白质的生物学特征和进化多样性,这些信息更多代表整个自然界所有的蛋白质序列的共性,并被编码到该模块输出的张量中,因为该蛋白质语言模型是无监督的,所以在本发明中直接使用即可,输入突变序列后即可得到编码后的张量II。
蛋白质语言模型会先将突变序列编码为向量,且编码过程会将氨基酸彼此之间的联系融入其中。该蛋白质语言模型是在拥有2.5亿条序列的UniRef50数据库上进行预训练,训练时蛋白质语言模型会根据部分氨基酸被遮掩的序列编码和完整序列编码的差异来调整参数。
所述结构编码器的输入为突变序列和野生型结构,结构编码器使用开源的无监督模型对突变序列折叠成野生型结构的概率评估,输出包含蛋白质结构信息的张量III,张量III为长度为L的一维输出向量,该张量III即为无监督模型的打分,该打分旨在评估该序列折叠成给定蛋白质野生型结构的能力,更高的分数意味着该突变序列包含的突变比其他的更有利。
在结构编码器中,对于野生型结构使用开源的esm-if1模型获得饱和单点突变评分矩阵,饱和单点突变评分矩阵的尺寸为L×20;
对突变序列进行one-hot编码,得到编码矩阵,编码矩阵的尺寸为L×20;
计算饱和单点突变评分矩阵和编码矩阵在每一个氨基酸位置的交叉熵,计算结果进行softmax后得到张量III,张量III中元素即为概率表征突变序列中对应位置的氨基酸是否为esm-if1模型认定的最优氨基酸。
具体地,在打分时,本发明使用esm-if1模型,因为esm-if1模型会针对给定结构对给定序列折叠成该结构的概率进行评估。突变序列和野生型序列在此处的评估结果的比值,便是我们使用的打分。
esm-if1会对更有可能折叠成原始结构的序列给出更高的评价,那么如果突变序列在esm-if1模型中得分更高,说明该突变更有利于序列折叠成原始结构,这便是一个有益的突变,那么被用在我们模型中的打分就会更高。
具体来说,如图3所示,序列中每个位置上所有可能的单点突变都会有相应的分数(该分数即对应的突变序列的得分),其分布会构成一个尺寸为L×20的矩阵(L为序列长度),该分布是饱和单点突变评分矩阵。
然后,对突变序列进行one-hot编码,得到编码矩阵,编码矩阵的尺寸为L×20,每一行代表该位置的氨基酸的元素为1,其余为0;
计算饱和单点突变评分矩阵和编码矩阵在每一个氨基酸位置的交叉熵,计算结果进行softmax后得到张量III,张量III是长度为L的一维输出向量,张量III中元素即为概率表征突变序列中对应位置的氨基酸是否为esm-if1模型认定的最优氨基酸。
所述注意力层的输入为代表蛋白质序列信息的张量IV,张量IV是张量I和张量II层归一化后拼合而成,张量I和张量II具有相同的尺寸,因此可以将直接对齐拼接并通过注意力机制变换为单个向量,张量IV的尺寸为L×512;
在注意力层中,张量IV在注意力机制下会得到序列注意力权重;
序列注意力权重的获得是深度学习中的注意力机制,编码序列信息的张量IV中每个氨基酸都会得到一个评估其与序列中其他氨基酸相关度大小的权重。该权重是由编码了蛋白质序列信息的张量IV和待学习参数矩阵计算得到的;
计算通式为:
其中,r
张量III在注意力机制下会得到结构注意力权重;
而结构注意力权重,则是编码了蛋白质结构信息的张量III和待学习参数矩阵一起构成结构注意力权重,被用于评估结构编码器的输出中每个氨基酸和其余氨基酸的关联程度;
结构注意力权重的计算通式为:
其中p’
将序列注意力权重和结构注意力权重的平均值作为联合注意力权重;联合注意力权重的计算通式为:
w= 注意力层根据联合注意力权重和张量IV进行加权求和输出聚合向量,聚合向量的尺寸为1x512; 聚合向量的表征为: 所述输出层的输入为聚合向量和无监督模型的打分,该无监督模型的打分即为张量III; 在输出层中,先对聚合向量采用ReLU函数处理,得到隐含向量; 根据隐含向量和无监督模型的打分使用Sigmoid函数计算动态权重,该动态权重表示在多大程度上信任无监督模型的打分。此处的无监督模型的打分是结构编码器中提到的突变序列折叠成给定蛋白质野生型结构的能力,即张量III; 使用线性层计算隐含向量的突变效果打分; 最后,将动态权重*突变效果打分与(1-动态权重)*无监督模型的打分之和作为输出层的输出。 使用基于蛋白质工程的集成序列与结构特征的深度学习模型的预测方法,因为训练数据的获取方式跟蛋白质有关,对于具有充足实验数据的蛋白质,直接使用实验数据作为训练数据; 对于实验数据不足甚至缺失的蛋白质,则使用数据增强策略获得训练数据,。 如图4所示,所以下面将分成直接使用实验数据的预测方法以及使用数据增强策略的预测方法。 对于具有充足实验样本的蛋白质,步骤为: 训练深度学习模型: 获取训练数据: 训练数据直接是已有的实验数据,包括蛋白质的野生型序列、野生型结构、突变内容以及突变后评估蛋白质特质的数值打分; 突变内容是指野生型序列中哪个位置的氨基酸突变为哪一种氨基酸,一条实验数据不一定是单点突变,也可以是多个点位突变的集成。在突变发生后,新的蛋白质会在热稳定性或某个功能上表现出差异。实验会用特定数值来评估对这些突变体的某些性质,打分的高低表示此性质的强弱。 划分训练集和测试集: 将训练数据中突变内容按8:2的比例划分为训练集和验证集; 训练模型: 野生型序列根据训练集中各突变内容生成突变序列集; 将突变序列集中的每个突变序列依次输入到深度学习模型中进行迭代训练; 每次迭代时:突变序列被局域编码器编码成具有同族蛋白进化信息的张量I; 突变序列被全局编码器编码成包含蛋白质共性生化特征和进化信息的张量II; 突变序列被结构编码器编码成包含蛋白质结构信息的张量III,张量III即为无监督模型的打分; 张量I和张量II分别层归一化后拼接成表征蛋白质序列信息的张量IV; 在注意力层中,张量IV在注意力机制下得到序列注意力权重; 张量III在注意力机制下得到结构注意力权重; 序列注意力权重和结构注意力权重的平均值作为联合注意力权重; 联合注意力权重和张量IV的加权求和得到的聚合向量就是注意力层的输出; 聚合向量输入到输出层,在输出层中,聚合向量先进行ReLU函数处理,得到隐含向量; 隐含向量和无监督模型的打分使用Sigmoid函数计算动态权重; 隐含向量在线性层中得到突变效果打分; 突变效果打分和无监督模型的打分在动态权重的分配下得到该突变序列的突变预测分值; 突变预测分值与对应的突变后评估蛋白质特质的数值打分计算损失函数,损失函数所采用的形式为均方根误差(MSE),并更新深度学习模型的参数,得到更新后的深度学习模型; 将突变序列集下一个突变序列输入到更新后的深度学习模型进行再训练,直至循环结束,得到训练后的深度学习模型; 使用验证集对训练后的深度学习模型进行验证得到验证后的深度学习模型; 目标突变预测: 给定目标突变内容,将目标突变序列输入到验证后的深度学习模型中得到突变预测分值。 由于定向进化仅需要借助深度学习来指导突变点位的选择,模型预测结果的绝对值便没有太大意义。将所有目标突变的预测结果根据打分进行排序,靠前的突变即被模型认为有更高的可能让蛋白质产生有益的变化。变化的具体方向很大程度上取决于用于训练的实验数据中突变打分衡量的蛋白质性质。 对于使用数据增强策略的预测方法,步骤为: 对于实验数据不足甚至缺失的蛋白质,则使用数据增强策略获得训练数据。 而数据增强策略对于有少量实验数据和缺失实验数据两种情形。 对于有少量实验数据的情形; 对于无监督模型的选择,则是先使用各无监督模型分别对已有实验数据中的所有突变进行预测,选择预测结果与实验结果的排序相关性最高的无监督模型; 然后通过选择的无监督模型通过给定突变内容获得该蛋白质大量的低点位突变数据; 并对各低点位突变数据进行打分,得到突变后评估蛋白质特质的数值打分; 然后使用低点位突变数据和对应的突变后评估蛋白质特质的数值打分作为训练数据对深度学习模型进行预训练; 接着,使用具有的实验数据对预训练后的深度学习模型进行二次训练; 训练完成后对高维突变进行预测; 给定高维突变内容,并将高维突变序列输入到二次训练后的深度学习模型中得到预测结果。 对于实验数据缺失的蛋白质的情形,无监督模型的选择根据蛋白质以及蛋白质特质凭经验选择;通过给定突变内容获得该蛋白质大量的低点位突变数据; 将各低点位突变数据使用无监督模型进行打分,得到突变后评估蛋白质特质的数值打分; 低点位突变数据和对应的突变后评估蛋白质特质的数值打分作为训练数据;训练后获得的深度学习模型对高维度突变进行预测; 给定高维突变内容,并将高维突变序列输入到二次训练后的深度学习模型中得到预测结果。 本发明以GFP蛋白为例,进行了不同体量实验数据对模型的训练,并使用训练后的深度学习模型对2-8点位突变效果进行预测,结果如图5所示,其中,A-D图展示了在GFP蛋白上使用40,100,400,1084条单点位实验数据进行微调的模型对2-8点位突变效果的预测结果和实验结果的排序相关性,在每幅图中两个矩形条组中,右侧部分表示提前使用ESM-IF1无监督模型生成的数据进行预训练的预测结果与实验结果的排序相关性,左侧部分表示没有使用无监督模型生成数据仅使用实验数据进行训练的模型的预测结果与实验结果的排序相关性。 从图5中可以发现使用无监督模型产生的数据对模型进行预训练会让最终预测结果准确率大幅度提升,这种提升在实验数据较少时(数据量为40左右)更为明显。 同样,本发明还以GFP蛋白为例进行了无实验数据时使用无监督模型预测结果进行预训练的模型结果,如图6所示,A、B图展示了无实验数据时预训练模型和其使用的对应无监督模型对2-8点位突变效果的预测结果和实验结果的排序相关性。对于两个矩形条组中左侧部分为预训练模型的结果,右侧部分为无监督模型直接预测的结果。并且A图中使用的无监督模型为ESM-IF1,B图中使用的模型为ProGen2。 从图6中可看出当实验数据完全缺失时,仅使用无监督模型产生的结果进行训练的模型也可以达到较高的预测准确度。并且图6中A图和B图的差异证明选择不同的无监督模型会产生不同的结果。因此使用少量实验数据来筛选无监督模型对提升预测准确度有较大帮助,并且此筛选工作所需要的实验数据量远远小于其他监督模型所需要的数据量,是任何一个普通生化实验室都可以承担的。。 本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。 以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。